

Abstract
The successful treatment of malignant disease generally requires the use of multiple therapeutic agents that are coordinated in a spatiotemporal manner to enable synergy. Here, we have designed a porous silicon-based micro/nano composite (MNC) that is capable of simultaneously delivering chemotherapeutic agents and small interfering RNA (siRNA) to the lungs following intravenous injection. The pores of the silicon microparticles were loaded with BRAF siRNA-containing liposomes, while the surface was conjugated with docetaxel-encapsulated polymeric nanoparticles. The synergistic antitumor effect of the MNC was demonstrated in vitro in melanoma cells and in vivo using a mouse model for melanoma lung metastasis. The MNC displayed superior therapeutic efficacy and increased accumulation in metastatic melanoma lesions in the lungs in comparison to combination therapy with liposomes and polymers. The results indicate that the MNC could be used as an effective delivery vehicle for simultaneous enrichment of multiple therapeutic agents in the lungs.
Keywords: biomaterials, drug delivery, polymeric materials, silicon microdisks, siRNA delivery
Graphical abstract
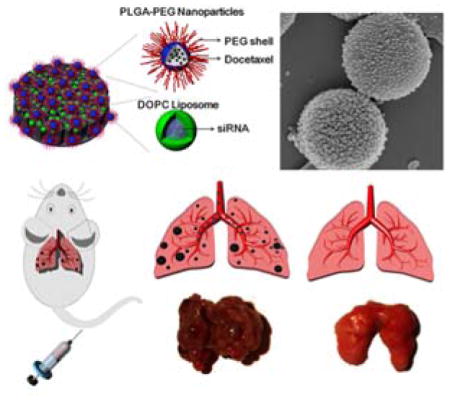
We design a novel porous silicon-based micro/nano composite (MNC) that is capable of simultaneously delivering chemotherapeutic agents and small interfering RNA (siRNA) to the lungs following intravenous injection. The MNC displayed superior therapeutic efficacy and increased accumulation in metastatic melanoma lesions in the lungs in comparison to combination therapy with liposomes and polymeric nanoparticles.
1. Introduction
The past decade has witnessed a substantial growth in the use of nanotechnology for the treatment of various diseases.[1, 2] Indeed, a major advantage of nanotherapeutics in comparison to conventional drugs is increased tumor accumulation due to reduced renal clearance and the enhanced permeability and retention (EPR) effect.[2, 3] Nevertheless, systemically injected nanoparticles are rapidly taken up by the mononuclear phagocyte system, which predominantly consists of resident macrophages in the liver and spleen.[4] Therefore, nanoparticles generally display limited retention in the lungs, as illustrated in a study in which ~3% of intravenously injected poly(lactide-co-glycolide) (PLGA) nanoparticles were detected in lung tissue after 24 h.[5] The treatment of life-threatening pulmonary conditions, such as cancer lung metastasis, necessitates the development of strategies to increase the accumulation of therapeutic agents in lung tissue.
Modulation of the size and shape of particles is a strategy that can be used to improve biodistribution. For instance, it has been shown that non-spherical particles display increased retention in the lungs.[6, 7] In particular, porous silicon microdisks have previously been used for the delivery of drug-loaded nanoparticles to tumor tissue.[8] These porous silicon particles display enhanced accumulation in the lungs due to their micrometer size and discoidal structure, which also facilitate interactions with diseased endothelium.[7, 9] These microdisks have been used for the delivery of small interfering RNA (siRNA)-loaded liposomes or chemotherapy-loaded micelles.[10, 11] However, combination therapy with different types of nanoparticles has not been realized due to the limited loading capacity of the silicon particles. In fact, the size of the silicon pores is usually restricted in order to maintain stability.
Here, we have designed a micro/nano composite (MNC) that is capable of simultaneously delivering two types of nanoparticles to lung tissue. Specifically, the MNC consists of a discoidal porous silicon microparticle loaded with 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) liposomes and conjugated to PLGA-polyethylene glycol (PEG) nanoparticles (Figure 1a). As a consequence of this polymeric coating, PEG chains protrude from the particle surface, thus endowing the MNC with stealth-like properties that further reduce liver and spleen uptake. Previously, DOPC liposomes have emerged as efficient delivery vehicles for small interfering RNA (siRNA),[11] while PLGA-PEG nanoparticles have been reported as suitable carriers for chemotherapeutic agents.[12, 13] In this study, we have combined these nanoparticles and therapeutic agents into a single platform by fully utilizing both the pores and surface of the silicon microparticles. In addition to increasing loading capacity, this arrangement permits the implementation of combination therapy. In summary, the microparticle component of the MNC was designed to lodge in the small capillaries of the lungs and gradually release the polymeric nanoparticles and liposomes as the silicon material degrades (Figure 1b). Subsequently, the nanoparticles can infiltrate cancerous lesions by utilizing the EPR effect and enter into cancer cells through endocytosis. Accordingly, at different stages following intravenous injection, the MNC combines the distinct advantages of microparticles and nanoparticles.
Figure 1.

Design of the micro/nano composite (MNC). a) Schematic illustration of the MNC fabrication process. A porous silicon microdisk was modified with 3-aminopropyltriethoxysilane (APTES) and loaded with small interfering RNA (siRNA)-containing 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) liposomes through sonication. Docetaxel-encapsulated poly(lactide-co-glycolide) (PLGA)-polyethylene glycol (PEG) nanoparticles were then conjugated to the surface of the silicon microdisk. b) Schematic illustrating the therapeutic use of the MNC. The MNC was designed to accumulate in metastatic melanoma lesions in the lungs following intravenous injection. Gradual degradation of the silicon material triggers the release of liposomes and polymeric nanoparticles, which can then enter cancer cells through endocytosis. EDC, 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide; sulfo-NHS, N-hydroxysulfosuccinimide.
