

Abstract
The gut hormone ghrelin is widely beneficial in many disease states. However, ghrelin exists in two distinctive isoforms, each with its own metabolic profile. In Parkinson's Disease (PD) acylated ghrelin administration is neuroprotective, however, the role of des‐acylated ghrelin remains unknown. In this study, we wanted to identify the relative contribution each isoform plays using the MPTP model of PD. Chronic administration of acylated ghrelin in mice lacking both isoforms of ghrelin (Ghrelin KO) attenuated the MPTP‐induced loss on tyrosine hydroxylase (TH) neuronal number and volume and TH protein expression in the nigrostriatal pathway. Moreover, acylated ghrelin reduced the increase in glial fibrillary acidic protein and Ionized calcium binding adaptor molecule 1 microglia in the substantia nigra. However, injection of acylated ghrelin also elevated plasma des‐acylated ghrelin, indicating in vivo deacetylation. Next, we chronically administered des‐acylated ghrelin to Ghrelin KO mice and observed no neuroprotective effects in terms of TH cell number, TH protein expression, glial fibrillary acidic protein and ionized calcium binding adaptor molecule 1 cell number. The lack of a protective effect was mirrored in ghrelin‐O‐acyltransferase KO mice, which lack the ability to acylate ghrelin and consequently these mice have chronically increased plasma des‐acyl ghrelin. Plasma corticosterone was elevated in ghrelin‐O‐acyltransferase KO mice and with des‐acylated ghrelin administration. Overall, our studies suggest that acylated ghrelin is the isoform responsible for in vivo neuroprotection and that pharmacological approaches preventing plasma conversion from acyl ghrelin to des‐acyl ghrelin may have clinical efficacy to help slow or prevent the debilitating effects of PD.
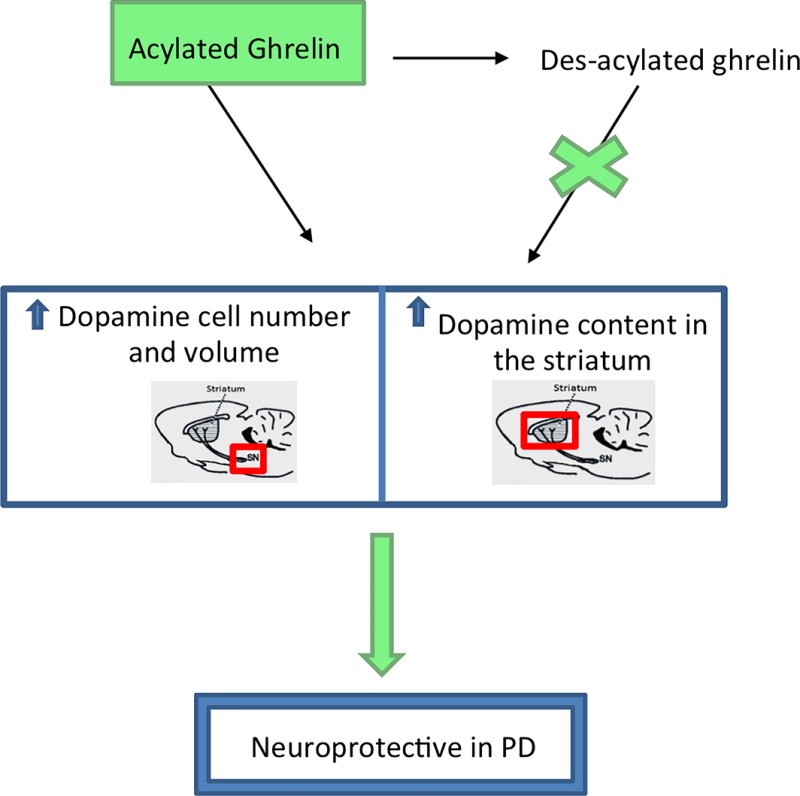
Ghrelin exists in the plasma as acyl and des‐acyl ghrelin. We determined the form responsible for in vivo neuroprotection in a mouse model of Parkinson's disease. Although exogenous acyl ghrelin is deacylated in situ to des‐acyl, only acyl ghrelin was neuroprotective by attenuating dopamine cell loss and glial activation. Acyl ghrelin is a therapeutic option to reduce Parkinson's Disease progression.
Cover Image for this issue: doi: 10.1111/jnc.13316.
Keywords: acylated ghrelin, corticosterone, des‐acylated ghrelin, dopamine, GOAT, Parkinson's disease
Abbreviations used
- DA
dopamine
- GFAP
glial fibrillary acidic protein
- GHSR1a
growth hormone secretagogue receptor 1a
- GLP1
glucagon‐like peptide 1
- GOAT
ghrelin‐O‐acyltransferase
- IBA1
ionized calcium binding adaptor molecule 1
- MPTP
1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine
- NEFA
non‐esterified fatty acid
- PD
Parkinson's disease
- PFA
paraformaldehyde
- SN
substantia nigra
- TH
tyrosine hydroxylase
Parkinson's disease (PD) is the second most common neurodegenerative disease characterized by rigidity, tremor and an overall lack of smooth motor control. These symptoms are because of a generalized lack of dopamine secreted from nerve terminals in the striatum. The cell bodies of these dopamine neurons are located in the substantia nigra (SN) and collectively this pathway is known as the nigrostriatal pathway. The cause of this debilitating disease remains unknown and for this reason treatment involves symptom management as opposed to halting disease progression.
In recent years, efforts have focussed on discovering endogenous neuroprotective mediators of SN dopamine neurons primarily because such treatments have the potential to reach clinical settings faster. An example of this is the gut hormone glucagon‐like peptide 1 (GLP1) that controls blood glucose. It was discovered that, in addition to regulating glucose homeostasis, GLP1 also targets receptors on SN dopamine neurons to prevent neurodegeneration in mouse models of PD (Bertilsson et al. 2008; Li et al. 2009). Since this initial discovery, the GLP1 agonist Exenatide has proven to be clinically effective in reducing disease progression (Aviles‐Olmos et al. 2013).
We have focussed on another gut hormone ghrelin, which also targets the brain to regulate metabolism and neuroprotection. Ghrelin is a hormone produced from the stomach and released into the bloodstream. It plays a role in maintaining body weight and adiposity. Importantly, ghrelin exists in the plasma in two biologically distinct states, acyl and des‐acyl ghrelin with des‐acyl ghrelin being the dominant form in the blood (Takagi et al. 2013). The enzyme ghrelin‐O‐acyltransferase (GOAT) acylates proghrelin in the endoplasmic reticulum after which a large proportion is rapidly converted to des‐acyl ghrelin in the plasma by acyl‐protein thioesterase 1 (Satou et al. 2010). GOAT is found in the gastrointestinal tract, predominantly in the stomach (Yang et al. 2008) where it is co‐localized with ghrelin producing cells (Sakata et al. 2009). Acylated ghrelin binds to the growth hormone secretagogue receptor 1a (GHSR) located in many different brain regions including the hypothalamus, hippocampus, substantia nigra, and olfactory bulb (Mani et al. 2014). However, des‐acyl ghrelin acts through an unknown receptor.
Plasma ghrelin levels are elevated just prior to meal initiation and a single injection of acylated ghrelin enhances food intake in both rodents and humans (Wren et al. 2000, 2001; Cummings et al. 2001). Ghrelin also has many non‐metabolic roles, such as reducing anxiety and stress (Spencer et al. 2012), enhancing learning and memory (Diano et al. 2006) and neuroprotection in PD (Andrews et al. 2009; Moon et al. 2009). Chronic administration of acylated ghrelin is protective in a mouse model of PD (Andrews et al. 2009) an effect mediated through the GHSR. Mice lacking both des‐acyl and acyl ghrelin (Ghrelin KO mice) showed enhanced neurodegeneration in a mouse model of PD (Andrews et al. 2009), although in this model it is impossible to ascribe the protective effect to acyl or des‐acyl ghrelin. Indeed, in normal mice chronic acylated ghrelin administration is neuroprotective (Andrews et al. 2009), however, as acylated ghrelin is readily deacetylated the protective effect could have been because of acylated ghrelin or des‐acyl ghrelin (that was deacetylated in situ). Thus, the question remains: is des‐acyl ghrelin neuroprotective in a mouse model of PD? This is an extremely important question that must be answered before ghrelin can be considered for clinical trials in PD.
