

Abstract
14-3-3 proteins regulate biological processes by binding to phospho-Ser or phospho-Thr motifs of cellular proteins. Among them, 14-3-3ε is crucial for antiviral immunity by mediating the cytosol-to-mitochondrial-membrane translocation of the pathogen sensor RIG-I. Here we show that the NS3 protein of dengue virus (DV) binds to 14-3-3ε and prevents RIG-I translocation to the adaptor MAVS, thereby blocking antiviral signaling. Intriguingly, a highly conserved phosphomimetic RxEP motif in NS3 is essential for 14-3-3ε binding. A recombinant mutant DV deficient in 14-3-3ε binding is impaired in RIG-I antagonism and elicits a markedly augmented innate immune response and enhanced T cell activation. Our work reveals a novel phosphomimetic-based mechanism for viral antagonism of 14-3-3-mediated immunity, which may guide the rational design of therapeutics.
INTRODUCTION
Dengue virus (DV) is responsible for ~390 million infections annually, which can lead to dengue fever or the potentially lethal dengue hemorrhagic fever or shock syndrome. Four serotypes of DV exist and infection by one serotype only confers long-lasting immunity to that particular serotype. Currently, there are no FDA-approved therapies against DV infection. A tetravalent vaccine candidate recently completed two phase III clinical trials but showed weak to moderate protection against DV serotype 2 (DV2)1, 2. Hence, there is a pressing need to better understand dengue pathogenesis to aid the design of broadly effective vaccines and antivirals.
Germline-encoded pattern recognition receptors (PRRs) are key components of the innate immune system. They detect microbial nucleic acids or structural components and subsequently trigger an antiviral response3, 4. Among the PRRs, RIG-I (retinoic acid-inducible gene-I) has emerged as a key sensor of many RNA viruses including DV, by recognizing cytosolic viral RNA species harboring a 5′ tri- or di-phosphate moiety and/or poly(U-UC) motifs5, 6. Viral RNA binding triggers a conformational change in RIG-I, allowing K63-linked ubiquitination at its N-terminal caspase activation and recruitment domains (2CARD) mediated by the E3 ubiquitin ligase TRIM257–9. Ubiquitination of RIG-I facilitates its tetramerization, and the activated RIG-I tetramer subsequently translocates from the cytosol to MAVS, found at the outer mitochondrial membrane, mitochondrial-associated membranes (MAMs), and peroxisomes10–12. MAVS assembles a multi-protein signaling complex that leads to IRF3 or IRF7 activation to induce the expression of type-I IFNs, proinflammatory cytokines, and IFN-stimulated genes (ISGs)13, 14. Recently, the mitochondrial-targeting chaperone protein 14-3-3ε has been identified as a crucial mediator of the redistribution of RIG-I from the cytosol to mitochondrion-associated MAVS by forming a ‘translocon’ complex with RIG-I and TRIM25, ultimately triggering an antiviral response15.
DV has evolved to evade both innate and adaptive immune responses, allowing it to replicate unchecked and to disseminate16. DV suppresses both type-I IFN induction and IFN-α or -β receptor (IFNAR) signal transduction through a variety of strategies17. Specifically, DV NS5 protein blocks IFNAR signaling by inducing STAT2 degradation18, while DV NS2B-NS3 protease complex cleaves stimulator of interferon genes (STING)19, 20, an adaptor downstream of cytosolic DNA sensors. However, how DV escapes innate immune detection by RIG-I is unknown.
Here, we uncover that the NS3 protein of DV binds to 14-3-3ε using a highly conserved phosphomimetic motif, blocking the translocation of RIG-I to mitochondria and thereby antiviral signaling. A recombinant DV encoding a mutant NS3 protein deficient in 14-3-3ε binding loses the ability to antagonize RIG-I and elicits an augmented innate immune response and enhanced T cell activation.
RESULTS
The NS3 protein of DV interacts with 14-3-3ε
We hypothesized that NS3 and NS5, two major IFN-antagonistic proteins of DV, inhibit the innate host defense via unidentified mechanisms. To address this, we sought to identify novel cellular interaction partners of NS3 and NS5 by utilizing affinity purification and mass spectrometry (MS) analysis of defined domains of both viral proteins: the NS3 protease and helicase domains (FLAG-NS3-Pro and FLAG-NS3-Hel), as well as the NS5 methyltransferase and polymerase domains (FLAG-NS5-MTase and FLAG-NS5-Pol). MS analysis showed that 14-3-3ε was specifically present in complex with FLAG-NS3-Pro, but not with the other domains (Supplementary Fig. 1a and data not shown).
We first confirmed that c-myc-tagged 14-3-3ε specifically bound to NS3-Pro, but not to NS3-Hel (Fig. 1a). In agreement with our MS results, FLAG-14-3-3ε interacted specifically with NS3 (fused to Glutathione S-transferase; GST-NS3), but not GST-NS5 or GST alone (Fig. 1b). Furthermore, GST-NS3 interacted specifically with 14-3-3ε, but not with 14-3-3σ, which shares ~75% homology with 14-3-3ε (Fig. 1c). Moreover, exogenously expressed NS3 of DV strongly interacted with 14-3-3ε, whereas the NS3 proteins of Hepatitis C virus (HCV) and Yellow Fever virus (YFV), also Flaviviridae members, did not bind 14-3-3ε (Fig. 1d). Importantly, NS3 efficiently formed a complex with endogenous 14-3-3ε during DV infection (Fig. 1e). Confocal microscopy showed that 14-3-3ε was expressed throughout the cytoplasm, whereas DV NS3, as previously reported, formed perinuclear cytoplasmic speckles, which are indicative of DV replication complexes at ER-derived membranes21. NS3 partially co-localized with 14-3-3ε in these perinuclear bodies, which also co-stained with NS4A, a key component of the DV replication complex22 (Fig. 1f and Supplementary Fig. 1b). Finally, GST-NS3 immobilized on agarose beads, but not GST, efficiently bound to bacterially-purified recombinant (r) 14-3-3ε, demonstrating a direct interaction (Fig. 1g). These results indicate that NS3 and 14-3-3ε bind directly to each other and form a complex during DV infection.
Figure 1. The NS3 protein of DV interacts with 14-3-3ε.
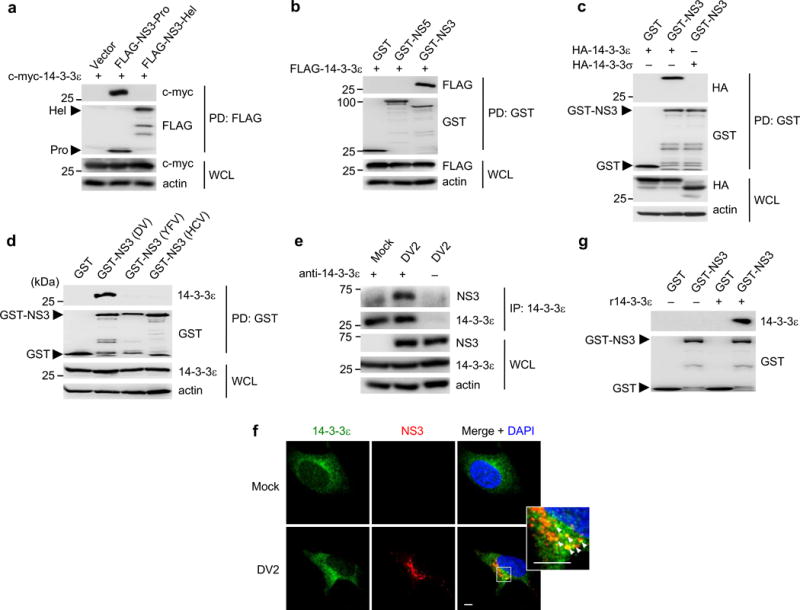
(a) Binding of c-myc-tagged 14-3-3ε and FLAG-tagged NS3-Pro or NS3-Hel, assessed in HEK293T cells by FLAG-pulldown (PD) and IB with anti-c-myc and anti-FLAG antibodies. (b) Binding of FLAG-14-3-3ε and GST, GST-NS5, or GST-NS3, assessed in HEK293T cells by GST-PD and IB with anti-FLAG and anti-GST antibodies. (c) Binding of HA-tagged 14-3-3ε or 14-3-3σ and GST or GST-NS3, assessed in HEK293T cells by GST-PD and IB with anti-HA antibody. (d) Binding of endogenous 14-3-3ε and GST, or GST-NS3 of DV2 (strain NGC), YFV (strain 17D), or HCV (strain Con1) in transfected HEK293T cells, assessed by GST-PD and IB with anti-14-3-3ε antibody. (e) Huh7 cells were mock-infected or infected with DV2 NGC (MOI 1) for 28 h. WCLs were subjected to immunoprecipitation (IP) with anti-14-3-3ε antibody, followed by IB with anti-NS3 and anti-14-3-3ε antibodies. (f) Huh7 cells were mock-infected, or infected with DV2 (strain NGC) at MOI 0.2 for 24 h. Cells were stained for endogenous 14-3-3ε (green) and NS3 (red) and imaged by confocal microscopy. Nuclei were stained with DAPI (blue). Scale bar; 5 μm. (g) In vitro binding of recombinant (r) 14-3-3ε and purified GST or GST-NS3, determined by IB with anti-14-3-3ε antibody. Data are representative of at least 2 independent experiments (a–g).