In order to demonstrate the therapeutic efficacy of this platform, a mouse model of melanoma lung metastasis was employed. In fact, the lungs are the most common site for melanoma metastasis.[14] Notably, the prevailing genetic alteration in metastatic melanoma is the BRAF V600E mutation, which is strongly correlated with reduced survival rates.[15] Although BRAF inhibitors can reduce tumor vascularity and induce cancer cell apoptosis,[16] resistance to such inhibitors is frequently encountered.[17] For instance, studies have revealed response rates of approximately 40–60% for inhibitors such as vemurafenib, dabrafenib, and encorafenib, while the response rates are even lower in the case of trametinib.[18] The identified mechanisms of drug resistance in such studies include secondary mutations in BRAF, reactivation of mitogen-activated protein kinase (MAPK), and activation of alternate malignant pathways. In this regard, a proposed strategy for overcoming drug resistance is the use of combination therapy that can suppress tumor growth through multiple mechanisms.[19] For example, the United States Food and Drug Administration (FDA) has approved concurrent treatment with BRAF and MEK inhibitors[20] and clinical trials combining docetaxel and BRAF inhibitor are ongoing.[21] In light of the urgent need to develop effective treatment strategies for melanoma lung metastasis, the MNC was devised to facilitate the synergistic anticancer efficacy of BRAF siRNA and docetaxel.
2. Results and Discussion
2.1. Characteristics of the MNC
Synthesization of the MNC requires a step-by-step process that can be broadly divided into three separate components: a silicon microdisk, siRNA-loaded liposomes, and docetaxel-loaded PLGA-PEG nanoparticles. Photolithography and electrochemical etching of silicon wafers was used to fabricate porous silicon microdisks with a size of 2.6 μm × 0.7 μm, a pore size of 50–60 nm, and a zeta potential of ~25 mV (Figure 2a and Table 1). Previous studies have described the loading of siRNA-containing liposomes in porous silicon microdisks.[11] Here, a loading capacity of ~115 μg siRNA per billion MNCs (~ 2 mg) was achieved (Table 1). Scanning electron microscopy (SEM) revealed that silicon microdisks were successfully coated with PLGA-PEG nanoparticles (Figure 2b–c), which displayed a size and zeta potential of ~87 nm and −30 mV, respectively (2d–f and Table 1). The resulting docetaxel concentration was ~804 μg per billion MNCs (Table 1).
Figure 2.

Characterization of the MNC. a–c) Scanning electron microscopy (SEM) images of the MNC. a) Front side (top) and back side (bottom) of a porous silicon microdisk; b) front side (top) and back side (bottom) of a porous silicon microdisk coated with PLGA-PEG nanoparticles; c) front side (top) and back side (bottom) of a liposome-loaded porous silicon microdisk coated with PLGA-PEG nanoparticles. Scale bar, 0.6 μm. d) Chemical structure of the PLGA-PEG polymer. e) SEM images of PLGA-PEG nanoparticles. Scale bar, 0.4 μm. f) Size distribution of PLGA-PEG nanoparticles. g,h) Cumulative docetaxel release from the MNC. g) Full release profile from day 0–5; h), 24 h release profile. i,j) Cumulative siRNA release from the MNC. Full release profile from day 0–12; i) 24 h release profile; j) release profile from day 1–12. Results are presented as mean ± SD of three measurements. k) Western blot showing the transfection efficiency of a commercial transfection reagent (INTERFERin), DOPC liposomes, and the MNC. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control. Values above the bands represent the relative reduction in protein amount. Lip, DOPC liposomes; MEK, mitogen-activated protein kinase; p-MEK, phosphorylated MEK; Scr, scrambled siRNA.
Table 1.
Characterizations of the micro/nano composite (MNC).
| Size | Zeta potential (mV) | Docetaxel loading | siRNA loading | |
|---|---|---|---|---|
| Silicon microdisk | 2.6 μm * 0.7 μm | 25.3 ± 0.9 | N/A | N/A |
| PLGA-PEG nanoparticle | 87.48 ± 1.82 nm | −30.3 ± 0.5 | 52.61 ± 0.52 μg/mg | N/A |
| MNC/Lip- Braf&NP- docetaxel | 2.6 μm * 0.7 μm | −38.8 ± 0.9 | 803.55 ± 53.50 μg/billion | 114.90 ± 11.76 μg/billion |
Lip, liposomes; NP, polymeric nanoparticle; PEG, polyethylene glycol; PLGA, poly(lactic-co-glycolic acid).
The release of docetaxel and siRNA from the MNC was evaluated in vitro in buffers with a pH of 5.0 and 7.4. The results demonstrate that the release profile of docetaxel was similar at both pH values (Figure 2g–h). In particular, a burst release of 70% was observed during the first 24 h, after which the drug was slowly released, implying a diffusion-controlled release mechanism.[22] Exposing tumors to high levels of docetaxel in a short period of time could be beneficial for eliminating the bulk of cancer cells, while any remaining cells would be subject to lower concentrations of docetaxel released over a prolonged period of time. In contrast to docetaxel, siRNA release was dependent on pH (Figure 2i–j). In the first 24 h, ~16% and 50% of siRNA was released at a pH of 5.0 and 7.4, respectively. After 12 days, ~50% of the siRNA had been released at acidic pH and ~70% at neutral pH. The reason for pH-related differences in siRNA release kinetics is likely due to the accelerated degradation of silicon in alkaline environments,[23] thereby indicating a degradation-controlled release mechanism. Indeed, SEM images reveal that the MNC was gradually degraded in phosphate buffered saline (PBS) (Figure S1, Supporting Information).
The transfection efficiency of the MNC was evaluated by measuring protein levels of BRAF, phosphorylated-BRAF (p-BRAF), MEK (downstream effector), and phosphorylated-MEK (p-MEK) in A375 melanoma cells. Exposure to the MNC caused the expression levels of BRAF, p-BRAF and p-MEK to decrease by 60%, 50%, and 50%, respectively (Figure 2k). Notably, these values were comparable to those achieved with a commercial transfection reagent (Figure 2k).
Colocalization of siRNA and chemotherapeutics in the MNC was assessed using confocal microscopy. Figure 3a demonstrates that coumarin 6 (fluorescent small molecule equivalent of docetaxel)-loaded PLGA-PEG nanoparticles were predominantly distributed on the top and side surfaces of the silicon microdisk, while AF555-labeled siRNA-loaded liposomes were present throughout the MNC. This pattern of localization was further confirmed in images of the MNC positioned on its side (Figure S2a, Supporting Information). Moreover, the localization of siRNA and coumarin 6 in A375 cells was determined following exposure to the MNC. The results show that both compounds were colocalized after 0.5 h, after which they gradually separated as a consequence of drug release (Figure 3b). The cellular uptake efficiency of the MCN is shown in Figure S2b, Supporting Information.