There are numerous reports showing that des‐acyl ghrelin acts independently of both acyl ghrelin and of GHSR. For example, des‐acyl ghrelin administration prevents cell death in cultured neurons exposed to oxygen and glucose deprivation in the presence of a GHSR antagonist (Chung et al. 2008; Hwang et al. 2009). In vivo, des‐acyl ghrelin administration exhibits a vasodilator response (Ku et al. 2015) and both acylated and des‐acyl ghrelin are protective after transient focal ischaemia reperfusion (Hwang et al. 2009). In microglia exposed to amyloid beta, des‐acyl ghrelin counteracted the activation of the pro‐inflammatory cytokine IL‐6, whereas acylated ghrelin had no effect (Bulgarelli et al. 2009). We hypothesized that elevated des‐acyl ghrelin will prevent nigrostriatal degeneration in a mouse model of PD.
Methods
Animals
All experiments were approved by the Monash University Animal Ethics Committee and conducted in compliance with the Committee's guidelines. Mice were kept at standard laboratory conditions with free access to food and water at 23°C in a 12 h light/dark cycle unless otherwise stated. Male Ghrelin and GOAT KO mice (approximately 8–10 weeks old) on a C57/Bl6 background were attained from Regeneron Pharmaceuticals (Tarrytown, NY, USA) and bred in the Monash Animal Services.
Experimental protocol
GOAT KO mice, which contain naturally high circulating des‐acyl ghrelin levels but negligible acylated ghrelin as described previously (Zhao et al. 2010), were injected with 1‐methyl4‐phenyl‐1,2,3,6,‐tetrahydropyridine (MPTP) (30 mg/kg, i.p.) dissolved in saline over two consecutive days. Control mice received sterile saline. Animals were culled 7 days later and perfused for immunohistochemical analysis or fresh tissue collection for western blot or high‐performance liquid chromatography (HPLC) analysis.
Ghrelin KO mice that produce neither acylated nor des‐acylated ghrelin (Wortley et al. 2004, 2005) were used to examine the role each distinct form of ghrelin plays in PD progression when each isoform was returned. The use of Ghrelin KO mice allows us to directly assess the impact of exogenous acyl or des‐acyl ghrelin without endogenous acyl or des‐acyl ghrelin acting as a confounding variable. Mice were treated chronically (14 days) with des‐acyl ghrelin (1 mg/kg) or acylated ghrelin (1 mg/kg). This dose was chosen based upon previous research showing that acylated ghrelin elicits a feeding response (Briggs et al. 2013). To prevent mice from consuming excess calories during this process, they were injected during the light phase and food was removed. Six hours later food was reintroduced. Previous studies (Andrews et al. 2009) indicate that if calories are consumed after injection of acyl ghrelin then no neuroprotective effect is observed, presumably because the actions of ghrelin are negated by the consumption of food and the associated metabolic feedback of glucose and insulin. After 7 days of treatment mice received MPTP treatment (30 mg/kg, i.p.) or sterile saline over two consecutive days. Seven days after these injections mice were sacrificed (45 min after final injection) and perfused for immunohistochemistry or fresh tissue collection for western blot analysis.
Immunohisochemistry
All mice were deeply anaesthetized and perfused with 0.05% phosphate‐buffered saline followed by 4% paraformaldehyde. Brains were collected and post‐fixed overnight in paraformaldehyde then transferred into a 30% sucrose solution. After 48 h in sucrose 30 μm coronal sections of the entire substantia nigra were collected and stored in cryoprotectant (30% glycerol and 20% ethylene glycol in 0.1 M PB) at −20°C until staining was performed.
Before the primary antibody was added, the tissue was thoroughly washed using 0.1 M PB, and endogenous peroxidase activity was blocked with 1% H2O2 in 0.1 M PB for 15 min and washed again. The tissue was then blocked using 4% normal horse serum followed by a secondary mouse blocking step using AffiniPure goat anti‐mouse IgG (H+L) (1 : 200; Jackson Immuno‐Research, West Grove, PA, USA) to prevent non‐specific binding of mouse antibodies in mouse tissue. The sections were then incubated with the primary antibody, either anti‐tyrosine hydroxylase (TH) (mouse, 1 : 5000; Millipore Corporation, Bedford, MA, USA) and anti‐ionized calcium binding adaptor molecule 1 (IBA1) (rabbit, 1 : 1000; Wako Life Sciences, CA, USA) or anti‐TH (mouse, 1 : 5000; Millipore Corporation) and anti‐glial fibrillary acidic protein (GFAP) (rabbit, 1 : 1000; Dako, Carpinteria, CA, USA), diluted in 0.1 M PB + 0.3% Triton‐X + 4% normal horse serum and incubated at 4°C overnight. After this incubation the tissue was washed thoroughly and incubated in the corresponding secondary antibody 1 : 400 goat anti‐mouse IgG (H+L) Alexa Fluor 488 (Invitrogen, Carlsbad, CA, USA) and 1 : 400 goat anti‐rabbit IgG (H+L) Alexa Fluor 594 (Invitrogen) for fluorescent staining for 90 min at 22°C. The tissue was subsequently washed, mounted onto slides and cover slipped using anti‐fade media. Specificity of the primary antibody was confirmed with secondary antibody omission.
Stereological investigation of cell number and volume
We used design‐based stereology to quantify the number of microglia and astrocytes in the SN as well as the number (optical fractionator probe) and volume (nucleator probe) of the TH neurons. The cells were visualized using a Zeiss microscope (Jena, Germany) with a motorized stage and a MicroFibre digital camera connected to the computer and analysed using the StereoInvestigator software (MicroBrightField, Williston, VT, USA). The entire SN was cut at 30 μm (allowing for a 20 μm optical dissector within each section) with systematic sampling of every fifth section, the first sample set was chosen at random.
We estimated the total TH cells in the SN by counting a fraction of the total cells using randomly positioned counting frames controlled by StereoInvestigator. Guard zones, which account for section damage and prevent oversampling error, were set at 10% of the thickness of the section. The width (X) and height (Y) of the counting frame was 40.2 μm producing a counting frame area (XY) of 1616 μm2. The dissector height (Z) was 20 μm to create a dissector volume (XYZ) of 32320 μm3. To be included as within the counting frame, the cells had to touch the green inclusion border and not come into contact with the red exclusion border of the sampling grid. An acceptable cell estimation had a coefficient of error (CE; using the Gunderson method) less than or equal to 0.1 with a smoothness factor m = 1. This value is used to estimate sampling precision, which is independent of natural biological variance, whereby the closer the value is to 0 the less uncertainty there is in the estimate.
Concurrent to counting cell number we also measured cell volume using the well‐defined nucleator method. The nucleator generates orthogonal lines at the midpoint of the cell, whereby markers are placed at the intersection between these lines and the cell boundary. The cell was volume calculated by taking the third power of these measurements.