14-3-3ε is critical for controlling DV replication
14-3-3ε mediates the redistribution of RIG-I from the cytosol to mitochondria for MAVS interaction and antiviral signaling. Infection studies demonstrated that 14-3-3ε is required for an effective antiviral response against Sendai virus (SeV), vesicular stomatitis virus (VSV), and HCV, all of which are sensed by RIG-I15. We found that ectopically expressed 14-3-3ε also suppressed the replication of DV strains representing all four serotypes (DV1-4), but had no effect on herpes simplex virus-1 (HSV-1), a DNA virus (Supplementary Fig. 1c–e). Cells in which 14-3-3ε was ectopically expressed showed enhanced IFN-sensitive response element (ISRE) promoter activation upon DV infection, indicating that the IFN response plays a role in 14-3-3ε-mediated control of DV replication (Supplementary Fig. 1f). When we depleted 14-3-3ε in Huh7 cells using short interfering RNA, we observed cytotoxic effects (data not shown), impeding assessment of DV replication. In contrast, studies using K562 myeloid cells, which better tolerated 14-3-3ε depletion, showed that knockdown of 14-3-3ε significantly enhanced DV replication as compared to non-targeting control siRNA (Supplementary Fig. 1g), supporting a role for 14-3-3ε in controlling DV replication.
Proteolysis-independent inhibition of RIG-I by NS2B-NS3
NS3 together with its cofactor NS2B forms the DV protease complex, and for most flaviviruses it is believed that proteolytic activity of the viral protease is essential for antagonizing host immunity. For example, DV NS2B-NS3 has been shown to cleave STING19, 20, while NS3-NS4A of HCV cleaves MAVS23, 24. Therefore, we first asked whether NS3 in complex with NS2B can cleave 14-3-3ε. Overexpression of wild-type NS2B-NS3 [NS2B-NS3(WT)] did not result in any cleavage products of co-expressed 14-3-3ε (Supplementary Fig. 2a and data not shown). In accord, endogenous 14-3-3ε protein amounts were unchanged during DV infection (Fig. 1e), strongly suggesting that DV NS2B-NS3 does not cleave 14-3-3ε. NS2B-NS3 did not cleave TRIM25 or RIG-I either, but readily cleaved STING (Supplementary Fig. 2a).
We also generated a catalytically-inactive mutant of NS2B-NS3 [NS2B-NS3(S135A)]25 which, in contrast to NS2B-NS3(WT), was unable to cleave itself or STING (Supplementary Fig. 2b). Both NS2B-NS3(WT) and NS2B-NS3(S135A) potently suppressed IFN-β induction mediated by RIG-I 2CARD, the constitutively active signaling module of RIG-I (Fig. 2a,b). In contrast, HCV NS3-NS4A(WT), but not its catalytically-inactive mutant NS3-NS4A(S139A), suppressed RIG-I 2CARD-mediated IFN-β induction (Fig. 2a). Importantly, NS3 alone, which has no proteolytic activity26 (Supplementary Fig. 2b), blocked RIG-I 2CARD- and SeV-induced IFN-β promoter activation as potently as NS2B-NS3(WT) and NS2B-NS3(S135A) (Fig. 2b,c). NS3 overexpression also markedly inhibited IRF3 dimerization and ISG induction triggered by SeV infection (Fig. 2d,e). Crucially, ectopic expression of NS3 did not inhibit SeV infection, ruling out that the dampening effect of NS3 on RIG-I signaling is due to less efficient SeV replication (Supplementary Fig. 2c). Furthermore, NS3 suppressed IFN-β promoter activation mediated by RIG-I 2CARD, but not MAVS, supporting that NS3 acts at the level of RIG-I (Fig. 2f). These results indicate that DV NS3 inhibits RIG-I- and 14-3-3ε-mediated signaling in a cleavage-independent manner.
Figure 2. NS2B-NS3 inhibits RIG-I activation independent of proteolytic activity.

(a) IFN-β luciferase activity in HEK293T cells transfected with GST or GST-RIG-I(2CARD) together with vector, DV NS2B-NS3(WT) or NS2B-NS3(S135A), or HCV NS3-NS4A(WT) or NS3-NS4A(S139A), normalized to pGK-β-gal. Viral protein expressions were determined by IB. The results are expressed as means ± SD (n = 3). (b) IFN-β luciferase activity in HEK293T cells transfected with GST or GST-RIG-I(2CARD) together with vector, or increasing amounts of DV NS2B-NS3(WT), NS2B-NS3(S135A) or NS3, normalized to pGK-β-gal. The results are expressed as means ± SD (n = 3). (c) HEK293T cells, transfected with vector, DV NS2B-NS3(WT), NS2B-NS3(S135A) or NS3, were infected with SeV (50 HAU/ml) for 18 h. Luciferase assay and IB were performed as in (a). The results are expressed as means ± SD (n = 3). (d) Native PAGE and IB with anti-IRF3 in HEK293T cells transfected with GST or GST-NS3 and infected with SeV (50 HAU/ml) for 16 h. WCLs were further used for SDS-PAGE and IB with the indicated antibodies. (e) Similar to (d), except WCLs were immunoblotted with the indicated antibodies at 22 h after SeV infection. Expression of PP1γ, which is not an ISG, served as control. (f) IFN-β luciferase activity in HEK293T cells transfected with FLAG-MAVS or RIG-I(2CARD) together with GST or GST-NS3, normalized to pGK-β-gal. The results are expressed as means ± SD (n = 3). *p< 0.05; **p< 0.005, unpaired t-test. NS; not significant. Data are representative of at least 2 independent experiments (a–f).
NS3 prevents binding of RIG-I to 14-3-3ε
To gain further insights into the mechanism of RIG-I antagonism, we asked whether NS3 blocks the K63-linked ubiquitination of RIG-I, interferes with the 14-3-3ε-RIG-I-TRIM25 complex formation, or inhibits RIG-I translocation to mitochondria, all of which are critical steps of RIG-I activation. Efficient ubiquitination of RIG-I upon SeV infection was detected in both GST and GST-NS3 co-expressing cells (Fig. 3a). Consistently, we detected robust ubiquitination of endogenous RIG-I during DV infection, indicating that DV does not inhibit RIG-I K63-ubiquitination (Fig. 3b). We found that SeV infection robustly triggered both 14-3-3ε and TRIM25 binding to RIG-I; however, expression of GST-NS3, but not GST, profoundly reduced RIG-I binding to 14-3-3ε, but not to TRIM25 (Fig. 3c). In agreement, DV infection induced endogenous RIG-I-TRIM25 complex formation as efficiently as SeV infection (Fig. 3d). However, the interaction between 14-3-3ε and RIG-I during DV infection was minimal and comparable to mock-infected cells (Fig. 3d). This indicates that NS3 does not affect TRIM25 binding and RIG-I K63-ubiquitination, but instead specifically blocks the RIG-I-14-3-3ε interaction. In agreement, RIG-I, but not TRIM25, interacted with r14-3-3ε in vitro, suggesting that the RIG-I-14-3-3ε interaction is direct, whereas TRIM25 binds to 14-3-3ε indirectly (Supplementary Fig. 3a). DV NS3 constitutively bound to 14-3-3ε and with much greater binding capacity than RIG-I; in contrast, RIG-I binding to 14-3-3ε was very low in uninfected cells, and was induced upon SeV infection (Supplementary Fig. 3b). A competitive binding assay showed that NS3 inhibited the RIG-I-14-3-3ε interaction in a dose-dependent manner (Fig. 3e). Fractionation studies showed that RIG-I failed to translocate to the mitochondrial fraction during DV infection and was present almost exclusively in the cytosolic fraction, similar to mock-infected cells. In contrast, in SeV-infected cells, RIG-I was abundant in the mitochondrial fraction, along with MAVS, indicating its translocation (Fig. 3f). Furthermore, ectopic expression of GST-NS3 diminished RIG-I amounts in the mitochondrial fraction of SeV-infected cells (Supplementary Fig. 3c). Overexpression of 14-3-3ε rescued the cytosol-to-mitochondria translocation of RIG-I during DV infection (Supplementary Fig. 3d), supporting that the defect in RIG-I translocation in DV-infected cells is due to 14-3-3ε antagonism. These data indicate that NS3 blocks 14-3-3ε from interacting with the activated RIG-I-TRIM25 complex, preventing RIG-I from translocating to mitochondria to initiate signaling.