Figure 3.

Colocaliztion of liposomes and PLGA-PEG nanoparticles. a,b) Confocal laser scanning microscopy (CLSM) images of the MNC. a) Columns from left to right show coumarin 6-loaded PLGA-PEG nanoparticle (green), AF555 siRNA-loaded liposomes (red), brightfield images of the MNC, and colocalization of liposomes and PLGA-PEG nanoparticles. b) Uptake of the MNC in A375 human melanoma cells. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Scale bar, 30 μm.
2.2. Synergistic Anticancer Activity In Vitro
The therapeutic efficacy of the MNC was assessed in A375 melanoma cells. A375 cells were used for in vitro experiments, while luciferase expressing highly metastatic A375 cells (A375SM-Luc) were used for in vivo experiments. DNA sequence analysis confirmed that both cell lines carried the BRAF V600E mutation (Figure S3, Supporting Information). Prior to loading the MNC with siRNA-containing liposomes, the anticancer activity of three different BRAF siRNA sequences was measured, and the most potent one was used throughout the study (Figure S4, Table S1, Supporting Information). Cells were then treated with MNCs containing various siRNA to docetaxel ratios (1:4.2, 1:1.1, and 1:0.6) and cell viability was measured (Figure 4a–c). The corresponding half maximal inhibitory concentration (IC50) values are listed in Table 2. Calculations of the combination index demonstrated that siRNA to docetaxel dose ratios of 1:4.2 and 1:0.6 were synergistic (combination index: 0.4–0.6), while a dose ratio of 1:1.1 displayed synergy only at low growth inhibitory rates (Figure 4d). In light of these results, a siRNA to docetaxel dose ratio of 1:4.2 was used for further experiments. In addition, cell viability measurements revealed that A375 cells and A375SM-Luc cells responded similarly to MNCs (Figure S5, Supporting Information).
Figure 4.

Synergistic therapeutic efficacy of the MNC in vitro. a–c) Viability of A375 human melanoma cells exposed to the MNC loaded with various ratios of BRAF siRNA to docetaxel (Doc): a) 1:4.2, b) 1:1.1, and c) 1:0.6. Data are presented as the mean ± SD of six replicates. d) Combination index for the MNC. e) Western blot analysis showing the effect of the MNC on MEK and extracellular signal-regulated kinase (ERK) protein levels in response to MNCs (siRNA, 100 nM; docetaxel, 100 nM). GAPDH was used as a loading control. Values above the bands represent the relative reduction in protein amount. NP, PLGA-(PEG) nanoparticles; p-ERK, phosphorylated-ERK.
Table 2.
Half maximal inhibitory concentration (IC50) values for the MNC loaded with various BRAF siRNA to docetaxel ratios.
| IC50 | Microdisk/NP-docetaxel | Microdisk/Lip-BRAF | Microdisk/Lip-BRAF&NP-docetaxel |
|---|---|---|---|
| 1:4.2 | 128.1 nM docetaxel | N/A | 12.9 nM siRNA + 54.3 nM docetaxel |
| 1:1.1 | 68.0 nM docetaxel | N/A | 27.0 nM siRNA + 29.7 nM docetaxel |
| 1:0.6 | 50.2 nM docetaxel | N/A | 48.2 nM siRNA + 28.9 nM docetaxel |
To investigate the mechanism of synergy, the expression of various proteins in the BRAF pathway was evaluated by Western blot. A375 cells were exposed to MNCs containing either BRAF siRNA or docetaxel and to MNCs containing both agents. The results show that BRAF siRNA reduced the levels of phosphorylated extracellular signal-regulated kinase (p-ERK), while docetaxel increased the levels of this protein (Figure 4e). When both agents were combined, p-ERK levels were similar to that of control cells. It is possible that a reduction in p-ERK in response to BRAF siRNA could improve the anticancer activity of docetaxel, since the activation of ERK has previously been linked to docetaxel drug resistance.[24] Moreover, treatment with BRAF siRNA caused a modest reduction in the levels of MEK and p-MEK, while simultaneous treatment with docetaxel dramatically reduced the protein levels of this oncogene (Figure 4e). Taken together, the synergy observed with BRAF siRNA and docetaxel could be due to a dramatic reduction in MEK and p-MEK levels and a normalization of pERK levels.
2.3. Preferential Accumulation of MNCs in Lung Tissue
The biodistribution of intravenously injected AF647-labeled siRNA-loaded liposomes, lipophilic carbocyanine (DiR)-loaded PLGA-PEG nanoparticles, and MNCs was measured in mice bearing A375SM melanoma lung metastases. The heart, liver, spleen, lungs, and kidneys were collected and the fluorescence intensity of each organ was measured ex vivo. The use of liposomes as a delivery vehicle resulted in siRNA retention in the kidneys after 6 h, while no signal was detected in any of the organs after 24 h (Figure 5a, Figure S6, Supporting Information). On the contrary, when siRNA-liposomes were loaded into the MNC, siRNA was detected in the liver and lungs after 6 h and predominantly in the lungs after 24 h (Figure 5a, Figure S6, Supporting Information). Quantitative analysis revealed that the siRNA amount in the lungs was 3.6-fold (6 h) and 5.5-fold (24 h) higher when using the MNC (Figure 5b). Notably, the accumulation of siRNA increased in all organs as a result of microparticle delivery, presumably due to reduced renal clearance of siRNA (Figure 5a–b, Figure S6, Supporting Information). Since siRNA delivered with liposomes was barely detectable in any organs after 24 h, it is difficult to assess whether the MNC increased the relative accumulation of siRNA in the lungs versus other organs. However, the deposition of siRNA/g tissue was the highest in the lungs when using the MNC (Figure 5b), which is typically not the case for liposomal delivery.[25]
Figure 5.