Western blot
Fresh tissue samples of the substantia nigra and striatum were processed for western blot analysis. Briefly, tissue was immersed in radioimmunoprecipitation buffer (50 mM Tris, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 1% Triton X 100) containing a proteinase inhibitor (Sigma, St Louis, MO, USA) and sonicated. The samples were subsequently centrifuged (10 000 rpm, 10 min, 4°C) to remove cellular debris and the supernatant collected. Protein concentration was measured using a bicinchoninic acid kit (Pierce, Rockford, IL, USA) according to kit instructions. The samples were then standardized and mixed with Laemmli buffer and boiled for 5 min. Samples were loaded onto 10% acrylamide gels, separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred to a polyvinylidene difluoride membrane. The blots were then blocked for 1 h in tris‐buffered saline solution, containing 0.1% Tween‐20 (TBST) and 5% bovine serum albumin, and incubated overnight at 4°C in TBST and 5% bovine serum albumin with the following antibodies: anti‐TH (mouse, 1 : 1000; Millipore Corporation) with anti‐β actin (rabbit, 1 : 1000; Abcam, Cambridge, UK) used as a control. After thorough washing in TBST the blots were incubated with their corresponding secondary antibodies: goat anti‐mouse IgG (Light Chain Specific) and goat anti‐rabbit IgG (H+L)‐horseradish peroxidase conjugate (1 : 10 000; Jackson Immuno‐Research). The blots were then visualized using the chemiluminescence method (ECL; Amersham Pharmacia Biotech, Piscataway, NJ, USA) using the acquisition and analysis ImageLab Software, version 4.1; Bio‐Rad Laboratories, Hercules, CA, USA.
High‐performance liquid chromatography
Using HPLC we identified, separated and quantified dopamine and 3,4‐Dihydroxyphenylacetic acid (DOPAC) in the striatum. Striatal tissue was rapidly dissected and snap frozen at approximately −70°C. The tissue was then sonicated in 0.4 mL cold 0.1 M perchloric acid containing an internal standard. The samples were centrifuged and dopamine, DOPAC and the internal standard in the supernatant were extracted on alumina at pH 8.4, eluted in 0.1 M perchloric acid, separated by reverse‐phase HPLC and detected using electrochemical detection. The concentration of dopamine and DOPAC was calculated in reference to the internal and external standards. Protein concentration was determined from the centrifuged pellet of each sample using the Lowry method. The concentrations of dopamine and DOPAC are expressed as ng/mg of protein present (mean ± SEM).
Plasma analysis
Trunk blood was collected at time of death during the light cycle (approximately 45 min after injection) and collected into EDTA tubes pre‐treated with pefabloc (SC Roche Applied Science, Mannheim, Germany) to achieve a concentration of 1 mg/mL. The blood was collected during the light cycle as corticosterone levels are cyclic in nature and levels will be lowest during the light phase (Malisch et al. 2008). This low baseline allowed us to accurately detect any minute differences that resulted from the injection of acylated/des‐acylated ghrelin. All samples were collected at the same time (approximately 1400). The blood was subsequently centrifuged and acidified using HCl (final concentration 0.05 N). The plasma was analysed for: Acylated ghrelin and Des‐acyl ghrelin Enzyme‐Linked Immunoassay Kits (Mitsubishi Chemical Medicine, Tokyo, Japan), non‐esterified fatty acid (NEFA) (Wako Life Sciences), Triglycerides (Roche, Basel, Switzerland/Hitachi, Indiana, USA), Corticosterone (Abnova, Taipei City, Taiwan) and Blood glucose (Sigma). All measurements were obtained following kit instructions.
Statistical analysis
All data are represented as mean ± standard error of the mean (SEM). Two‐way anova with a Bonferroni post hoc was used to determine statistical significance, unless otherwise stated in the Figure Legend. p < 0.05 was considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Results
Acylated ghrelin restricts TH loss after MPTP administration
To examine the neuroprotective properties of des‐acyl ghrelin, we first confirmed that acyl ghrelin attenuated SN dopamine (DA) cell loss in ghrelin KO mice. Administration of acylated ghrelin significantly elevated circulating plasma concentration for acylated (Fig. 1a) and des‐acylated (Fig. 1b) ghrelin, indicating in vivo deacetylation has occurred. There was no significant change in body weight as a result of chronic acylated ghrelin administration (Figure S1a). Administration of MPTP reduced body weight (Figure S1a) and elevated NEFA, Triglycerides and Corticosterone (Fig. 1c, d and r) in the plasma, as well as IBA1 and GFAP in the SN. Microglia (IBA1+ cells) and astrocytes (GFAP+ cells) are activated during cellular damage and are responsible for minimizing overall dopaminergic cell loss (Kohutnicka et al. 1998). A greater number of microglia and astrocytes present indicate a greater amount of cellular damage. Both microglia and astrocytes were significantly elevated post‐MPTP administration with a significant protective effect of acylated ghrelin on microglial numbers only, although there was a trend for acyl ghrelin to reduce GFAP positive astrocytes (Fig. 1g and h). This was concomitant with a significant reduction in the number and size of TH neurons (enzyme marker of dopamine neurons) in the SN post‐MPTP administration, which was significantly attenuated with chronic acylated ghrelin (Fig. 1e, f and k). Acylated ghrelin also attenuated the MPTP‐induced decrease in TH levels in the SN and Striatum (Fig. 1l–n). HPLC analysis of dopamine and DOPAC revealed a significant reduction with MPTP administration (Fig. 1o and p). Acylated ghrelin prevented the increase in the DOPAC:DA ratio observed after MPTP administration (Fig. 1q). Although these results collectively indicate that acylated ghrelin is neuroprotective in PD, data from Fig. 1(b) indicate that injection of acylated ghrelin significantly increases des‐acyl ghrelin. Therefore, it is reasonable to assume that some neuroprotection may have come from elevated des‐acyl ghrelin.
Figure 1.
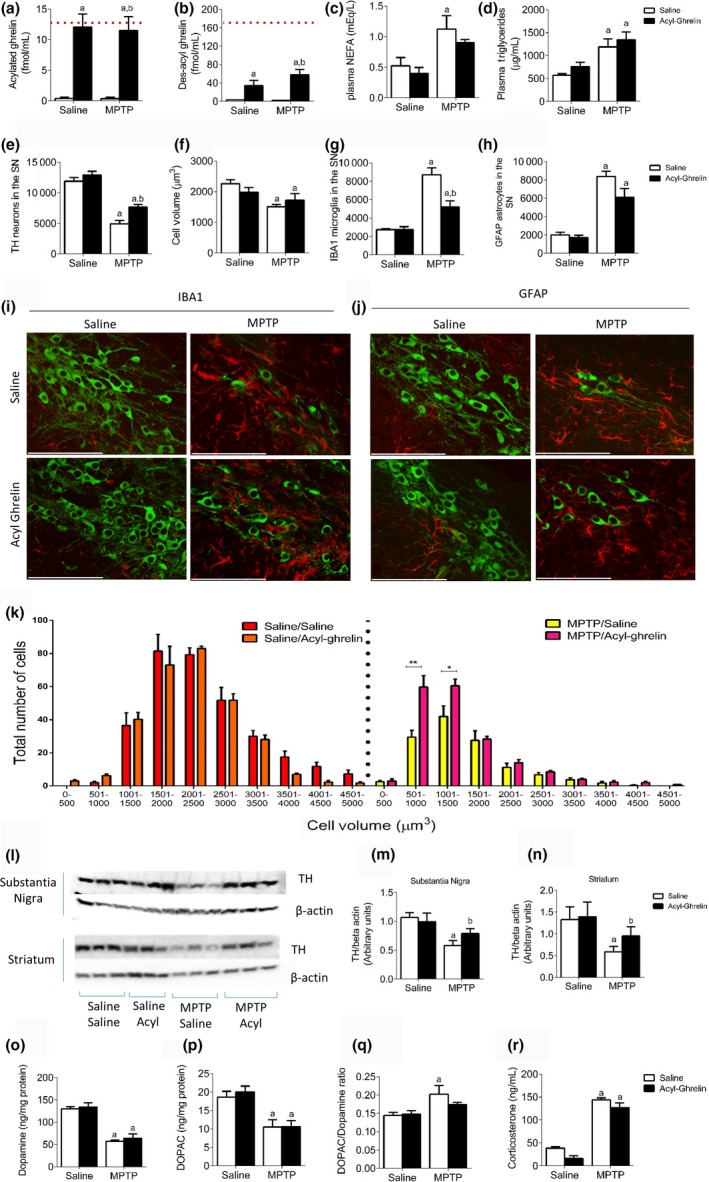
Neuroprotective effects in Ghrelin KO mice reinstated with acylated ghrelin. (a and b) Plasma analysis of acyl and des‐acyl ghrelin show an elevation in both acylated and des‐acyl ghrelin after acylated ghrelin administration. The red dotted line indicates average circulating levels of acyl and des‐acyl ghrelin in wild‐type mice. (c and d) Plasma non‐esterified fatty acid and Triglyceride levels are elevated post‐MPTP administration. (e) Stereological quantification of tyrosine hydroxylase (TH) neurons in the SN showing chronic acylated ghrelin is protective in Ghrelin KO mice. (f) Overall cell volume showed a significant reduction with MPTP regardless of injection. Stereological quantification of ionized calcium binding adaptor molecule 1 (IBA1) (g) and GFAP (h) shows elevated levels following MPTP, which is attenuated in mice treated with acylated ghrelin. (i + j) Representative images showing MPTP induced (i) microglial and (j) astrocyte activation in the SN (green = TH and red = (i) IBA1 or (j) GFAP). (k) When TH cells were separated and plotted based on number distribution, mice treated with MPTP and acylated ghrelin had a significant effect on smaller volume (500–1500 μmᵌ) cells compared to those not treated with acylated ghrelin. (l) Representative western blot images of TH and beta actin in the SN and Striatum. Quantification of TH levels in the SN (m) and Striatum (n) reveals an attenuated loss of TH in MPTP treated with MPTP and acylated ghrelin compared to MPTP alone. (o and p) MPTP significantly reduced both dopamine and DOPAC with no effect of acylated ghrelin. (q) Acylated ghrelin reduced the elevation of the DOPAC:dopamine ratio in MPTP‐treated mice compared saline alone. (r) Plasma corticosterone levels are significantly elevated in response to MPTP regardless of injection. a, significant compared to Saline/saline‐treated mice and b, significant compared to Saline/MPTP‐treated mice. *p < 0.05, **p < 0.01 compared to MPTP/Saline. Data are represented as mean ± SEM (n = 4–8, two‐way anova,). Scale bar = 100 μm.