Figure 3. NS3 inhibits binding of RIG-I to 14-3-3ε, preventing the translocation of activated RIG-I to mitochondria.
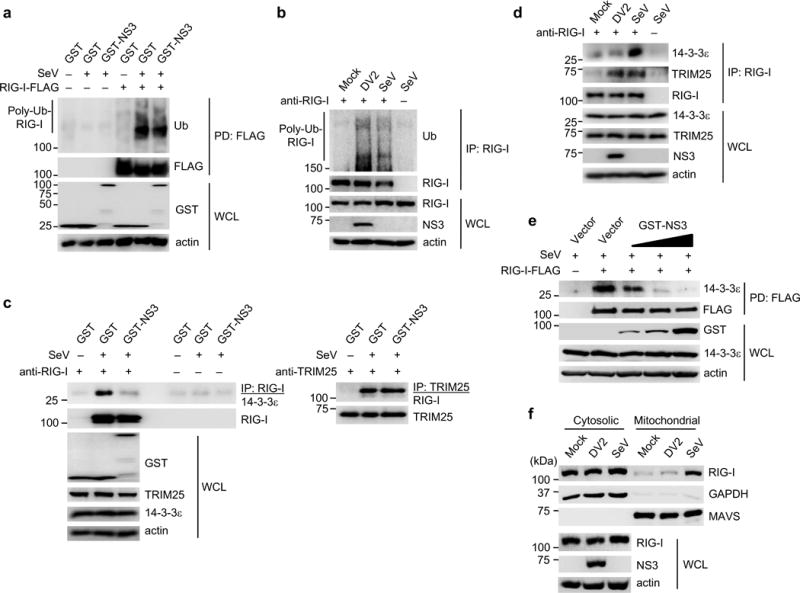
(a) Ubiquitination of RIG-I-FLAG in transfected HEK293T cells that were mock-infected or infected with SeV (50 HAU/ml) for 19 h. WCLs were subjected to FLAG-PD, followed by IB with anti-ubiquitin (Ub) and anti-FLAG. (b) Ubiquitination of endogenous RIG-I in Huh7 cells that were mock-infected, infected with DV2 NGC (MOI 1) or SeV (50 HAU/ml) for 18 h. IP with anti-RIG-I was performed, followed by IB with anti-Ub and anti-RIG-I. (c) Binding of endogenous RIG-I, TRIM25 and 14-3-3ε in transfected HEK293T cells that were infected with SeV (50 HAU/ml) for 23 h. WCLs were subjected to IP with anti-RIG-I (left) or anti-TRIM25 (right), followed by IB with anti-14-3-3ε, anti-TRIM25 or anti-RIG-I. The data shown are from the same experiment. (d) Huh7 cells were mock-infected, or infected with DV2 NGC (MOI 1) or SeV (50 HAU/ml) for 18 h. IP with anti-RIG-I was performed, followed by IB with anti-14-3-3ε, anti-TRIM25 or anti-RIG-I. (e) HEK293T cells were transfected with RIG-I-FLAG together with vector or increasing amounts of GST-NS3. 48 h later, cells were infected with SeV (50 HAU/ml) for 20 h, and FLAG-PD was performed. (f) Cytosol-mitochondria fractionation of WCLs from Huh7 cells that were mock-infected, infected with DV2 NGC (MOI 1) or SeV (50 HAU/ml) for 22 h. IB was performed with anti-RIG-I, anti-MAVS and anti-GAPDH. RIG-I and NS3 expressions were determined in the WCL. Data are representative of at least 2 independent experiments (a–f).
NS3 binds to 14-3-3ε using a phosphomimetic motif
To identify the binding site of 14-3-3ε in NS3-Pro, we constructed NS3-Pro truncation fragments and tested them for their abilities to bind endogenous 14-3-3ε by Co-IP (Fig. 4a and Supplementary Fig. 4a). Full-length GST-NS3, GST-NS3(1–92) and GST-NS3(43–92), but not other NS3 fragments, efficiently interacted with 14-3-3ε.
Figure 4. NS3 binds to 14-3-3ε using a phosphomimetic RxEP motif.

(a) Binding of GST-fused NS3 truncations to endogenous 14-3-3ε was assessed in transfected HEK293T cells by GST-PD and IB with anti-14-3-3ε antibody. (b) Amino acid sequence of the canonical motif (green) of cellular 14-3-3-binding proteins as well as the NS3 region harboring the 14-3-3-binding motif (green) from DV (serotypes 1-4), WNV, YFV and HCV. Viral strains are indicated in parenthesis. (c) (Left) Ribbon representation of the crystal structure of the NS3 protein of DV4 with Pro domain shown in red, linker in grey, and Hel domain in blue. The RLEP motif (arrow) is illustrated in green. (Right) Closed up view of the RLEP motif. (d) Bioinformatics analysis of 3280 DV NS3 protein sequences. The most common residue for each position is shown as the consensus sequence (bottom) and is represented in blue in the bar graph. Polymorphisms for each position are represented by different colors. One polymorphism for each Glu66 and Pro67 in the RLEP motif was identified, as indicated in parenthesis. (e) Binding of GST, GST-NS3, or the indicated GST-NS3 mutants and endogenous 14-3-3ε was assessed in transfected HEK293T cells by GST-PD and IB with anti-14-3-3ε antibody. (f) Cytosol-mitochondria fractionation of WCLs from transfected HEK293T cells that were mock-infected or infected with SeV (50 HAU/ml) for 20 h. Data are representative of at least 2 independent experiments (a, e, f).
A hallmark of many cellular proteins that bind to 14-3-3 family members is the presence of a canonical high-affinity binding motif, Rxx(pS/pT)xP, where × denotes any residue and pS/pT indicates a phosphorylated Ser or Thr residue27. Ser/Thr phosphorylation in Rxx(pS/pT)xP is essential for 14-3-3 binding, as dephosphorylation of this residue abrogates 14-3-3 interaction28. A closer examination of NS3(43–92), which is sufficient for 14-3-3ε binding (Fig. 4a), revealed a “compact” 64-RxEP-67 motif resembling the cellular motif but harboring a charged residue, Glu66, in place of pSer or pThr (Fig. 4b). Based on the crystal structure of the DV NS3 protein29, the 64-RxEP-67 motif is exposed and found on a surface distal to the catalytic site (Fig. 4c and Supplementary Fig. 4b). The DV2 strains NGC and 16681 harbor a 64-RIEP-67 motif, while DV1, 3 and 4 have a similar motif, 64-RLEP-67 (Fig. 4b), suggesting that the 64-RxEP-67 motif is conserved across DV strains. Indeed, bioinformatics analysis aligning more than 3000 NS3 sequences derived from fully-sequenced DV1–4 strains showed that the 64-RxEP-67 motif is conserved in all except two analyzed NS3 sequences, while adjacent residues show substantially more polymorphisms (Fig. 4d).
Since Glu is a negatively charged residue which is often used to mimic phospho-Ser or phospho-Thr in molecular biology experiments, we hypothesized that DV NS3 may utilize the phosphomimetic Glu66 in 64-RxEP-67 for 14-3-3ε binding. To test this, we transplanted the corresponding motif from DV1, 3 and 4 (RLEP), or WNV (RLDP), both harboring phosphomimetic residues (Glu66 or Asp66), into full-length NS3 derived from DV2 (NGC strain, which contains RIEP). We also transplanted the corresponding motif from YFV (KLIP), harboring an uncharged hydrophobic residue at position 66 (Ile66), into DV2 NS3. Chimeric NS3 proteins containing RLEP and RLDP [NS3(RLEP) and NS3(RLDP)], which harbor Glu66 or Asp66, were both able to bind 14-3-3ε, while NS3(KLIP) showed strongly diminished binding (Supplementary Fig. 4c). This suggests that Asp66, which is also a phosphomimetic residue, can substitute for Glu66, and that Ile65 and Leu65 are functionally equivalent.
To corroborate the importance of the phosphomimetic Glu66 in DV NS3 for 14-3-3ε binding, we replaced Glu66 with Asp (Asp66; acidic residue similar to Glu), Gln (Gln66; similar side chain as Glu but uncharged), or Lys (Lys66; positively charged). Whereas mutation of Glu66 to Asp66 did not affect NS3-14-3-3ε binding, introduction of Gln66 or Lys66 diminished 14-3-3ε binding, further strengthening that the phosphomimetic Glu66 (or Asp66) is critical for 14-3-3ε interaction (Fig. 4e). Furthermore, additional mutation of Arg64 to Lys64 [NS3(KIKP)] led to a near-complete loss of 14-3-3ε binding, demonstrating the importance of Glu66 and Arg64 for NS3 interaction with 14-3-3ε (Fig. 4e). Interestingly, introduction of the 64-RxEP-67 motif into the NS3 protein of YFV, which is unable to bind 14-3-3ε (Fig. 1d), did not rescue 14-3-3ε binding (data not shown), suggesting that additional residues besides the 64-RxEP-67 motif or the overall conformation of NS3 are important for 14-3-3ε binding. Collectively, this indicates that DV NS3 binds 14-3-3ε using a compact RxEP motif that contains a phosphomimetic (Glu66) residue.