Biodistribution of the MNC in nude mice bearing A375SM melanoma lung metastases. a) Biodistribution of the MNC loaded with DOPC liposomes containing AF647-labeled siRNA or PLGA-PEG nanoparticles containing lipophilic carbocyanine (DiR). Top lane in each image (left to right): heart, liver, and spleen; bottom lane in each image (left to right): lungs and kidneys. b) Quantitative biodistribution of MNCs loaded with liposomes. c) Quantitative biodistribution of MNCs loaded with PLGA-PEG nanoparticles. d) Ratio of lung to liver and liver to spleen accumulation of MNCs. Results are presented as mean ± SD (n=3). e–h) Flow cytometry analysis of various particles in lung tissue (24 h post-injection). A375 human melanoma cells were differentiated from mouse cells using the APC-Cy7-labeled human leukocyte antigen (HLA)-ABC antibody. e) AF555-siRNA-loaded DOPC liposomes; f) MNC with AF555-siRNA-loaded DOPC liposomes; g) coumarin 6-loaded PLGA-PEG nanoparticles; h) MNC with coumarin 6-loaded PLGA-PEG nanoparticles. SSC, side scatter.
Moreover, lung accumulation of DiR was markedly increased when the MNC was used as a delivery vehicle (Figure 5a). In particular, delivery with polymeric nanoparticles resulted in high accumulation of DiR in the liver, while delivery with the MNC caused high retention of DiR in lung tissue. As a result of nanoparticle conjugation to the MNC, the fluorescent signal in the lungs increased 2-fold and 3.2-fold after 6 h and 24 h, respectively (Figure 5c). In fact, the MNC dramatically improved the ratio of lung to liver and lung to spleen retention (Figure 5d). For example, the ratio of lung to liver DiR accumulation for PLGA-PEG nanoparticles and MNCs after 24 h was 0.53 and 1.37, respectively. Collectively, these experiments demonstrate that the MNC can be used to achieve preferential accumulation of siRNA and small molecules in the lungs.
Next, mice were treated with AF555-tagged siRNA-loaded liposomes, coumarin 6-loaded PLGA-PEG nanoparticles, and MNCs. After 24 h, lung tissues were digested and analyzed with flow cytometry in order to determine the amount of siRNA and coumarin 6 in normal cells (negative for human leukocyte antigen, HLA) and cancer cells (positive for HLA). The results demonstrate that 0.008% and 0.772% of normal cells and cancer cells, respectively, were positive for siRNA when liposomes were used as a delivery system (Figure 5e). Notably, much higher amounts of siRNA were detected in cells as a result of MNC injection. In this case, 1.71% of normal cells and 8.97% of cancer cells were positive for siRNA (Figure 5f), representing a ten-fold increase in particle accumulation in metastatic lesions. Following administration of PLGA-PEG nanoparticles, 1.38% of normal cells and 21.2% of cancer cells were positive for coumarin 6 (Figure 5g). These levels increased to 3.46% (normal cells) and 40.1% (cancer cells) when the MNC was used as a delivery system (Figure 5h). In summary, these results indicate that the MNC can improve the accumulation of therapeutic agents in lung tissue and especially in melanoma lung metastases.
2.4. Therapeutic Efficacy of the MNC In Vivo
The therapeutic efficacy of the MNC was assessed in mice bearing A375SM-Luc melanoma lung metastases. The results clearly demonstrate that the MNC outperformed all other treatments, including combination therapy with siRNA-loaded liposomes and docetaxel-loaded PLGA-PEG nanoparticles. In particular, treatment with the MNCs resulted in a reduction in metastatic burden (Figure 6a, Figure S7, Supporting Information), a marked improvement in survival time (Figure 6b), and a dramatic decrease in the number of metastatic nodules (Figure 6c–d). Moreover, synergy between BRAF siRNA and docetaxel was clearly evident, as microdisks loaded with either agent alone were less effective than microdisks loaded with both agents. For example, 81 days after treatment initiation, 87.5% of the mice were alive in the MNC group, while corresponding survival percentages in the microdisk/docetaxel and microdisk/BRAF siRNA groups were 50% and 0%, respectively (Figure 6b). Moreover, combination treatment with docetaxel-loaded nanoparticles and BRAF siRNA-liposomes caused a similar reduction in metastatic burden as docetaxel-loaded nanoparticles alone (Figure 6a, Figure S7, Supporting Information), demonstrating that therapeutic synergy could only be obtained when the MNC was utilized. In addition, histological sections of lung tissue revealed that metastatic lesions could not be detected in mice treated with MNCs (Figure 6e, Figure S8, Supporting Information). Importantly, animal body weight was not affected in response to treatment (Figure S9, Supporting Information). To confirm that the MNC was capable of reducing BRAF protein levels in vivo, immunohistochemistry analysis of lung tissue was performed 72 h after particle injection. Indeed, the levels of BRAF were markedly reduced in the MNC and microdisk/BRAF siRNA treatment groups (Figure 6f). To further confirm the therapeutic efficacy of the delivery system, the anticancer activity of MNCs was evaluated in a more aggressive context. Specifically, cancer cells were inoculated into mice at a younger age, consequently resulting in higher tumor growth rates.[26] The results reveal a similar trend in the reduction of metastatic nodules in response to therapy, although the overall tumor burden was increased in younger mice (Figure S10, Supporting Information).
Figure 6.

Anticancer efficacy of the MNC in vivo. a) Anticancer efficacy of the MNC in a lung metastasis mouse model of A375SM human melanoma cells. Animals received weekly intravenous injections of the MNC for four weeks (docetaxel, 4 mg/kg; BRAF siRNA, 1 mg/kg). Melanoma cells were transfected with a luciferase gene and therapeutic efficacy was assessed through real-time monitoring of bioluminescence. b) Survival curves of mice bearing melanoma lung metastasis (n=8; Log-rank test, P<0.0001). Day 0 indicates treatment initiation. c) Number of pulmonary surface metastases 35 days after treatment initiation. Data is presented as the mean ± SD (n=4). d) Images of lungs 35 days after treatment initiation. e) Histological sections of the lungs 35 days after treatment initiation. Tissues were stained with haematoxylin and eosin. Scare bar, 200 μm. f) Immunohistochemistry of BRAF in melanoma lung metastases 35 days after treatment initiation. Scale bar, 50 μm.