Chronic des‐acyl administration is not neuroprotective in PD
To determine the relative neuroprotective potential of des‐acyl ghrelin in vivo, we chronically administered Ghrelin KO mice (lacking both acylated and des‐acylated ghrelin) with des‐acyl ghrelin. The use of Ghrelin KO mice allowed us to directly assess the impact of the exogenously administered des‐acyl ghrelin without confounding changes in the acyl to des‐acyl ghrelin ratio. Plasma analysis shows negligible acylated ghrelin levels and a higher than average circulating plasma des‐acyl concentration (Fig. 2a and b). This indicates that des‐acyl ghrelin cannot be converted back to its acylated counterpart and that any observed effects are solely because of elevated des‐acyl ghrelin. We analysed various metabolic markers in the plasma and found elevated NEFA (Fig. 2c) and Triglycerides (Fig. 2d) with no change in blood glucose (Figure S1d) in response to MPTP.
Figure 2.
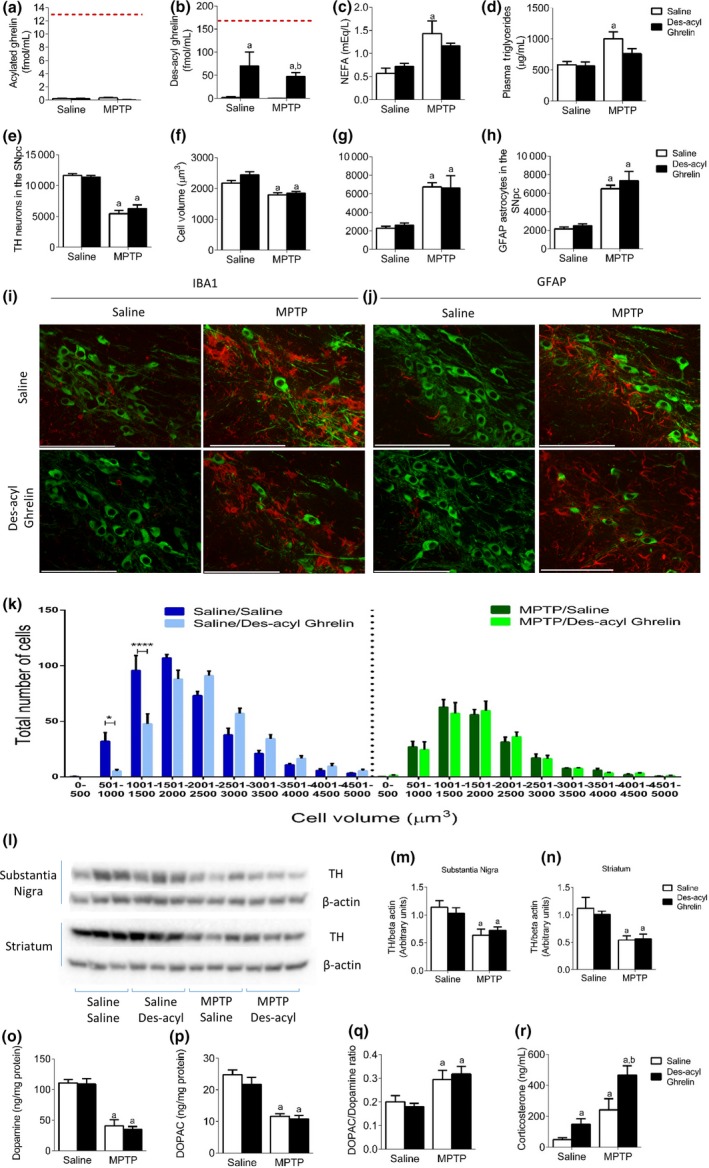
No neuroprotective action in Ghrelin KO mice re‐instated with des‐acyl ghrelin. (a and b) Plasma analysis of acyl and des‐acyl ghrelin show an elevation in des‐acyl ghrelin after injection and no change in acylated ghrelin levels. The red dotted line indicates average circulating levels of acyl and des‐acyl ghrelin in wild‐type mice. (c and d) Plasma non‐esterified fatty acid and Triglyceride levels are elevated post‐MPTP administration. (e) Stereological quantification of tyrosine hydroxylase (TH) neurons in the SN showing a significant reduction after MPTP administration but no effect with chronic des‐acyl ghrelin. (f) Overall cell volume showed a significant reduction with MPTP regardless of injection. Stereological quantification of ionized calcium binding adaptor molecule 1 (IBA1) (g) and GFAP (h) shows elevated levels following MPTP, with no difference between chronic saline and chronic des‐acyl ghrelin. (i + j) Representative images showing MPTP induced (i) microglial and (j) astrocyte activation in the SN (green = TH and red = (I) IBA1 or (j) GFAP). (k) TH cell distribution shows no difference comparing MPTP saline and MPTP des‐acyl ghrelin. (l) Representative western blot images of TH and beta actin in the SN and Striatum. Quantification of TH levels in the SN (m) and Striatum (n) reveals a significant loss of TH in MPTP‐treated mice with no effect of des‐acyl ghrelin. (o and p) MPTP significantly reduced both dopamine and DOPAC with no effect of des‐ acyl ghrelin. (q) MPTP treatment significantly elevated the DOPAC:dopamine ratio regardless of treatment. (r) Plasma corticosterone levels are significantly elevated in response to both des‐acylated ghrelin and MPTP with a cumulative effect when co‐administered. a, significant compared to Saline/saline‐treated mice and b, significant compared to Saline/MPTP‐treated mice. *p < 0.05, ****p < 0.0001 significant compared to Saline/Saline. Data are represented as mean ± SEM (n = 6, two‐way anova). Scale bar = 100 μm.