Functional studies showed that whereas NS3(WT) potently inhibited SeV-induced RIG-I-14-3-3ε binding, NS3(KIKP) did not affect their interaction (Supplementary Fig. 4d). Consistently, NS3(WT), but not NS3(KIKP), potently blocked RIG-I translocation to mitochondria (Fig. 4f). Furthermore, expression of NS3(WT), but not NS3(KIKP), significantly suppressed SeV-mediated IFN-β transcriptional activation and ISG protein expressions (Supplementary Fig. 4e,f). This shows that a mutant DV NS3 protein that is deficient in 14-3-3ε binding is unable to inhibit the cytosol-to-mitochondria translocation of RIG-I and to suppress antiviral gene expression.
WNV NS3 harbors a phosphomimetic RLDP motif
As WNV NS3 harbors a motif (64-RLDP-67) similar to the one found in DV NS3, in which Asp66 could act as a phosphomimetic for 14-3-3ε binding, we first tested the ability of NS3 from two different WNV strains (NY99 and Kunjin) to interact with 14-3-3ε. WNV NS3 efficiently bound to endogenous 14-3-3ε (Supplementary Fig. 5a). In line with this, ectopic expression of WNV NS3 robustly inhibited RIG-I 2CARD-mediated IFN-β promoter activation (Supplementary Fig. 5b). Furthermore, as seen with DV NS3, replacing Arg64 and Asp66 in WNV NS3 with lysine residues [WNV NS3(KIKP)] strongly diminished its interaction with 14-3-3ε (Supplementary Fig. 5c). Our results indicate that the NS3 protein of both DV and WNV target 14-3-3ε to inhibit RIG-I signaling, and further reinforce the importance of a phosphomimetic motif in the viral NS3 protein for 14-3-3ε binding.
DV2(KIKP) elicits a stronger innate immune response
To determine the physiological relevance of the disruption of 14-3-3ε-mediated RIG-I translocation by NS3, we sought to construct a recombinant DV encoding NS3(KIKP). Because NS3, as part of the protease complex, processes the viral polyprotein and is therefore essential for DV replication, we first assessed if NS2B-NS3(KIKP) retains proteolytic activity. Similar to NS2B-NS3(WT), NS2B-NS3(KIKP) was able to cleave itself and STING (Supplementary Fig. 6a). Using reverse genetics, we introduced the R64K and E66K mutations in an infectious clone originally derived from DV2 1668130, 31, engineering a recombinant mutant DV encoding NS3(KIKP) [subsequently referred to as DV2(KIKP)].
DV2(KIKP) exhibited a slightly reduced (~1-log) replication capacity compared to the parental virus [DV2(WT)] in Vero cells, which are deficient in type-I IFN responses32, (Supplementary Fig. 6b,c), suggesting that DV2(KIKP) is slightly attenuated independent of type-I IFNs. In Huh7 cells, which have an intact type-I IFN response, DV2(WT) and DV2(KIKP) exhibited similar replication at 24 h after infection (Supplementary Fig. 6d); however, at 72 h post-infection, the replication of DV2(KIKP) was strongly suppressed, while DV2(WT) replicated efficiently (Fig. 5a). In contrast, DV2(KIKP) replicated almost as efficiently as DV2(WT) in Huh7.5 cells, a sub-cell line of Huh7 naturally harboring a RIG-I mutant protein that is defective in TRIM25 binding and thus RIG-I activation33, 34 (Fig. 5a).
Figure 5. DV2(KIKP) is attenuated in replication and elicits an enhanced innate immune response.
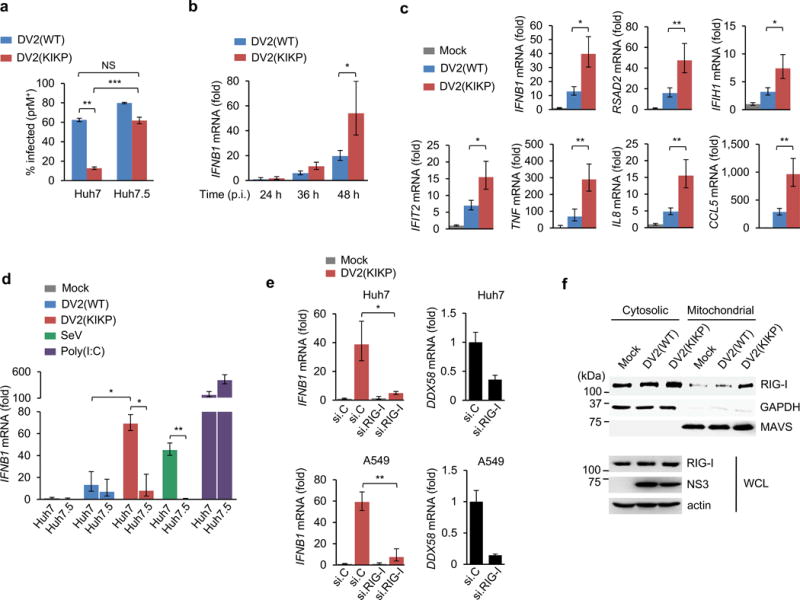
(a) Huh7 or Huh7.5 cells were infected with DV2(WT) or DV2(KIKP) (both MOI 0.01). 72 h later, cells were harvested for intracellular prM staining and flow cytometry analysis. The results are expressed as means ± SD (n = 3). (b,c) Huh7 cells were infected with DV2(WT) or DV2(KIKP) (both MOI 0.5). At 24 – 48 h (b) or 48 h (c) post-infection, total RNA was extracted and transcript levels of indicated genes were determined by qRT-PCR. Values were normalized to GAPDH and shown as fold levels compared to mock-infected cells. The results are expressed as means ± SD (n = 3). (d) Huh7 or Huh7.5 cells were infected with DV2(WT) or DV2(KIKP) (both MOI 0.5) or SeV (50 HAU/ml), or transfected with high-molecular-weight poly(I:C) (2 μg/ml) and harvested 48 h later for qRT-PCR as described in (b). The results are expressed as means ± SD (n = 3). (e) IFNB1 and DDX58 transcripts in Huh7 (upper) or A549 (lower) cells that were transfected with RIG-I-specific siRNA (si.RIG-I) or non-targeting siRNA (si.C) and infected with DV2(KIKP) (MOI 0.5) for 48 h, as determined by qRT-PCR. The results are expressed as means ± SD (n = 3). (f) Cytosol-mitochondria fractionation and IB analysis of Huh7 cells that were mock-infected, or infected with DV2(WT) or DV2(KIKP) (both MOI 0.8) for 20 h. *p< 0.05; **p< 0.005, unpaired t-test. Data are representative of 2 (a, d–f) or 3 (b, c) independent experiments.
DV2(KIKP) induced higher IFNB1 transcript amounts than DV2(WT), especially at late time points (Fig. 5b). DV2(KIKP) also elicited greater IFN-β protein production than DV2(WT) and VSV, but less efficiently than Newcastle Disease virus (NDV), which is known to robustly induce IFN-β production (Supplementary Fig. 6e). Crucially, the enhanced IFN-inducing ability of DV2(KIKP) was not due to higher amounts of viral RNA (Supplementary Fig. 6f). DV2(KIKP) also elicited significantly higher transcripts amounts of ISGs, proinflammatory cytokines and chemokines than DV2(WT) (Fig. 5c). In agreement, DV2(KIKP), but not DV2(WT), robustly induced ISG protein expression in neighboring non-infected cells (Supplementary Fig. 6g). To test if the higher IFN-β induction by DV2(KIKP) was indeed mediated by RIG-I, we compared the IFNB1 transcript amounts of Huh7 and Huh7.5 following DV2(WT) or DV2(KIKP) infection. DV2(WT) triggered low IFNB1 induction in both cells; in contrast, DV2(KIKP) infection strongly induced IFNB1 expression in Huh7, but not Huh7.5 cells, similar to infection with SeV, a virus sensed specifically by RIG-I (Fig. 5d). Depletion of RIG-I also strongly reduced IFNB1 induction triggered by DV2(KIKP), further strengthening that DV2(KIKP) induces innate signaling in a RIG-I-dependent manner (Fig. 5e). Importantly, we verified that DV2(WT) and DV2(KIKP) were both able to effectively degrade STAT2 (Supplementary Fig. 6h). Mechanistically, DV2(KIKP), but not DV2(WT), efficiently induced RIG-I translocation to mitochondria (Fig. 5f). Collectively, this indicates that DV2(KIKP) fails to antagonize RIG-I and thus elicits enhanced cytokine and ISG induction.