3. Conclusion
We have developed a silicon-based micro/nano composite that permits simultaneous delivery of chemotherapeutic agents and siRNA to lung tissue. The composite was loaded with BRAF siRNA and docetaxel, and therapeutic efficacy was evaluated in A375 melanoma cells and in a mouse model of melanoma lung metastasis. The results reveal that superior synergistic anticancer efficacy can be obtained both in vitro and in vivo by using this delivery system. In particular, treatment with the composite reduced the tumor burden, decreased the number of metastatic nodules, and dramatically prolonged survival in comparison to monotherapy or combination therapy with liposomes and polymers. Figure S11 (Supporting Information) depicts a schematic for the proposed mechanism of synergy. On the molecular level, several studies have demonstrated that cancer cells can acquire resistance to BRAF inhibitors,[27] primarily through reactivation of MEK.[28] Here, docetaxel was shown to inhibit the MEK pathway, potentially sensitizing cells to BRAF inhibition. Furthermore, we also show that BRAF siRNA reversed docetaxel-induced overexpression of ERK, which has previously been linked to cancer cell survival.[29] Consequently, the combination of these therapeutic agents can act in a synergistic manner to prevent cancer cells from acquiring resistance to therapy.
Moreover, therapeutic synergy is also highly dependent on coordinated spatiotemporal delivery of therapeutic agents. The results demonstrate that the micro/nano composite was capable of simultaneously delivering siRNA and chemotherapy agents to metastatic melanoma lesions in the lungs. Indeed, the microparticle component is designed to preferentially accumulate in lung vasculature following intravenous injection. Once the composite is lodged in pulmonary vessels, the nanoparticle component can infiltrate cancerous lesions by exploiting the EPR effect. On the contrary, when polymeric nanoparticles and liposomes were freely injected, the former caused accumulation in the liver and spleen, while the latter resulted in siRNA retention in the kidneys. Consequently, treatment with separate delivery vehicles failed to achieve synergistic anticancer efficacy, highlighting the importance of colocalized delivery of combination therapy.
Taken together, the composite is an efficient platform for treating melanoma lung metastasis, since it enables synergistic antitumor activity both on the molecular and systemic level. In light of these results, it is likely that the micro/nano composite could also be used to treat other pulmonary conditions. Additionally, minor modifications to the composite could enable delivery of various therapeutic agents, thereby providing endless opportunities for combination therapy.
4. Experimental Section
Materials
PLGA-PEG-COOH was synthesized as previously described.[13] Materials were acquired from the following sources: PLGA (50:50, carboxylate end group, inherent viscosity 0.20 dL/g) from Lactel Absorbable Polymers (Pelham, AL, USA); NH2-PEG-COOH (MW2000) from Laysan Bio, Inc. (Arab, AL, USA); docetaxel (>99%) from LC Laboratories (Woburn, MA, USA); sulfo-NHS from Thermo Fisher Scientific, Inc; DiR dye from Life Technologies; DOPC from Avanti Polar Lipids, Inc. (Alabaster, AL, USA); Allstar Neg. siRNA AF555 and AF 647 from Qiagen (Germantown, MD, USA); fetal bovine serum (FBS) from Atlas Biologicals (Fort Collins, CO, USA); luciferin potassium salt from Gold Biotechnology Inc. (St. Louis, MO, USA); all antibodies from Cell Signaling Technology; cell counting kit-8 (CCK-8) from Dojindo Meolecular Tehcnologies, Inc. (Santa Clara, CA, USA); PBS, Dulbecco’s Modified Eagle Medium (DMEM), trypsin, penicillin/streptomycin solution, and anti-BRAF siRNA from GE Healthcare Life Sciences (Pittsburgh, PA, USA); all other chemicals from Sigma-Aldrich.
Preparation and characterization of the MNC
Porous silicon microdisks were fabricated by photolithography and electrochemical etching and modified with 3-aminopropyltriethoxysilane (APTES) as previously described.[30] Docetaxel-loaded PLGA-PEG nanoparticles were prepared by the nanoprecipitation method as previously reported.[31] Briefly, PLGA-PEG-COOH and docetaxel (weight ratio 20:1) were dissolved in acetone at a polymer concentration of 5 mg/mL. The solution was added drop-wise into ultrapure water with an oil-water ratio of 1:2 under vigorous stirring. After 6 h, the suspension was washed and centrifuged at 2000 rpm (20 min, 4 °C) and then at 12,000 rpm (20 min, 4 °C). The same procedure was used to synthesize fluorescent coumarin-6- or DiR-loaded PLGA-PEG nanoparticles, where docetaxel was replaced by 1 wt% coumatin-6 or DiR. siRNA-loaded DOPC liposomes were prepared as previously described.[20,21] In brief, siRNA (anti-BRAF, Neg. siRNA AF555, or AF647, 1 mg/mL in H2O), DOPC (20 mg/mL in t-butanol), Tween 20 (1.2‰ v/v in H2O), and tert-butanol with a volume ratio of 1:0.5:0.5:42 were mixed using a vortex. The mixture was freeze-dried in vacuum, hydrated in PBS, and sonicated at 4°C for a few minutes.
siRNA-loaded liposomes (70 μg siRNA) were then added to 1 billion vacuum-dried APTES-modified silicon microdisks and sonicated for several min at 4°C. The liposome-loaded microdisks were then washed with PBS and centrifuged two times at 4500 rpm. The PLGA-PEG nanoparticles (200 mg PLGA-PEG) were activated with 1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC, 60 mg) and N-hydroxysulfosuccinimide (sulfo-NHS, 48 mg) in PBS for 30 min and then mixed with liposome-loaded microdisks for 3 h (final volume: 20 mL). The MNCs were washed and centrifuged three times with water at 2000 rpm. Prior to performing experiments, the composites were sterilized with UV irradiation overnight.
The size and zeta potential of the particles were measured with a Zetasizer (Zetasizer Nano, Malvern). SEM characterization was performed with an ultra-high resolution scanning electron microscope (SEM 230, NovaNano). Western blot was performed with 4%–15% Bio-Rad Mini-Protean TGX Precast Gels following standard protocols.
Controlled release profile
The MNCs were dispersed in buffers with different pH values (acetate buffer, pH 5.0; PBS buffer, pH 7.4) containing 0.1% v/v Tween-80 to improve docetaxel solubility. The MNCs were placed in an orbital shaker (120 rpm, 37 °C) and the solution was centrifuged (10,000 rpm, 20 min) at designated time intervals. The pellet was then resuspended in fresh buffer to continue the drug release process. Half of the supernatant was extracted with dichloromethane and dissolved in a solution of acetonitrile and water (mobile phase, 1:1 volume ratio). The solution was then filtered through 0.45 mm polyvinylidene fluoride (PVDF) membranes for high-performance liquid chromatography (HPLC) analysis. The docetaxel amount in the column effluent was detected at 230 nm using ultraviolet–visible spectroscopy. The remaining supernatant was added to a black 96-well plate and fluorescent intensity was measured to determine the amount of AF555 siRNA.