MPTP administration reduced dopamine cell number (Fig. 2e) and volume (Fig. 2f and k), and elevated IBA‐ and GFAP‐positive cells (Fig. 2g and h). However, there was no protective effect of des‐acyl ghrelin injection in terms of TH cell loss or gliosis response. TH expression in the SN and Striatum (Fig. 2l–n) were reduced post‐MPTP administration with no protective effect of des‐acyl ghrelin. HPLC analysis of dopamine and DOPAC revealed a significant reduction with MPTP administration with no effect of des‐acyl administration (Fig. 2o and p). Des‐acyl ghrelin did not alter the DOPAC:DA ratio (Fig. 2q). Plasma corticosterone was elevated in response to either des‐acyl ghrelin administration or MPTP (Fig. 2r).
GOAT mice show no differences in response to MPTP
As shown above des‐acylated ghrelin did not attenuate SN TH cell number or cell volume loss in a mouse model of PD. To strengthen these observations, we employed a genetic mouse model that exhibits increased endogenous des‐acyl ghrelin and no acylated ghrelin throughout the animal's entire lifespan. Mice lacking the enzyme GOAT have high levels of des‐acyl ghrelin and negligible acylated ghrelin (Fig. 3a and b) and thereby provide an ideal model to test the role of endogenously high des‐acyl ghrelin. We also analysed various metabolic markers in the plasma and found elevated NEFA (Fig. 3c) and Triglycerides (Fig. 3d) with no change in blood glucose (Figure S1f) in response to MPTP. MPTP administration significantly reduced the number and size of TH neurons (Fig. 3e, f and k) concurrent with elevated GFAP and IBA1 cells in both GOAT WT and KO mice, with no overall effect of genotype (Fig 3g and h). These results were mirrored in the analysis of TH protein levels in both the SN and Striatum (Fig. 3l–n). HPLC analysis of dopamine and DOPAC revealed a significant reduction with MPTP administration with no effect of genotype (Fig. 3o and p). Genotype did not alter the DOPAC:DA ratio (Fig. 3q). Interestingly, corticosterone levels were elevated in GOAT KO mice and with MPTP indicating a stress response to the elevated levels of des‐acyl ghrelin (Fig 3r).
Figure 3.
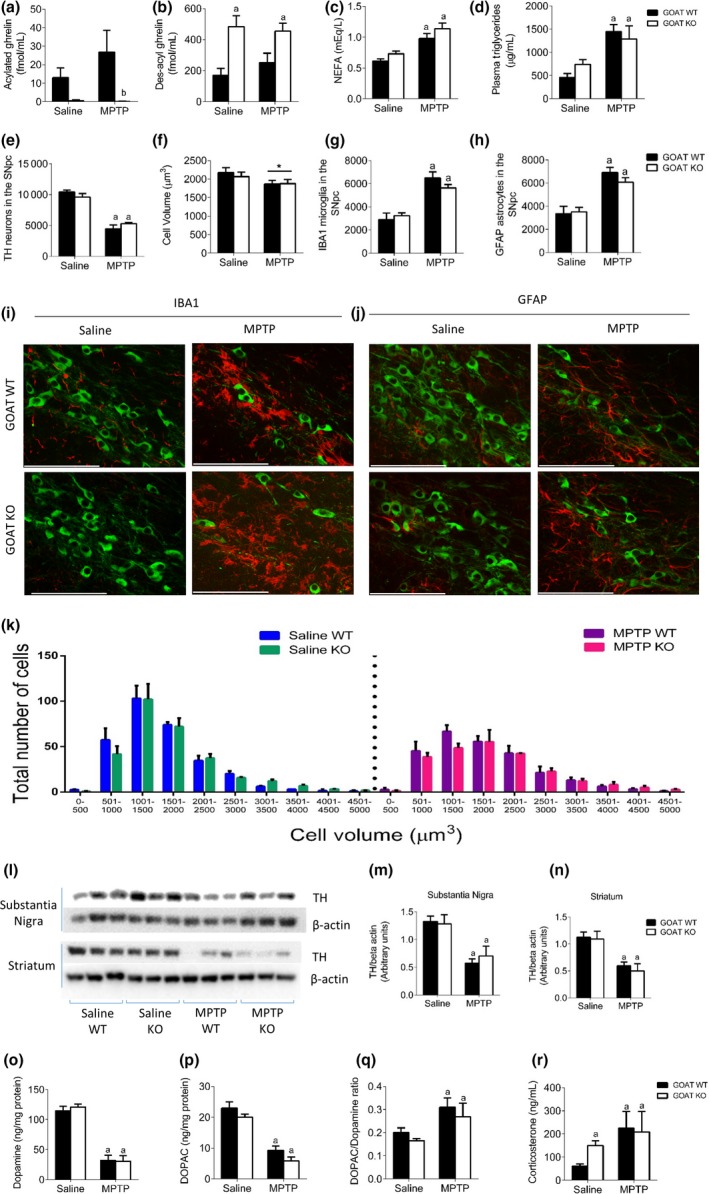
No significant difference between ghrelin‐O‐acyltransferase (GOAT) WT and KO mice after MPTP exposure. (a and b) Plasma analysis of acyl and des‐acyl ghrelin show an elevation in des‐acyl ghrelin with negligible acylated ghrelin in GOAT KO mice. Both acylated and des‐acylated ghrelin are present in GOAT WT mice. (c and d) Plasma non‐esterified fatty acid and Triglyceride levels are elevated post‐MPTP administration. (e) Stereological quantification of tyrosine hydroxylase (TH) neurons in the SN showing no effect of genotype in response to MPTP. (f) Overall cell volume showed a significant reduction with MPTP regardless of genotype. Stereological quantification of ionized calcium binding adaptor molecule 1 (IBA1) (g) and GFAP (h) shows elevated levels following MPTP, independent of genotype. (i + j) Representative images showing MPTP induced (i) microglial and (j) astrocyte activation in the SN (green = TH and red = (i) IBA1 or (j) GFAP). (k) TH cell distribution was not different between genotypes. (l) Representative western blot images of TH and beta actin in the SN and Striatum. Quantification of TH levels in the SN (m) and Striatum (n) reveals a significant loss of TH post‐MPTP administration with no significant effect of genotype. (o and p) MPTP significantly reduced both dopamine and DOPAC with no effect of genotype. (q) MPTP treatment significantly elevated the DOPAC:dopamine ratio regardless of genotype. (r) Plasma corticosterone levels are significantly elevated in response to MPTP and also in saline‐treated GOAT KO mice. a, significant compared to GOAT WT saline‐treated mice and b, significant compared to GOAT WT MPTP‐treated mice. Data are represented as mean ± SEM (n = 6–7, two‐way anova). Scale bar = 100 μm.
Discussion
Recently, many studies have investigated the beneficial effects of ghrelin administration to combat a multitude of diseases including stroke (Liu et al. 2009), epilepsy (Lucchi et al. 2013), encephalomyelitis (Theil et al. 2009), and Alzheimer's disease (Dhurandhar et al. 2013). However, these studies either focused exclusively on the beneficial effects of acylated ghrelin or simply did not consider des‐acyl ghrelin as a potential protective agent. Ghrelin exists in two biologically distinct forms (acylated and des‐acylated ghrelin) and research into both of these isoforms is necessary before appropriate therapeutics can be generated. In humans des‐acyl ghrelin is the most abundant form where circulating ghrelin is composed of > 90% des‐acyl ghrelin and less than 10% acylated ghrelin (Patterson et al. 2005). This ratio can be explained by the comparatively shorter half‐life of acylated ghrelin (Akamizu et al. 2005) and active deacetylation of acyl ghrelin into des‐acyl ghrelin (Tong et al. 2013). Because acylated ghrelin can be converted into des‐acyl ghrelin in the blood, there is the possibility that many physiological actions of acylated ghrelin are in fact caused by elevated des‐acyl ghrelin.