DV2(KIKP) elicits enhanced activation of immune cells
Although the liver is commonly involved during DV infection, mononuclear phagocytes are thought to be the primary in vivo cell targets for DV replication35. Whereas IFNB1 transcript amounts in primary human CD14+ monocytes were below the detection limit for both DV2(WT) and DV2(KIKP) (data not shown), we detected robust gene expression of proinflammatory cytokines and IL-6 protein secretion upon infection with DV2(KIKP), but not DV2(WT) (Fig. 6a,b).
Figure 6. DV2(KIKP) elicits stronger immune responses in primary human mononuclear phagocytes and T cells as compared to DV2(WT).
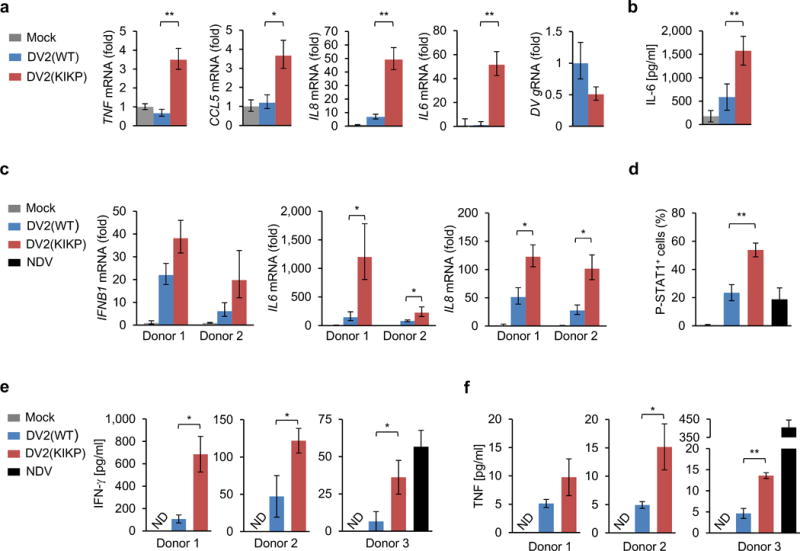
(a) Primary CD14+ monocytes were infected with DV2(WT) or DV2(KIKP) (both MOI 1) for 24 h. RNA transcript amounts of the indicated genes were determined by qRT-PCR. The results are expressed as means ± SD (n = 3). (b) Primary CD14+ monocytes were infected as in (a). Supernatants were harvested and analyzed for IL-6 by ELISA. The results are expressed as means ± SD (n = 4). (c – f) Primary moDCs were infected with DV2(WT) or DV2(KIKP) (both MOI 1), or NDV (MOI 0.5), and then co-cultured with syngeneic naïve pan T cells at 1:1 ratio. 72 h later, moDCs were analyzed by qRT-PCR (c) and T cells were analyzed for intracellular staining of phosphorylated STAT1 (pY701) (d). 96 h later, supernatants were analyzed for IFN-γ (e) or TNF (f) protein secretion by ELISA. The results are expressed as means ± SD (n = 3, except (d) where n = 5). ND; not detectable. *p< 0.05; **p< 0.005, unpaired t-test. Data are representative of 2 (a–d) or 3 (e–f) independent experiments, and each experiment was performed with a different donor.
Type-I IFN antagonism by DV contributes to the inefficient priming of T cells36. To test if DV2(KIKP) elicits an enhanced T cell response compared to DV2(WT), we first infected primary human monocyte-derived dendritic cells (moDCs) with DV2(WT) or DV2(KIKP). Both viruses induced comparable expression levels of the costimulatory marker CD86 (Supplementary Fig. 6i). However, in infected moDCs that had been co-cultured with syngeneic naïve pan T cells, DV2(KIKP) elicited a trend of higher IFNB1 transcripts and significantly higher IL6 and IL8 mRNA amounts as compared to DV2(WT) (Fig. 6c). Furthermore, DV2(KIKP) triggered STAT1 phosphorylation (pSTAT1) in primary T cells more efficiently than both DV2(WT) and NDV, indicative of augmented IFNAR activation by DV2(KIKP) (Fig. 6d and Supplementary Fig. 6j). In accord, co-cultures of T cells and DV2(KIKP)-infected moDCs secreted higher amounts of IFN-γ and TNF than co-cultures infected with DV2(WT) (Fig. 6e,f). TNF production induced by DV2(KIKP), however, was moderate as compared to NDV (Fig. 6f). Together, this indicates that DV2(KIKP) efficiently triggers innate immune responses in human hepatocytes and primary human mononuclear phagocytes, which in turn leads to stronger T cell activation.
DISCUSSION
Membrane-bound organelles play important roles in the orchestration of antimicrobial immunity by serving as platforms for the assembly of multi-protein signaling complexes. Hence, upon PAMP recognition, cytosolic PRRs must relocate from the sites of PAMP detection to these organelles – a highly regulated process that requires specific trafficking factors. However, it has been unknown whether viral pathogens target specific trafficking proteins to perturb PRR translocation. Our findings show that whereas RIG-I is initially ubiquitinated and activated during DV infection, NS3 disrupts its interaction with 14-3-3ε, blocking the translocation of RIG-I to mitochondrion-localized MAVS. Thus, unlike HCV NS3-NS4A, DV NS2B-NS3 inhibits RIG-I-mediated signaling in a cleavage-independent manner, highlighting the importance of proteolysis-independent functions of viral proteases for manipulating the host response.
The chemical similarity of phosphomimetic residues (Glu or Asp) and phospho-Ser or phospho-Thr is well established, leading investigators to commonly utilize Glu or Asp substitutions to mimic site-specific phosphorylation in molecular biology experiments. Mutational analysis showed that Glu66 in 64-RxEP-67 of DV NS3 is critical for 14-3-3ε binding, providing the first example of a naturally occurring phosphomimetic utilized in an infectious disease mechanism. Using a phosphomimetic residue for 14-3-3ε-binding may provide advantages to DV such as the lack of requirement for a kinase, be it viral or cellular, and the uniform “phosphorylation state” of all NS3 molecules. Interestingly, the synthetic peptide R18, which is commonly used to block 14-3-3 binding to cellular proteins, also harbors a phosphomimetic motif (WLDLE), in which D and E engage the same residues in 14-3-3 proteins that are engaged by the pSer or pThr-containing motifs of cellular proteins37. Thus, in addition to canonical phosphorylated motifs, short phosphomimetic sequences – either synthetic or naturally occurring in a viral pathogen – mediate efficient 14-3-3 binding. Additional studies are necessary to determine if DV NS3 antagonizes also other functions of 14-3-3ε or other 14-3-3 family members.
DV is known to potently suppress the innate immune system, a prerequisite for successful virus replication and pathogenesis. Our studies using a mutant DV that is deficient in 14-3-3ε binding and RIG-I antagonism showed that disabling a specific viral IFN-antagonistic mechanism leads to viral growth attenuation and efficient activation of innate and adaptive immunity. We propose that systematically disabling immune evasion mechanisms while preserving viral functions may allow for the rational design of live-attenuated dengue vaccines. Furthermore, our detailed characterization of the NS3-14-3-3ε interaction may also guide the design of small molecule inhibitors for antiviral therapy. In summary, our work unveils a key immune evasion mechanism of DV, which provides a framework for rational vaccine design and antiviral development.
METHODS
Cell Culture and Viruses
HEK293T, Vero, A549 (all purchased from ATCC), and NHLF cells (Clonetics) as well as Huh7 (provided by M. Farzan, Scripps Institute Florida), Huh7.5 (provided by J. Jung, University of Southern California), and HEK293T ISRE-luciferase cells38 were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES and 1% penicillin-streptomycin (Gibco). BHK-21 cells (provided by M. Farzan, Scripps Institute Florida) were propagated in Minimum Essential Medium Alpha (MEM-α) supplemented with 10% FBS, 10 mM HEPES and 1% penicillin-streptomycin. Huh7 cells stably expressing vector or HA-tagged 14-3-3ε were generated by retroviral transduction as previously described39 and propagated in DMEM supplemented with 1 μg/ml of puromycin (Gibco).