Cell culture
A375 cells were obtained from ATCC. A375SM cells and A375SM-Luc cells were obtained from Dr. Isaiah Fidler’s laboratory (Houston, Texas, United States). Cells were cultured in DMEM with 10% (v:v) FBS and 1% (v:v) penicillin/streptomycin solution in an incubator at 37 °C with 5% CO2.
Confocal microscopy
A375 cells were seeded in 4-well chamber slides (LAB-TEK, Nagle Nunc, IL, USA) overnight at a density of 2×104 cells/well. Cells were then incubated with MNCs (0.1 billion/mL) for various time periods. The cells were washed twice with pre-warmed PBS, fixed with 70% ethanol for 20 min, washed twice with PBS, and stained with 4′,6-diamidino-2-phenylindole (DAPI) for 45 min. Finally, cells were washed twice with PBS and imaged with confocal microscopy.
In vitro cytotoxicity
A375 cells were seeded in 96-well plates (Costar, IL, USA) at a density of 3×103 cells/well. After 12 h, cells were exposed to MNCs for 72 h and cell viability was measured with the CCK-8 assay according to the manufacturer’s instructions.
Animal model
Animal studies were performed in accordance with the guidelines of the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals following protocols approved by the Institutional Animal Care and Use Committee (IACUC). Athymic nude mice were purchased from Charles River Laboratories (Boston, MA, USA). A375SM-Luc cells were harvested from exponential cultures and injected through the tail vein (106 cells/mouse) of six-week-old mice or ten-week-old mice. The formation of lung metastases was monitored using a bioluminescence imaging system (IVIS 200, Caliper).
Biodistribution
Biodistribution studies of the intravenously injected particles (AF647-labeled siRNA, 1 mg/kg; DiR, 0.2 mg/kg; MNCs, 15 billion/kg) were initiated 6 weeks after injection of A475SM-Luc cells. The heart, liver, spleen, lungs and kidneys were collected 6 h or 24 h after particle injection. Fluorescence intensity was quantified using IVIS 200 and normalized to tissue weight.
Flow cytometry (Fortessa, BD FACS) was used to determine the percentage of fluorescent particles in cancer cells and normal cells harvested from lung tissue. Briefly, 24 h after particle injection, lungs were minced and digested in DMEM/F12 medium containing 300 U/mL collagenase for 1.5 h at 37°C (100 mg tissue/mL medium). The solution was then filtered through a 40 μm mesh to obtain a single-cell suspension. Dead cells and A375SM cells were stained with CYTOX® Blue (Life Technologies) and APC-Cy7 Anti-human HLA-ABC antibody (BD Bioscience), respectively.
In vivo anticancer efficacy
Treatment was initiated two weeks after injection of A375SM-Luc cells. Based on bioluminescence imaging, mice were divided into groups with equal tumor burden. Randomly assigned treatment groups received weekly intravenous injections of particles or PBS (control group) for a duration of four weeks (BRAF siRNA, 1 mg/kg; docetaxel, 4 mg/kg). Tumor volume was quantified weekly using bioluminescent imaging (IVIS 200). At day 22 (tumor inoculation in six-week-old mice) or day 35 (tumor inoculation in ten-week-old mice) after treatment initiation, a subset of mice was sacrificed in order to count the number of metastatic nodules in the lungs. The lungs were then fixed with formalin, embedded in paraffin, and histological sections were stained with hematoxylin and eosin.
For immunohistochemistry (IHC) analysis, mice were administered with particles six weeks after injection of A375SM cells. The lungs were harvested 72 hours after particle injection and fixed and stained following standard IHC protocols using a monoclonal BRAF antibody and a peroxidase-labeled secondary antibody. Random images were captured with an optical microscope.
Supplementary Material
Acknowledgments
This work was supported by the Houston Methodist Research Institute, and the Ernest Cockrell Jr. Distinguished Endowed Chair (M.F.), and the US Department of Defense (W81XWH-09-1-0212) (M.F.), the National Institute of Health (U54CA143837, U54CA151668) (M.F.), Department of Defense grant W81XWH-12-1-0414 (M.F.) and the State of Texas CPRIT grant RP121071 (M.F. and H.S.). We thank Dr. Kemi Cui, Dr. Jianhua James Gu, and Dr. Xukui Wang for providing experimental facilities.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Yu Mi, Department of Nanomedicine, Houston Methodist Research Institute, 6670 Bertner Ave., Houston, TX 77030, USA
Dr. Chaofeng Mu, Department of Nanomedicine, Houston Methodist Research Institute, 6670 Bertner Ave., Houston, TX 77030, USA
Joy Wolfram, Department of Nanomedicine, Houston Methodist Research Institute, 6670 Bertner Ave., Houston, TX 77030, USA.