In this study we wanted to look at the relative contributions of each isoform. To do this, we chronically injected acyl or des‐acyl ghrelin into Ghrelin KO mice. These mice have negligible levels of both acylated and des‐acylated ghrelin for their entire lifespan and by re‐introducing exclusively one isoform we can determine the effects of one without the interference of the other. The results from chronic injections of acylated ghrelin were consistent with previous literature showing the neuroprotective properties of acyl ghrelin post‐MPTP administration (Jiang et al. 2008; Andrews et al. 2009; Moon et al. 2009). However, when we measured both acyl and des‐acyl ghrelin in the plasma there was an elevation in both acylated and des‐acylated ghrelin. Hence, the neuroprotective properties could be because of either acylated or des‐acylated ghrelin. However, neuroprotective properties were not seen in mice that were chronically treated with des‐acyl ghrelin, indicating that the beneficial effects are solely because of acylated ghrelin. To definitively show that des‐acyl ghrelin is not neuroprotective, we used a genetic approach involving GOAT knockout mice. GOAT acylates ghrelin, and thus GOAT KO mice have negligible acylated ghrelin and elevated des‐acyl ghrelin throughout their entire lifespan. This allows us to analyse the physiological consequences of a deficiency of ghrelin acylation. Indeed, there was no neuroprotective effect between genotypes in these mice, confirming that des‐acyl ghrelin is not neuroprotective in PD.
Interestingly, des‐acyl ghrelin can be neuroprotective in in vitro settings. Administration of des‐acyl ghrelin prevents cell death in cultured neurons during oxygen and glucose deprivation (Chung et al. 2008; Hwang et al. 2009). Des‐acyl ghrelin also counteracted the activation of pro‐inflammatory cytokine IL‐6 in microglia exposed to amyloid beta (Bulgarelli et al. 2009). In vivo studies show a mixed response to des‐acyl ghrelin. Recently des‐acyl ghrelin, but not acylated ghrelin, has elicited vasodilatory actions in the cerebral endothelium in a model of stroke. This effect was independent of GHSR, potentially acting through a novel des‐acyl ghrelin receptor in cerebral arteries (Ku et al. 2015). In another study using GHSR KO mice, des‐acyl ghrelin increased the pilocarpine‐evoked seizure threshold, thus acting as an anticonvulsant, in WT but not KO mice (Portelli et al. 2015). In a study examining the role of each isoform during myocardial injury, there was a protective therapeutic action of acylated ghrelin that was significantly weaker than des‐acyl ghrelin (Li et al. 2006). This raises the possibility that des‐acyl ghrelin may be protective in some diseases/treatments but not in others. This might reflect differences in expression of the unidentified des‐acyl ghrelin receptor, although this requires experimental proof. However, our studies show that in an in vivo setting des‐acyl ghrelin does not have neuroprotective effects in a mouse model of PD.
Our data show that chronic des‐acylated ghrelin administration elevates the stress hormone, corticosterone. Both age and stress play a role in the development of PD. During ageing cortisol levels are elevated (Deuschle et al. 1998), and this increase has been linked with memory deficits in the elderly (Lupien et al. 1998). Interestingly, cortisol is elevated in PD patients compared with healthy age‐matched controls (Hartmann et al. 1997). Either chronic stress or corticosterone administration have detrimental effects on motor control during the reaching test in animals (Metz et al. 2005), indicating an association between corticosterone and abnormal motor behaviour. In animal models of depression, chronic stress enhances the pro‐inflammatory environment (You et al. 2011). Acylated ghrelin has also been linked to the stress axis. Intraperitoneal injection of acylated ghrelin produces a significant dose‐dependent increase in serum corticosterone levels 1 h after injection (Asakawa et al. 2001). In mice that received chronic immobilization stress there was a stress‐related increase in plasma corticosterone and acylated ghrelin levels (Meyer et al. 2014), implying that acylated ghrelin via the actions of corticosterone influence the stress axis. Ghrelin KO mice are more anxious after acute stress, an effect reversed with administration of acyl ghrelin (Spencer et al. 2012). In these studies, we did not see any effect of chronic acyl ghrelin on plasma corticosterone, as was observed in the studies cited above, in response to MPTP. We believe that this discrepancy could be because of the lack of an acute stress event in these studies or because of the time frame of plasma collection after injection. Comparsion to saline controls reveals that plasma corticosterone was already significantly higher in the MPTP‐treated group, which may have also limited the ability of acyl ghrelin to further significantly increase corticosterone.
Moreover, these studies do not consider des‐acyl ghrelin and since acylated ghrelin is readily deacetylated, the effects in the above studies could be at least partly because of des‐acyl ghrelin. Indeed mice receiving chronic des‐acyl ghrelin, but not acylated ghrelin, had an elevated corticosterone response, which was further exacerbated when the stressor MPTP was administered. Stress‐induced corticosterone release is detrimental to the nigrostriatal system by increasing oxidative stress products (Kim et al. 2005) as well as reducing striatal dopamine levels (Ahmad et al. 2010) making the system more susceptible to degeneration. Collectively our results suggest that elevated levels of des‐acyl ghrelin may negate any neuroprotective effect of des‐acyl ghrelin on dopaminergic neurons by the elevation of the stress hormone corticosterone. Indeed, this may explain differences between in vivo and in vitro studies examining the neuroprotective treatment of des‐acyl ghrelin (Chung et al. 2008; Bulgarelli et al. 2009; Hwang et al. 2009). For example the lack of a physiological response in vitro, which would include increased corticosterone, to des‐acyl ghrelin treatment may allow experimentally observed in vitro neuroprotection. There are certainly other possibilities that could explain the lack of a neuroprotective effect of des‐acyl. For example studies show that the ratio of ghrelin peptides can influence the outcome on energy homeostasis and body composition (Epelbaum et al. 2010). Also, des‐acyl ghrelin has no effect on microglia or GFAP cell number in this study, which will also contribute to the lack of effect observed. Inflammation has been researched intensively in PD. Perhaps most relevant, chronic non‐steroidal anti‐inflammatory (NSAID) use (including ibuprofen, indomethacin, naproxen and diflunisal) reduces the risk for the development of PD in a retrospective study (Chen et al. 2003). In our study MPTP‐induced neurotoxicity increased the number of microglia and astrocytes in the nigrostriatal pathway, with a significant protective effect of chronic acylated ghrelin, but not des‐acyl administration. This result directly implies that exogenous acyl ghrelin directly reduces gliosis in a mouse model of PD, contributing to the neuroprotective effect. Astrocytes and microglia release deleterious cytokines, which results in oxidative damage to proteins, DNA and lipids in addition to impairing dopaminergic neurons in the nigrostriatal pathway. Acylated ghrelin has been shown in many studies to act as an anti‐inflammatory agent. Acylated ghrelin inhibits the release of pro‐inflammatory cytokines IL‐1β, IL‐6 and Tumour necrosis factor alpha after Lipopolysaccharide administration in peripheral macrophages (Dixit et al. 2004) and T‐cells (Waseem et al. 2008), and also reduces the pro‐inflammatory responses and NFκB activation in endothelial cells (Li et al. 2004). Exogenous administration of acylated ghrelin in rodents reduces inflammation in Alzheimer's Disease (Dhurandhar et al. 2013), colitis (Gonzalez‐Rey et al. 2006), and in mouse dopaminergic neurons acyl ghrelin attenuates Lipopolysaccharide‐induced release of IL‐6 (Beynon et al. 2013). This is clinically relevant as inflammation is documented in PD patients in post‐mortem samples (Ouchi et al. 2005) and in vivo PET imaging studies (Ikawa et al. 2011).
Overall, we show that acylated ghrelin is an ideal therapeutic target to reduce PD progression by reducing the gliosis response and attenuating dopaminergic cell loss in the nigrostriatal dopaminergic system. Des‐acyl ghrelin, on the other hand, was not neuroprotective and also induced a heightened stress response by elevating corticosterone levels, which may negate any neuroprotective potential. Furthermore, our studies suggest that pharmacological approaches preventing plasma conversion from acyl ghrelin to des‐acyl ghrelin may have clinical efficacy to help slow or prevent the debilitating effects of PD.