C6/36 cells (provided by M. Farzan, Scripps Institute Florida) were cultured in Eagle’s Minimum Essential Medium (EMEM) supplemented with 10% FBS and 1% penicillin-streptomycin, and grown at 28°C. K562 cells (provided by M. Farzan, Scripps Institute Florida) and primary CD14+ monocytes were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% FBS, 100 U/ml L-glutamine and 1% penicillin-streptomycin. In addition, 1 mM sodium pyruvate was added for culturing moDCs and T cells. Cell lines were not tested for mycoplasma contamination. DV2 NGC, DV1 276 RK1, DV2 16681, DV3 BC188/97 and DV4 814699 were propagated in C6/36 cells. SeV (Cantell) was purchased from Charles River Laboratories. NDV-GFP and VSV-GFP were a gift from A. Garcia-Sastre (Icahn School of Medicine at Mount Sinai) and S. Whelan (Harvard), respectively. HSV-1 was provided by D. Knipe (Harvard).
Plasmids and Transfections
pQCXIP-NS2B/3-HA and pQCXIP-NS3-HA were generated by subcloning NS2B-NS3 (containing NS2B and NS3) or NS3 of DV2 (strain NGC) into pQCXIP vector using NotI and BamHI sites. GST-NS3 and GST-NS5 were generated by subcloning NS3 or NS5 of DV2 (strain NGC) into pEBG vector between BamHI and ClaI. Similarly, NS3 of YFV (strain 17D, kindly provided by R. Kuhn, Purdue University), NS3 of HCV (strain Con1, kindly provided by Z. Chen, University of Texas Southwestern Medical Center) and NS3 of WNV (strain NY99 or Kunjin, kindly provided by M. Diamond, Washington University in St. Louis) were subcloned into the pEBG vector. pEF-BOS-FLAG-NS3-Pro (aa 1–179), pEF-BOS-FLAG-NS3-Hel (aa 169–618), pEF-BOS-FLAG-NS3 (full-length), pEF-BOS-FLAG-NS5-MTase (aa 1–319) and pEF-BOS-FLAG-NS5-Pol (aa 297–901) were generated by subcloning into pEF-BOS-FLAG vector using NotI and SalI sites. 14-3-3ε (Uniprot: P62258-1) was purchased as a cDNA clone and subcloned into pEF-BOS, pCAGGS and pQCXIP vectors with an N-terminal FLAG, c-myc and HA tag, respectively. HA-tagged 14-3-3σ was provided by S. Inoue (University of Tokyo) and has been described40. pQCXIP-STING-HA was generated by subcloning STING (clone ID 5762441, Thermo Scientific) into pQCXIP vector using NotI and BamHI sites. The plasmids encoding the HCV NS3-NS4A protease complex (pcDNA3-FLAG-NS3-NS4A) and its S139A catalytically-inactive mutant were a kind gift of Z. Chen23. Plasmids encoding GST-RIG-I(2CARD), RIG-I-FLAG, TRIM25-FLAG and FLAG-MAVS have been described previously7, 41. The DV NS3 truncation mutants GST-NS3(1–92), GST-NS3(93–168), GST-NS3(43–92), GST-NS3(82–168), GST-NS3(63–168), and GST-NS3(43–168) were generated by PCR using GST-NS3 full-length as template. All constructs were sequenced to verify 100% agreement with the original sequence. Transfections were performed using the calcium phosphate method, or with TurboFectin 8.0 (Origene), Lipofectamine and Plus reagent, or Lipofectamine 2000 (all Life Technologies) according to the manufacturer’s instructions. High molecular weight poly(I:C) complexed with LyoVec (Invivogen) was used to transfect cells at 2 μg/ml to stimulate MDA5 activation.
14-3-3ε and RIG-I Knockdown Experiments
siRNAs targeting 14-3-3ε (siGENOME SMARTpool M-017302-03-0005) or RIG-I (siGENOME SMARTpool M-012511-01-0005) as well as a non-targeting control siRNA were purchased from Dharmacon. K562 cells were seeded into 12-well plates and transfected with 300 nM siRNA using Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer’s instructions. Knockdown of endogenous 14-3-3ε was determined by western blot analysis. Similarly, Huh7 or A549 cells were seeded into 6-well plates and transfected with 100 nM siRNA using Lipofectamine RNAiMAX. Knockdown of endogenous RIG-I (DDX58) was determined by qRT-PCR.
Antibodies and Reagents
For western blot analysis, the following antibodies were used: anti-FLAG (1:2,000, M2, Sigma), anti-HA (1:2,000, HA-7, Sigma), anti-GST (1:2,000, GST-2, Sigma), anti-c-myc (1:2,000, 9E10, Covance), anti-β-actin (1:2,000, AC-15, Sigma), anti-RIG-I (1:2,000, Alme-1, Adipogen), anti-TRIM25 (1:1,000, 2/EFP, BD Biosciences), anti-ubiquitin (1:500, P4D1, Santa Cruz), anti-PP1γ (1:2,000, A300-906A, Bethyl Laboratories), anti-ISG15 (1:1,000, F-9, Santa Cruz), anti-ISG54 (1:1,000, 25-735, ProSci), anti-STAT2 (1:1,000, C-20, Santa Cruz), anti-14-3-3ε (1:1,000, 8C3, Santa Cruz), anti-NS3 (1:2,000, E1D8, kindly provided by E. Harris, University of California, Berkeley), anti-NS3 (1:2,000, GT2811, Genetex), anti-MAVS (1:2,000, AT107, Enzo), anti-GAPDH (1:2,000, CS204254, Millipore), anti-IRF3 (1:500, sc-9082, Santa Cruz), anti-SeV (1:1,000, PD029, MBL). All antibodies had been validated by manufacturer. For immunoprecipitation of 14-3-3ε, 3 μg of anti-14-3-3ε (11648-2-AP, Proteintech) was used per sample. For flow cytometry analysis, anti-prM (2H2, Merck Millipore) was conjugated to DyLight 633 using a commercial kit (Thermo Scientific) and used to detect DV-infected cells. Anti-CD14-FITC (M5E2), anti-CD86-FITC (FUN-1), anti-pSTAT1(pY701)-Alexa 647 (4a) and isotype control antibodies were purchased from BD Biosciences. ELISA kits for IFN-γ (Biolegend), IL-6 and IFN-β (both PBL Assay Science), and TNF (Affymetrix) were purchased and ELISA was performed according to the manufacturer’s instructions.
Luciferase Reporter Assay
HEK293T cells were seeded into 12-well plates. The following day, cells were transfected with 200 ng IFN-β luciferase construct42 and 300 ng β-gal-expressing pGK-β-gal43_ENREF_7, together with 100 ng - 1 μg of plasmid encoding effector protein or empty vector. To stimulate IFN-β promoter activity, 2 ng of GST-RIG-I(2CARD) or 30 ng of FLAG-MAVS was co-transfected, or cells were infected with SeV (50 HAU/ml) 48 hours after transfection7. Cells were harvested and assayed for luciferase activity (Promega). Luciferase values were normalized to β-galactosidase activity to control for transfection efficiency. For ISRE-luciferase assays, HEK293T ISRE-luciferase reporter cells were seeded into 6-well plates and transfected with 3 μg of vector or FLAG-14-3-3ε. 48 h later, cells were re-seeded into 24-well plates and subsequently infected with DV2 NGC (MOI 5). Cells were assayed for luciferase activity as described above and normalized to corresponding mock-infected cells.
Pull-down Assay, Co-Immunoprecipitation, and Immunoblot Analysis
HEK293T or Huh7 cells were lysed in NP-40 buffer (50 mM HEPES pH 7.4, 150 nM NaCl, 1% [vol/vol] NP-40, protease inhibitor cocktail [Sigma]) and centrifuged at 13,000 rpm for 20 min. GST or FLAG pull-down, Co-IP, and western blot analyses were performed as previously described7, 39. Elution of pull-down or Co-IP was performed by heating at 95°C for 5 min.
Large-scale Protein Purification and Mass Spectrometry
Five T175 flasks of HEK239T cells were transfected with pEF-BOS-FLAG-NS3-Pro, pEF-BOS-FLAG-NS3-Hel, pEF-BOS-FLAG-NS5-MTase or pEF-BOS-FLAG-NS5-Pol (each ~30 μg DNA per flask). Two days later, cells were lysed with NP-40 buffer supplemented with protease inhibitor cocktail (Sigma). Clarified lysates were mixed with a ~50% slurry of anti-FLAG-conjugated sepharose beads (Sigma) and incubated for 4 h at 4°C. After extensive washing of the beads with NP-40 lysis buffer, bound proteins were eluted and separated on a NuPAGE 4-12% Bis-Tris gradient gel (Life Technologies). Coomassie staining was performed and a ~30kDa band specifically present in the FLAG-NS3-Pro sample was excised and analyzed by ion-trap mass spectrometry at the Harvard Taplin Biological Mass Spectrometry facility. A total of 18 peptides (17 unique) of 14-3-3ε were detected by mass spectrometry.