Dr. Zaian Deng, Department of Nanomedicine, Houston Methodist Research Institute, 6670 Bertner Ave., Houston, TX 77030, USA
Prof. Tony Ye Hu, Department of Nanomedicine, Houston Methodist Research Institute, 6670 Bertner Ave., Houston, TX 77030, USA
Prof. Xuewu Liu, Department of Nanomedicine, Houston Methodist Research Institute, 6670 Bertner Ave., Houston, TX 77030, USA
Dr. Elvin Blanco, Department of Nanomedicine, Houston Methodist Research Institute, 6670 Bertner Ave., Houston, TX 77030, USA
Prof. Haifa Shen, Department of Nanomedicine, Houston Methodist Research Institute, 6670 Bertner Ave., Houston, TX 77030, USA. Department of Medicine, Weill Cornell Medical College, 1300 York Avenue, New York, NY 10065, USA
Prof. Mauro Ferrari, Department of Nanomedicine, Houston Methodist Research Institute, 6670 Bertner Ave., Houston, TX 77030, USA. Department of Cell and Developmental Biology, Weill Cornell Medical College, 1300 York Avenue, New York, NY 10065, USA
References
- 1.a) Bao G, Mitragotri S, Tong S. Annu Rev Biomed Eng. 2013;15:253. doi: 10.1146/annurev-bioeng-071812-152409. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ferrari M. Nat Rev Cancer. 2005;5:161. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]; c) Sanhai WR, Sakamoto JH, Canady R, Ferrari M. Nat Nanotechnol. 2008;3:242. doi: 10.1038/nnano.2008.114. [DOI] [PubMed] [Google Scholar]; d) Riehemann K, Schneider SW, Luger TA, Godin B, Ferrari M, Fuchs H. Angew Chem Int Ed. 2009;48:872. doi: 10.1002/anie.200802585. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kim BY, Rutka JT, Chan WC. New Engl J Med. 2010;363:2434. doi: 10.1056/NEJMra0912273. [DOI] [PubMed] [Google Scholar]; f) Wolfram J, Zhu M, Yang Y, Shen J, Gentile E, Paolino D, Fresta M, Nie G, Chen C, Shen H, Ferrari M, Zhao Y. Curr Drug Targets. 2015;16:1671. doi: 10.2174/1389450115666140804124808. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Torchilin VP. Nat Rev Drug Discov. 2005;4:145. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]; h) Liong M, Lu J, Kovochich M, Xia T, Ruehm SG, Nel AE, Tamanoi F, Zink JI. ACS nano. 2008;2:889. doi: 10.1021/nn800072t. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Oliva N, Unterman S, Zhang Y, Conde J, Song HS, Artzi N. Adv Healthcare Mater. 2015;4:1584. doi: 10.1002/adhm.201400778. [DOI] [PubMed] [Google Scholar]
- 2.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 3.a) Feng SS, Chien S. Chem Eng Sci. 2003;58:4087. [Google Scholar]; b) Ferrari M. Trends Biotechnol. 2010;28:181. doi: 10.1016/j.tibtech.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Mol Pharm. 2008;5:505. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Almeida JP, Chen AL, Foster A, Drezek R. Nanomedicine. 2011;6:815. doi: 10.2217/nnm.11.79. [DOI] [PubMed] [Google Scholar]; c) Wolfram J, Yang Y, Shen J, Moten A, Chen C, Shen H, Ferrari M, Zhao Y. Colloids Surf B. 2014;124:17. doi: 10.1016/j.colsurfb.2014.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Semete B, Booysen L, Lemmer Y, Kalombo L, Katata L, Verschoor J, Swai HS. Nanomedicine. 2010;6:662. doi: 10.1016/j.nano.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Huang X, Li L, Liu T, Hao N, Liu H, Chen D, Tang F. Acs Nano. 2011;5:5390. doi: 10.1021/nn200365a. [DOI] [PubMed] [Google Scholar]
- 7.Decuzzi P, Godin B, Tanaka T, Lee SY, Chiappini C, Liu X, Ferrari M. J Controlled Release. 2010;141:320. doi: 10.1016/j.jconrel.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 8.a) Godin B, Tasciotti E, Liu X, Serda RE, Ferrari M. Acc Chem Res. 2011;44:979. doi: 10.1021/ar200077p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tasciotti E, Liu X, Bhavane R, Plant K, Leonard AD, Price BK, Cheng MM, Decuzzi P, Tour JM, Robertson F, Ferrari M. Nat Nanotechnol. 2008;3:151. doi: 10.1038/nnano.2008.34. [DOI] [PubMed] [Google Scholar]; c) van de Ven AL, Kim P, Haley O, Fakhoury JR, Adriani G, Schmulen J, Moloney P, Hussain F, Ferrari M, Liu X, Yun SH, Decuzzi P. J Controlled Release. 2012;158:148. doi: 10.1016/j.jconrel.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wolfram J, Shen H, Ferrari M. J Controlled Release. 2015;219:406. doi: 10.1016/j.jconrel.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adriani G, de Tullio MD, Ferrari M, Hussain F, Pascazio G, Liu X, Decuzzi P. Biomaterials. 2012;33:5504. doi: 10.1016/j.biomaterials.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blanco E, Sangai T, Wu S, Hsiao A, Ruiz-Esparza GU, Gonzalez-Delgado CA, Cara FE, Granados-Principal S, Evans KW, Akcakanat A, Wang Y, Do KA, Meric-Bernstam F, Ferrari M. Mol Ther. 2014;22:1310. doi: 10.1038/mt.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Xu R, Huang Y, Mai J, Zhang G, Guo X, Xia X, Koay EJ, Qin G, Erm DR, Li Q, Liu X, Ferrari M, Shen H. Small. 2013;9:1799. doi: 10.1002/smll.201201510. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tanaka T, Mangala LS, Vivas-Mejia PE, Nieves-Alicea R, Mann AP, Mora E, Han HD, Shahzad MM, Liu X, Bhavane R, Gu J, Fakhoury JR, Chiappini C, Lu C, Matsuo K, Godin B, Stone RL, Nick AM, Lopez-Berestein G, Sood AK, Ferrari M. Cancer Res. 2010;70:3687. doi: 10.1158/0008-5472.CAN-09-3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noori Koopaei M, Khoshayand MR, Mostafavi SH, Amini M, Khorramizadeh MR, Jeddi Tehrani M, Atyabi F, Dinarvand R. Iranian J Pharm Res. 2014;13:819. [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng J, Teply BA, Sherifi I, Sung J, Luther G, Gu FX, Levy-Nissenbaum E, Radovic-Moreno AF, Langer R, Farokhzad OC. Biomaterials. 2007;28:869. doi: 10.1016/j.biomaterials.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jang S, Atkins MB. Lancet Oncol. 2013;14:e60. doi: 10.1016/S1470-2045(12)70539-9. [DOI] [PubMed] [Google Scholar]