Author contributions
J.A.B and Z.B.A designed experiments. J.A.B, V.V.S, M.D, J.E and M.B.L, performed experiments. J.A.B and Z.B.A wrote the manuscript.
Supporting information
Figure S1. Body weight and blood glucose graphs in Ghrelin KO mice reinstated with acylated or des‐acylated ghrelin and GOAT WT/KO mice.
Acknowledgements and conflict of interest disclosure
This work was supported by grants and fellowships from the Australian National Health and Medical Research Council to Z.B.A (546131, 1084344) and NIH NS056181 to J.E. The authors declare that there are no conflicts of interest.
All experiments were conducted in compliance with the ARRIVE guidelines.
References
- Ahmad A., Rasheed N., Banu N. and Palit G. (2010) Alterations in monoamine levels and oxidative systems in frontal cortex, striatum, and hippocampus of the rat brain during chronic unpredictable stress. Stress 13, 355–364. [DOI] [PubMed] [Google Scholar]
- Akamizu T., Shinomiya T., Irako T., Fukunaga M., Nakai Y., Nakai Y. and Kangawa K. (2005) Separate measurement of plasma levels of acylated and desacyl ghrelin in healthy subjects using a new direct ELISA assay. J. Clin. Endocrinol. Metab. 90, 6–9. [DOI] [PubMed] [Google Scholar]
- Andrews Z. B., Erion D., Beiler R. et al (2009) Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2‐dependent mitochondrial mechanism. J. Neurosci. 29, 14057–14065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakawa A., Inui A., Kaga T. et al (2001) A role of ghrelin in neuroendocrine and behavioral responses to stress in mice. Neuroendocrinology 74, 143–147. [DOI] [PubMed] [Google Scholar]
- Aviles‐Olmos I., Dickson J., Kefalopoulou Z. et al (2013) Exenatide and the treatment of patients with Parkinson's disease. J. Clin. Investig. 123, 2730–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertilsson G., Patrone C., Zachrisson O. et al (2008) Peptide hormone exendin‐4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson's disease. J. Neurosci. Res. 86, 326–338. [DOI] [PubMed] [Google Scholar]
- Beynon A. L., Brown M. R., Wright R., Rees M. I., Sheldon I. M. and Davies J. S. (2013) Ghrelin inhibits LPS‐induced release of IL‐6 from mouse dopaminergic neurones. J. Neuroinflammation 10, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs D. I., Lockie S. H., Wu Q., Lemus M. B., Stark R. and Andrews Z. B. (2013) Calorie‐restricted weight loss reverses high‐fat diet‐induced ghrelin resistance, which contributes to rebound weight gain in a ghrelin‐dependent manner. Endocrinology 154, 709–717. [DOI] [PubMed] [Google Scholar]
- Bulgarelli I., Tamiazzo L., Bresciani E., Rapetti D., Caporali S., Lattuada D., Locatelli V. and Torsello A. (2009) Desacyl‐ghrelin and synthetic GH‐secretagogues modulate the production of inflammatory cytokines in mouse microglia cells stimulated by beta‐amyloid fibrils. J. Neurosci. Res. 87, 2718–2727. [DOI] [PubMed] [Google Scholar]
- Chen H., Zhang S. M., Hernan M. A., Schwarzschild M. A., Willett W. C., Colditz G. A., Speizer F. E. and Ascherio A. (2003) Nonsteroidal anti‐inflammatory drugs and the risk of Parkinson disease. Arch. Neurol. 60, 1059–1064. [DOI] [PubMed] [Google Scholar]
- Chung H., Seo S., Moon M. and Park S. (2008) Phosphatidylinositol‐3‐kinase/Akt/glycogen synthase kinase‐3 beta and ERK1/2 pathways mediate protective effects of acylated and unacylated ghrelin against oxygen‐glucose deprivation‐induced apoptosis in primary rat cortical neuronal cells. J. Endocrinol. 198, 511–521. [DOI] [PubMed] [Google Scholar]
- Cummings D. E., Purnell J. Q., Frayo R. S., Schmidova K., Wisse B. E. and Weigle D. S. (2001) A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 50, 1714–1719. [DOI] [PubMed] [Google Scholar]
- Deuschle M., Weber B., Colla M., Depner M. and Heuser I. (1998) Effects of major depression, aging and gender upon calculated diurnal free plasma cortisol concentrations: a re‐evaluation study. Stress 2, 281–287. [DOI] [PubMed] [Google Scholar]
- Dhurandhar E. J., Allison D. B., van Groen T. and Kadish I. (2013) Hunger in the absence of caloric restriction improves cognition and attenuates Alzheimer's disease pathology in a mouse model. PLoS ONE 8, e60437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diano S., Farr S. A., Benoit S. C. et al (2006) Ghrelin controls hippocampal spine synapse density and memory performance. Nat. Neurosci. 9, 381–388. [DOI] [PubMed] [Google Scholar]
- Dixit V. D., Schaffer E. M., Pyle R. S., Collins G. D., Sakthivel S. K., Palaniappan R., Lillard J. W., Jr and Taub D. D. (2004) Ghrelin inhibits leptin‐ and activation‐induced proinflammatory cytokine expression by human monocytes and T cells. J. Clin. Investig. 114, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epelbaum J., Bedjaoui N., Dardennes R. et al (2010) Role of the ghrelin/obestatin balance in the regulation of neuroendocrine circuits controlling body composition and energy homeostasis. Mol. Cell. Endocrinol. 314, 244–247. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Rey E., Chorny A. and Delgado M. (2006) Therapeutic action of ghrelin in a mouse model of colitis. Gastroenterology 130, 1707–1720. [DOI] [PubMed] [Google Scholar]
- Hartmann A., Veldhuis J. D., Deuschle M., Standhardt H. and Heuser I. (1997) Twenty‐four hour cortisol release profiles in patients with Alzheimer's and Parkinson's disease compared to normal controls: ultradian secretory pulsatility and diurnal variation. Neurobiol. Aging 18, 285–289. [DOI] [PubMed] [Google Scholar]
- Hwang S., Moon M., Kim S., Hwang L., Ahn K. J. and Park S. (2009) Neuroprotective effect of ghrelin is associated with decreased expression of prostate apoptosis response‐4. Endocr. J. 56, 609–617. [DOI] [PubMed] [Google Scholar]
- Ikawa M., Okazawa H., Kudo T., Kuriyama M., Fujibayashi Y. and Yoneda M. (2011) Evaluation of striatal oxidative stress in patients with Parkinson's disease using [62Cu]ATSM PET. Nucl. Med. Biol. 38, 945–951. [DOI] [PubMed] [Google Scholar]
- Jiang H., Li L. J., Wang J. and Xie J. X. (2008) Ghrelin antagonizes MPTP‐induced neurotoxicity to the dopaminergic neurons in mouse substantia nigra. Exp. Neurol. 212, 532–537. [DOI] [PubMed] [Google Scholar]
- Kim S. T., Choi J. H., Chang J. W., Kim S. W. and Hwang O. (2005) Immobilization stress causes increases in tetrahydrobiopterin, dopamine, and neuromelanin and oxidative damage in the nigrostriatal system. J. Neurochem. 95, 89–98. [DOI] [PubMed] [Google Scholar]
- Kohutnicka M., Lewandowska E., Kurkowska‐Jastrzebska I., Czlonkowski A. and Czlonkowska A. (1998) Microglial and astrocytic involvement in a murine model of Parkinson's disease induced by 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP). Immunopharmacology 39, 167–180. [DOI] [PubMed] [Google Scholar]
- Ku J. M., Andrews Z. B., Barsby T. et al (2015) Ghrelin‐related peptides exert protective effects in the cerebral circulation of male mice through a nonclassical ghrelin receptor(s). Endocrinology 156, 280–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. G., Gavrila D., Liu X. et al (2004) Ghrelin inhibits proinflammatory responses and nuclear factor‐kappaB activation in human endothelial cells. Circulation 109, 2221–2226. [DOI] [PubMed] [Google Scholar]