Confocal Microscopy
Huh7 cells were grown on chamber slides or on cover slips in 24-well plates, and then infected with DV2 or SeV at indicated titers, or mock infected. Cells were harvested at the indicated time points and fixed with 4% (w/v) paraformaldehyde for 20 min, permeabilized with 0.2% (vol/vol) Triton-X-100 in PBS, and blocked with 10% (vol/vol) goat serum or FBS in PBS for 1 h. For immunostaining, anti-14-3-3ε (1:100, 11648-2-AP, Proteintech), anti-NS3 (1:1,000, GT2811 or GTX 124252, Genetex), anti-NS4A (1:500, GTX 124249, Genetex), anti-ISG54 (1:50, 12604-1-AP, Proteintech), anti-RIG-I (1:250, Alme-1, Adipogen), and anti-FLAG (all 1:2,000, M2 Sigma, ab1162 Abcam, and A190-101A Bethyl) were used, followed by incubation with secondary polyclonal antibodies conjugated to Alexa Fluor 488, Alexa Fluor 594, or Alexa Fluor 647 (Life Technologies or Abcam). Cells were mounted in DAPI-containing Vectashield (Vector Labs) to co-stain nuclei. All laser scanning images were acquired on an Olympus IX8I confocal microscope.
Direct Protein Interaction Assay
Bacterially-purified recombinant human 14-3-3ε protein (NP_006752.1) was purchased from Sino Biological. GST or GST-NS3 (DV2, strain NGC), expressed in HEK293T cells, was immobilized on glutathione-conjugated sepharose beads (G3907, Sigma) in NP-40 buffer and incubated with recombinant 14-3-3ε protein (final concentration of 10 μg/ml) for 2 h at 4°C. After extensive washing with NP-40 buffer, bound proteins were eluted from the beads with 2x Laemmli buffer and heated at 95°C for 5 min, followed by SDS-PAGE and western blot analysis. Similarly, TRIM25-FLAG and RIG-I-FLAG were purified from transfected HEK293T cells using anti-FLAG-conjugated sepharose beads (M2, Sigma) and tested for binding to recombinant 14-3-3ε.
Cytosol-Mitochondria Fractionation Assay
HEK293T or Huh7 cells were infected with DV or SeV at indicated titers, or mock infected. Importantly, conditions were chosen for these fractionation assays in which the upregulation of endogenous RIG-I protein amounts by the different stimuli tested was minimal, allowing direct assessment of RIG-I translocation. 20–24 hours later, a portion of cells was harvested for WCLs, and another portion for fractionation assay using a commercial mitochondria/cytosol fractionation kit (MIT1000, Merck Millipore) according to the manufacturer’s instructions. Briefly, cells were disrupted in Isotonic Mitochondrial Buffer using a Dounce homogenizer. Lysates were subjected to low-speed centrifugation to pellet nuclei and unbroken cells. Supernatant was subsequently centrifuged at 10,000 × g for 30 min at 4°C. The supernatant containing the cytosol and microsome fraction (‘cytosolic fraction’) as well as the pellet containing the enriched mitochondrial fraction were subjected to a bicinchoninic acid (BCA) assay. Equal amounts of protein were loaded for SDS-PAGE and analyzed by western blot. Anti-GAPDH and anti-MAVS western blot analyses served as controls.
Dengue Virus Infection and Flow Cytometry Analysis
Infection was performed based on a published protocol44. Briefly, ~1.5 × 105 cells per well were seeded into 24-well plates and allowed to adhere for 4 h. Virus diluted in 250 μl DMEM containing 2% FBS was incubated with the cells at 37°C for 1.5 h. At the indicated time points after infection, cells and/or supernatants were harvested. For K562 suspension cells, the infection was performed similarly except growth media was directly added to cells after infection. To detect DV-infected cells, cells were washed once in PBS, fixed in 1% (wt/vol) paraformaldehyde, permeabilized with 100 μl 0.1% saponin (Sigma), and then stained with anti-prM-DyLight 633 in 50 μl permeabilization buffer for ~40 min at 4°C. Subsequently, cells were washed with PBS and resuspended in 1% (wt/vol) paraformaldehyde before flow cytometry analysis on a FACS Calibur (BD Biosciences). Analysis was performed using FlowJo software (Tree Star). For Supplementary Fig. 1b–e, Huh7 cells were pre-transfected in 6-well plates for 48 h with 2.5 μg plasmid before re-seeding into 24-well plates for infection. Plaque assay using BHK-21 cells was performed as previously described39.
Bioinformatics analysis
NS3 protein sequences from full genome DV sequences were analyzed with NIAID Virus Pathogen Database and Analysis Resource (ViPR) online through the website at http://www.viprbrc.org.
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was extracted from cells using an RNA extraction kit (OMEGA Bio-Tek). Equal amounts of RNA (typically 10 – 100 ng) were used in an one-step qRT-PCR reaction (SuperScript III Platinum One-Step qRT-PCR kit with ROX, Life Technologies) with commercially available primers with FAM reporter dye for the indicated target genes (IDT). Expression level for each target gene was calculated by normalizing against 18S (for dendritic cells) or GAPDH (all other cells) using the ΔΔCT method and expressed as fold levels compared to mock-infected cells. For viral genomic RNA (gRNA), custom primers were designed against a region in NS4A (forward primer: AACAGAGAACACCCCAAGAC; reverse primer: ATGCTTCCCAATCCGAGATC; probe: CCGCAACCATGGCAAACGAGAT), normalized to GAPDH, and expressed as fold levels compared to DV2(WT) infection. All qRT-PCR reactions were run on a 7300 RT-PCR System or 7500 FAST RT-PCR System (both ABI).
Generation of a Mutant Dengue Virus Carrying a NS3(KIKP) protein
DV2(KIKP) was generated based on an infectious clone of DV2 16681, pD2/IC-30P, kindly provided by C. Huang (Center for Disease Control and Prevention) and described previously30, 31. PCR was used to generate mutant pD2/IC-30P harboring R64K and E66K mutations in the NS3 gene. The WT and mutant infectious clone plasmids were linearized by XbaI digestion and in vitro transcribed using the T7 promoter (RiboMAX Large Scale RNA Production System, Promega) with the addition of a m7G(5′)ppp(5′)A RNA cap structure analog (New England Biolabs). The in vitro transcribed RNA was purified using Micro Bio-Spin columns (Bio Rad) and transfected into Vero cells using Lipofectamine 2000. Viral supernatants were harvested and used to propagate the WT and mutant virus in Vero cells. Vero cells were further used to titer the recombinant viruses by serial dilution of the supernatant and staining with anti-prM antibody, followed by FACS analysis45.
DV Infection Studies in Primary Human Monocytes, moDCs and T Cells
Human peripheral blood or peripheral blood mononuclear cells (PBMCs) from unidentified healthy donors was purchased (HemaCare). This study was done in accordance with the ethical guidelines of Harvard Medical School. In the case of human peripheral blood, PBMCs were isolated using Ficoll-Hypaque (GE Healthcare) density gradient centrifugation. CD14+ monocytes were positively selected from PBMCs using anti-CD14 magnetic microbeads, and naïve pan T cells were negatively selected from PBMCs with magnetic microbeads, according to the manufacturer’s instructions (Miltenyi Biotec). The purity of CD14+ cells was routinely ~90%, as determined by anti-CD14-FITC staining (BD Biosciences) and flow cytometry analysis. For infection of monocytes, ~1.5 × 105 CD14+ monocytes per well were infected with DV2(WT) or DV2(KIKP) in a 96-well plate in 250 μl DMEM containing 2% FBS for 5 h, with occasional agitation.
To obtain naïve moDCs, CD14+ monocytes were cultured with 500 U/ml human GM-CSF and 1000 U/ml human IL-4 (both purchased from Peprotech) for ~6 days. For infection of moDCs, ~1 × 105 naïve moDCs per well were infected with DV2(WT), DV2(KIKP) or NDV at the indicated MOIs for 3 h in 96-well flat bottom plates. Subsequently, the media was replaced and ~1 × 105 syngeneic naïve pan T cells were co-cultured with the moDCs (1:1 ratio). 72 h later, adherent moDCs were analyzed by qRT-PCR after rinsing off T cells with PBS 3 times, or T cells were analyzed for intracellular staining of phosphorylated STAT1. Furthermore, supernatants of moDC-T cell co-cultures were analyzed for IFN-γ or TNF by ELISA (Biolegend or Affymetrix, respectively) 96 h after infection.
For intracellular pSTAT1 staining, T cells (~1 × 105) were fixed in 2% (wt/vol) paraformaldehyde, washed once with PBS, permeabilized with 100 μl Perm Buffer III (BD Biosciences) and then washed twice with PBS. Cells were blocked with 1 μg Human Fc block (BD Biosciences) in 25 μl PBS and subsequently stained with an anti-pSTAT1(pY701)-Alexa 647 antibody (BD Biosciences) by adding 5 μl of the antibody for 1 h at 4°C. All FACS data were acquired on a FACS Calibur (BD Biosciences) and analyzed using FlowJo (Tree Star) software.