- 15.a) Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Nature. 2002;417:949. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]; b) Kumar R, Angelini S, Czene K, Sauroja I, Hahka-Kemppinen M, Pyrhonen S, Hemminki K. Clin Cancer Res. 2003;9:3362. [PubMed] [Google Scholar]; c) Salama AK, Flaherty KT. Clin Cancer Res. 2013;19:4326. doi: 10.1158/1078-0432.CCR-13-0779. [DOI] [PubMed] [Google Scholar]; d) Ribas A, Flaherty KT. Nat Rev Clin Oncol. 2011;8:426. doi: 10.1038/nrclinonc.2011.69. [DOI] [PubMed] [Google Scholar]
- 16.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. New Engl J Med. 2010;363:809. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garber K. Nat Biotechnol. 2013;31:666. doi: 10.1038/nbt0813-666b. [DOI] [PubMed] [Google Scholar]
- 18.a) Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, Dummer R, Trefzer U, Larkin JM, Utikal J, Dreno B, Nyakas M, Middleton MR, Becker JC, Casey M, Sherman LJ, Wu FS, Ouellet D, Martin AM, Patel K, Schadendorf D. New Engl J Med. 2012;367:107. [Google Scholar]; b) McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, Ribas A, Hogg D, Hamid O, Ascierto PA, Garbe C, Testori A, Maio M, Lorigan P, Lebbe C, Jouary T, Schadendorf D, O’Day SJ, Kirkwood JM, Eggermont AM, Dreno B, Sosman JA, Flaherty KT, Yin M, Caro I, Cheng S, Trunzer K, Hauschild A. Lancet Oncol. 2014;15:323. doi: 10.1016/S1470-2045(14)70012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Tran MA, Gowda R, Sharma A, Park EJ, Adair J, Kester M, Smith NB, Robertson GP. Cancer Res. 2008;68:7638. doi: 10.1158/0008-5472.CAN-07-6614. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Coffee EM, Faber AC, Roper J, Sinnamon MJ, Goel G, Keung L, Wang WV, Vecchione L, de Vriendt V, Weinstein BJ, Bronson RT, Tejpar S, Xavier RJ, Engelman JA, Martin ES, Hung KE. Clin Cancer Res. 2013;19:2688. doi: 10.1158/1078-0432.CCR-12-2556. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Smalley KS, Flaherty KT. Br J Cancer. 2009;100:431. doi: 10.1038/sj.bjc.6604891. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Shannon AM, Telfer BA, Smith PD, Babur M, Logie A, Wilkinson RW, Debray C, Stratford IJ, Williams KJ, Wedge SR. Clin Cancer Res. 2009;15:6619. doi: 10.1158/1078-0432.CCR-08-2958. [DOI] [PubMed] [Google Scholar]
- 20.Queirolo P, Picasso V, Spagnolo F. Cancer Treat Rev. 2015;41:519. doi: 10.1016/j.ctrv.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 21.Gupta A, Love S, Schuh A, Shanyinde M, Larkin JM, Plummer R, Nathan PD, Danson S, Ottensmeier CH, Lorigan P, Collins L, Wise A, Asher R, Lisle R, Middleton MR. Ann Oncol. 2014;25:968. doi: 10.1093/annonc/mdu054. [DOI] [PubMed] [Google Scholar]
- 22.Kearney CJ, Mooney DJ. Nat Mater. 2013;12:1004. doi: 10.1038/nmat3758. [DOI] [PubMed] [Google Scholar]
- 23.a) Martinez JO, Evangelopoulos M, Chiappini C, Liu X, Ferrari M, Tasciotti E. J Biomed Mater Res A. 2014;102:3540. doi: 10.1002/jbm.a.35017. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ulin VP, Ulin NV, Soldatenkov FY, Semenov AV, Bobyl AV. Semiconductors. 2014;48:1211. [Google Scholar]
- 24.Jeong EK, Lee SY, Jeon HM, Ju MK, Kim CH, Kang HS. Int J Oncol. 2010;37:655. doi: 10.3892/ijo_00000714. [DOI] [PubMed] [Google Scholar]
- 25.a) Abrams MT, Koser ML, Seitzer J, Williams SC, DiPietro MA, Wang W, Shaw AW, Mao X, Jadhav V, Davide JP, Burke PA, Sachs AB, Stirdivant SM, Sepp-Lorenzino L. Mol Ther. 2010;18:171. doi: 10.1038/mt.2009.208. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chang YJ, Chang CH, Chang TJ, Yu CY, Chen LC, Jan ML, Luo TY, Lee TW, Ting G. Anticancer Res. 2007;27:2217. [PubMed] [Google Scholar]
- 26.Kjonniksen I, Storeng R, Pihl A, McLemore TL, Fodstad O. Cancer Res. 1989;49:5148. [PubMed] [Google Scholar]
- 27.a) Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, Sosman JA, Ribas A, Lo RS. Nature. 2010;468:973. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, Caponigro G, Hieronymus H, Murray RR, Salehi-Ashtiani K, Hill DE, Vidal M, Zhao JJ, Yang X, Alkan O, Kim S, Harris JL, Wilson CJ, Myer VE, Finan PM, Root DE, Roberts TM, Golub T, Flaherty KT, Dummer R, Weber BL, Sellers WR, Schlegel R, Wargo JA, Hahn WC, Garraway LA. Nature. 2010;468:968. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, Salton M, Dahlman KB, Tadi M, Wargo JA, Flaherty KT, Kelley MC, Misteli T, Chapman PB, Sosman JA, Graeber TG, Ribas A, Lo RS, Rosen N, Solit DB. Nature. 2011;480:387. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das Thakur M, Stuart DD. Clin Cancer Res. 2014;20:1074. doi: 10.1158/1078-0432.CCR-13-0103. [DOI] [PubMed] [Google Scholar]
- 29.a) Mhaidat NM, Thorne RF, Zhang XD, Hersey P. Mol Cancer Res. 2007;5:1073. doi: 10.1158/1541-7786.MCR-07-0059. [DOI] [PubMed] [Google Scholar]; b) Mhaidat NM, Zhang XD, Jiang CC, Hersey P. Clin Cancer Res. 2007;13:1308. doi: 10.1158/1078-0432.CCR-06-2216. [DOI] [PubMed] [Google Scholar]
- 30.Godin B, Chiappini C, Srinivasan S, Alexander JF, Yokoi K, Ferrari M, Decuzzi P, Liu X. Adv Funct Mater. 2012;22:4225. doi: 10.1002/adfm.201200869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamaly N, Fredman G, Subramanian M, Gadde S, Pesic A, Cheung L, Fayad ZA, Langer R, Tabas I, Farokhzad OC. Proc Natl Acad Sci USA. 2013;110:6506. doi: 10.1073/pnas.1303377110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.