- Li L., Zhang L. K., Pang Y. Z., Pan C. S., Qi Y. F., Chen L., Wang X., Tang C. S. and Zhang J. (2006) Cardioprotective effects of ghrelin and des‐octanoyl ghrelin on myocardial injury induced by isoproterenol in rats. Acta Pharmacol. Sin. 27, 527–535. [DOI] [PubMed] [Google Scholar]
- Li Y., Perry T., Kindy M. S. et al (2009) GLP‐1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc. Natl Acad. Sci. USA 106, 1285–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Chen L., Xu X., Vicaut E. and Sercombe R. (2009) Both ischemic preconditioning and ghrelin administration protect hippocampus from ischemia/reperfusion and upregulate uncoupling protein‐2. BMC Physiol. 9, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchi C., Curia G., Vinet J., Gualtieri F., Bresciani E., Locatelli V., Torsello A. and Biagini G. (2013) Protective but not anticonvulsant effects of ghrelin and JMV‐1843 in the pilocarpine model of Status epilepticus. PLoS ONE 8, e72716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien S. J., de Leon M., de Santi S. et al (1998) Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat. Neurosci. 1, 69–73. [DOI] [PubMed] [Google Scholar]
- Malisch J. L., Breuner C. W., Gomes F. R., Chappell M. A. and Garland T., Jr (2008) Circadian pattern of total and free corticosterone concentrations, corticosteroid‐binding globulin, and physical activity in mice selectively bred for high voluntary wheel‐running behavior. Gen. Comp. Endocrinol. 156, 210–217. [DOI] [PubMed] [Google Scholar]
- Mani B. K., Walker A. K., Lopez Soto E. J., Raingo J., Lee C. E., Perello M., Andrews Z. B. and Zigman J. M. (2014) Neuroanatomical characterization of a growth hormone secretagogue receptor‐green fluorescent protein reporter mouse. J. Comp. Neurol. 522, 3644–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz G. A., Jadavji N. M. and Smith L. K. (2005) Modulation of motor function by stress: a novel concept of the effects of stress and corticosterone on behavior. Eur. J. Neurosci. 22, 1190–1200. [DOI] [PubMed] [Google Scholar]
- Meyer R. M., Burgos‐Robles A., Liu E., Correia S. S. and Goosens K. A. (2014) A ghrelin‐growth hormone axis drives stress‐induced vulnerability to enhanced fear. Mol. Psychiatry 19, 1284–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon M., Kim H. G., Hwang L. et al (2009) Neuroprotective effect of ghrelin in the 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine mouse model of Parkinson's disease by blocking microglial activation. Neurotox. Res. 15, 332–347. [DOI] [PubMed] [Google Scholar]
- Ouchi Y., Yoshikawa E., Sekine Y., Futatsubashi M., Kanno T., Ogusu T. and Torizuka T. (2005) Microglial activation and dopamine terminal loss in early Parkinson's disease. Ann. Neurol. 57, 168–175. [DOI] [PubMed] [Google Scholar]
- Patterson M., Murphy K. G., le Roux C. W., Ghatei M. A. and Bloom S. R. (2005) Characterization of ghrelin‐like immunoreactivity in human plasma. J. Clin. Endocrinol. Metab. 90, 2205–2211. [DOI] [PubMed] [Google Scholar]
- Portelli J., Coppens J., Demuyser T. and Smolders I. (2015) Des‐acyl ghrelin attenuates pilocarpine‐induced limbic seizures via the ghrelin receptor and not the orexin pathway. Neuropeptides 51, 1–7. [DOI] [PubMed] [Google Scholar]
- Sakata I., Yang J., Lee C. E., Osborne‐Lawrence S., Rovinsky S. A., Elmquist J. K. and Zigman J. M. (2009) Colocalization of ghrelin O‐acyltransferase and ghrelin in gastric mucosal cells. Am. J. Physiol. Endocrinol. Metab. 297, E134–E141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satou M., Nishi Y., Yoh J., Hattori Y. and Sugimoto H. (2010) Identification and characterization of acyl‐protein thioesterase 1/lysophospholipase I as a ghrelin deacylation/lysophospholipid hydrolyzing enzyme in fetal bovine serum and conditioned medium. Endocrinology 151, 4765–4775. [DOI] [PubMed] [Google Scholar]
- Spencer S. J., Xu L., Clarke M. A., Lemus M., Reichenbach A., Geenen B., Kozicz T. and Andrews Z. B. (2012) Ghrelin regulates the hypothalamic‐pituitary‐adrenal axis and restricts anxiety after acute stress. Biol. Psychiatry 72, 457–465. [DOI] [PubMed] [Google Scholar]
- Takagi K., Legrand R., Asakawa A. et al (2013) Anti‐ghrelin immunoglobulins modulate ghrelin stability and its orexigenic effect in obese mice and humans. Nat. Commun. 4, 2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theil M. M., Miyake S., Mizuno M. et al (2009) Suppression of experimental autoimmune encephalomyelitis by ghrelin. J. Immunol. 183, 2859–2866. [DOI] [PubMed] [Google Scholar]
- Tong J., Dave N., Mugundu G. M., Davis H. W., Gaylinn B. D., Thorner M. O., Tschop M. H., D'Alessio D. and Desai P. B. (2013) The pharmacokinetics of acyl, des‐acyl, and total ghrelin in healthy human subjects. Eur. J. Endocrinol. 168, 821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waseem T., Duxbury M., Ito H., Ashley S. W. and Robinson M. K. (2008) Exogenous ghrelin modulates release of pro‐inflammatory and anti‐inflammatory cytokines in LPS‐stimulated macrophages through distinct signaling pathways. Surgery 143, 334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortley K. E., Anderson K. D., Garcia K. et al (2004) Genetic deletion of ghrelin does not decrease food intake but influences metabolic fuel preference. Proc. Natl Acad. Sci. USA 101, 8227–8232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortley K. E., del Rincon J. P., Murray J. D., Garcia K., Iida K., Thorner M. O. and Sleeman M. W. (2005) Absence of ghrelin protects against early‐onset obesity. J. Clin. Investig. 115, 3573–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wren A. M., Small C. J., Ward H. L. et al (2000) The novel hypothalamic peptide ghrelin stimulates food intake and growth hormone secretion. Endocrinology 141, 4325–4328. [DOI] [PubMed] [Google Scholar]
- Wren A. M., Seal L. J., Cohen M. A., Brynes A. E., Frost G. S., Murphy K. G., Dhillo W. S., Ghatei M. A. and Bloom S. R. (2001) Ghrelin enhances appetite and increases food intake in humans. J. Clin. Endocrinol. Metab. 86, 5992. [DOI] [PubMed] [Google Scholar]
- Yang J., Brown M. S., Liang G., Grishin N. V. and Goldstein J. L. (2008) Identification of the acyltransferase that octanoylates ghrelin, an appetite‐stimulating peptide hormone. Cell 132, 387–396. [DOI] [PubMed] [Google Scholar]
- You Z., Luo C., Zhang W., Chen Y., He J., Zhao Q., Zuo R. and Wu Y. (2011) Pro‐ and anti‐inflammatory cytokines expression in rat's brain and spleen exposed to chronic mild stress: involvement in depression. Behav. Brain Res. 225, 135–141. [DOI] [PubMed] [Google Scholar]
- Zhao T. J., Liang G., Li R. L. et al (2010) Ghrelin O‐acyltransferase (GOAT) is essential for growth hormone‐mediated survival of calorie‐restricted mice. Proc. Natl Acad. Sci. USA 107, 7467–7472. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Body weight and blood glucose graphs in Ghrelin KO mice reinstated with acylated or des‐acylated ghrelin and GOAT WT/KO mice.