Statistical analysis
Unpaired two-tailed Student’s t tests were used to compare differences between two unpaired experimental groups in all cases. A P value of <0.05 was considered statistically significant. No pre-specified effect size was assumed and in general 3 to 5 replicates for each condition was used, which was sufficient to demonstrate statistically significant differences.
Supplementary Material
Acknowledgments
We greatly thank C. Huang (Center for Disease Control and Prevention), E. Harris (University of California, Berkeley), M. Diamond (Washington University in St. Louis), Z. Chen (University of Texas Southwestern Medical Center), R. Kuhn (Purdue University), M. Farzan (Scripps Institute Florida), N. Hacohen (Massachusetts General Hospital), L. Gehrke (Harvard University), D. Knipe (Harvard University), S. Whelan (Harvard University) and A. García-Sastre (Icahn School of Medicine at Mount Sinai) for providing reagents. This study was supported in part by the US National Institutes of Health grant R01 AI087846, the Giovanni Armenise-Harvard Foundation, the William F. Milton Fund, and a John and Virginia Kaneb Fellowship (to M.U.G.).
Footnotes
AUTHOR CONTRIBUTIONS
Y.K.C. performed all aspects of this study. Y.K.C. and M.U.G. designed the study and wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at www.nature.com/ni/.
References
- 1.Villar L, et al. Efficacy of a Tetravalent Dengue Vaccine in Children in Latin America. The New England journal of medicine. 2014 doi: 10.1056/NEJMoa1411037. [DOI] [PubMed] [Google Scholar]
- 2.Capeding MR, et al. Clinical efficacy and safety of a novel tetravalent dengue vaccine in healthy children in Asia: a phase 3, randomised, observer-masked, placebo-controlled trial. Lancet. 2014;384:1358–1365. doi: 10.1016/S0140-6736(14)61060-6. [DOI] [PubMed] [Google Scholar]
- 3.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 4.Goubau D, Deddouche S, Reis e Sousa C. Cytosolic sensing of viruses. Immunity. 2013;38:855–869. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loo YM, Gale M., Jr Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goubau D, et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature. 2014;514:372–375. doi: 10.1038/nature13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gack MU, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 8.Peisley A, Wu B, Xu H, Chen ZJ, Hur S. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature. 2014;509:110–114. doi: 10.1038/nature13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeng W, et al. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315–330. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixit E, et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668–681. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horner SM, Liu HM, Park HS, Briley J, Gale M., Jr Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:14590–14595. doi: 10.1073/pnas.1110133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 13.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annual review of immunology. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belgnaoui SM, Paz S, Hiscott J. Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Current opinion in immunology. 2011;23:564–572. doi: 10.1016/j.coi.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 15.Liu HM, et al. The mitochondrial targeting chaperone 14–3–3epsilon regulates a RIG-I translocon that mediates membrane association and innate antiviral immunity. Cell host & microbe. 2012;11:528–537. doi: 10.1016/j.chom.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmid MA, Diamond MS, Harris E. Dendritic cells in dengue virus infection: targets of virus replication and mediators of immunity. Frontiers in immunology. 2014;5:647. doi: 10.3389/fimmu.2014.00647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Green AM, Beatty PR, Hadjilaou A, Harris E. Innate immunity to dengue virus infection and subversion of antiviral responses. Journal of molecular biology. 2014;426:1148–1160. doi: 10.1016/j.jmb.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashour J, Laurent-Rolle M, Shi PY, Garcia-Sastre A. NS5 of dengue virus mediates STAT2 binding and degradation. Journal of virology. 2009;83:5408–5418. doi: 10.1128/JVI.02188-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aguirre S, et al. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS pathogens. 2012;8:e1002934. doi: 10.1371/journal.ppat.1002934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu CY, et al. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS pathogens. 2012;8:e1002780. doi: 10.1371/journal.ppat.1002780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Apte-Sengupta S, Sirohi D, Kuhn RJ. Coupling of replication and assembly in flaviviruses. Current opinion in virology. 2014;9C:134–142. doi: 10.1016/j.coviro.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller S, Kastner S, Krijnse-Locker J, Buhler S, Bartenschlager R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. The Journal of biological chemistry. 2007;282:8873–8882. doi: 10.1074/jbc.M609919200. [DOI] [PubMed] [Google Scholar]
- 23.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meylan E, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 25.Khumthong R, Angsuthanasombat C, Panyim S, Katzenmeier G. In vitro determination of dengue virus type 2 NS2B-NS3 protease activity with fluorescent peptide substrates. Journal of biochemistry and molecular biology. 2002;35:206–212. doi: 10.5483/bmbrep.2002.35.2.206. [DOI] [PubMed] [Google Scholar]
- 26.Falgout B, Pethel M, Zhang YM, Lai CJ. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. Journal of virology. 1991;65:2467–2475. doi: 10.1128/jvi.65.5.2467-2475.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mhawech P. 14–3–3 proteins–an update. Cell research. 2005;15:228–236. doi: 10.1038/sj.cr.7290291. [DOI] [PubMed] [Google Scholar]
- 28.Yaffe MB, et al. The structural basis for 14–3–3:phosphopeptide binding specificity. Cell. 1997;91:961–971. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- 29.Luo D, et al. Crystal structure of the NS3 protease-helicase from dengue virus. Journal of virology. 2008;82:173–183. doi: 10.1128/JVI.01788-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kinney RM, et al. Construction of infectious cDNA clones for dengue 2 virus: strain 16681 and its attenuated vaccine derivative, strain PDK-53. Virology. 1997;230:300–308. doi: 10.1006/viro.1997.8500. [DOI] [PubMed] [Google Scholar]
- 31.Butrapet S, et al. Attenuation markers of a candidate dengue type 2 vaccine virus, strain 16681 (PDK-53), are defined by mutations in the 5′ noncoding region and nonstructural proteins 1 and 3. Journal of virology. 2000;74:3011–3019. doi: 10.1128/jvi.74.7.3011-3019.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Desmyter J, Melnick JL, Rawls WE. Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero) Journal of virology. 1968;2:955–961. doi: 10.1128/jvi.2.10.955-961.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gack MU, et al. Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:16743–16748. doi: 10.1073/pnas.0804947105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sumpter R, Jr, et al. permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. Journal of virology. 2005;79:2689–2699. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jessie K, Fong MY, Devi S, Lam SK, Wong KT. Localization of dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. The Journal of infectious diseases. 2004;189:1411–1418. doi: 10.1086/383043. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez-Madoz JR, Bernal-Rubio D, Kaminski D, Boyd K, Fernandez-Sesma A. Dengue virus inhibits the production of type I interferon in primary human dendritic cells. Journal of virology. 2010;84:4845–4850. doi: 10.1128/JVI.02514-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petosa C, et al. 14–3–3zeta binds a phosphorylated Raf peptide and an unphosphorylated peptide via its conserved amphipathic groove. The Journal of biological chemistry. 1998;273:16305–16310. doi: 10.1074/jbc.273.26.16305. [DOI] [PubMed] [Google Scholar]
- 38.Shapira SD, et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009;139:1255–1267. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan YK, Huang IC, Farzan M. IFITM proteins restrict antibody-dependent enhancement of dengue virus infection. PloS one. 2012;7:e34508. doi: 10.1371/journal.pone.0034508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Urano T, et al. Efp targets 14–3–3 sigma for proteolysis and promotes breast tumour growth. Nature. 2002;417:871–875. doi: 10.1038/nature00826. [DOI] [PubMed] [Google Scholar]
- 41.Wies E, et al. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity. 2013;38:437–449. doi: 10.1016/j.immuni.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin R, Genin P, Mamane Y, Hiscott J. Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7. Mol Cell Biol. 2000;20:6342–6353. doi: 10.1128/mcb.20.17.6342-6353.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hatzivassiliou E, Cardot P, Zannis VI, Mitsialis SA. Ultraspiracle, a Drosophila retinoic × receptor alpha homologue, can mobilize the human thyroid hormone receptor to transactivate a human promoter. Biochemistry. 1997;36:9221–9231. doi: 10.1021/bi963145k. [DOI] [PubMed] [Google Scholar]
- 44.Diamond MS, Edgil D, Roberts TG, Lu B, Harris E. Infection of human cells by dengue virus is modulated by different cell types and viral strains. Journal of virology. 2000;74:7814–7823. doi: 10.1128/jvi.74.17.7814-7823.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lambeth CR, White LJ, Johnston RE, de Silva AM. Flow cytometry-based assay for titrating dengue virus. Journal of clinical microbiology. 2005;43:3267–3272. doi: 10.1128/JCM.43.7.3267-3272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.