

Abstract
Cadherin subtype switching from E-cadherin to N-cadherin is associated with the epithelial-to-mesenchymal transition (EMT), a process required for invasion and dissemination of carcinoma cells. We found N-cadherin is expressed in human and mouse pancreatic intraepithelial neoplasia (PanIN), suggesting that N-cadherin may also play a role in early stage pancreatic cancer. To investigate the role of N-cadherin in mouse PanIN (mPanIN), we simultaneously activated oncogenic K-rasG12D and deleted the N-cadherin (Cdh2) gene in the murine pancreas. Genetic ablation of N-cadherin (N-cad KO) caused hyperproliferation, accelerated mPanIN progression, and early tumor development in K-rasG12D mice. Decreased E-cadherin and redistribution of β-catenin accompanied the loss of N-cadherin in pancreatic ductal epithelial cells (PDEC). Nuclear accumulation of β-catenin and its transcription co-activator Tcf4 led to activation of Wnt/β-catenin target genes. Unexpectedly, loss of N-cadherin in the K-rasG12D model resulted in increased mPanIN progression and tumor incidence. These in vivo results demonstrate for the first time that N-cadherin functions as a growth suppressor in the context of oncogenic K-ras.
Keywords: pancreatic intraepithelial neoplasia (PanIN), hyperproliferation, β-catenin, Wnt signaling, mouse models
Introduction
Pancreatic ductal adenocarcinoma (PDA) is the fourth leading cause of cancer death. In the United States approximately 48,000 patients are diagnosed with pancreatic cancer annually, and nearly an equal number will die from the disease (ACS, 2015). It is among the most lethal of all cancers, with a 5-year survival rate of only 5%. Activating point mutations in the KRAS oncogene are found in nearly 100% of PDA cases. Although KRAS mutations are important initiating events, it is clear that inactivation of the TP53 and p16/CDKN2A tumor suppressor genes, as well as SMAD pathway genes, are also important for tumor development and progression (1). A better understanding of the molecular changes driving PDA development is crucial for identifying potential therapeutic targets for this devastating disease.
PDA is thought to evolve through progression of precursor lesions, called pancreatic intraepithelial neoplasias (PanINs). PanINs are classified into four subgroups (1A, 1B, 2, and 3), based upon histological criteria, to describe the progression from hyperplastic to dysplastic ducts. PDA is characterized by genomic instability, and a large number of mutations and chromosomal abnormalities are found in each individual carcinoma (2, 3). Most human PDAs display mutations in 12 core signaling pathways including homophilic cell adhesion, KRAS and Wnt signaling pathways (3), but whether these pathways work in concert to enhance disease progression is unknown.
Classical cadherins are a family of cell surface glycoproteins that mediate calcium-dependent cell-cell adhesion primarily in a homophilic manner. Their adhesive function requires interaction with the actin cytoskeleton through catenins. It has been shown that cadherins play a role in late stage tumorigenesis (4). Specifically, cadherin subtype switching – from E-cadherin to N-cadherin – occurs during neoplastic cell invasion and metastasis and is associated with the epithelial-to-mesenchymal transition (EMT) (5, 6). Cadherin switching usually refers to a change in expression from E-cadherin to N-cadherin, but also includes situations in which E-cadherin levels do not change significantly but cells turn on N-cadherin expression. N-cadherin is not required for the EMT process itself, but it regulates the cell’s behavior following EMT including the cell’s migratory ability (7). This hypothesis is supported by genetic studies demonstrating that neoplastic cell behavior is sensitive to changes in N-cadherin expression levels (8–10).
A role for N-cadherin in the early stage of cancer development has not been studied. However, one study suggested that changes in E-cadherin might play a role in human PanIN development (11). E-cadherin was reduced at the membrane and increased in the cytoplasm in PanIN compared to normal ducts (11). Notably, β-catenin, a downstream effector of the Wnt signaling pathway, was found in the cytoplasm and nucleus of high-grade PanIN lesions. Furthermore, K-rasG12D-induced mPanINs express Tcf4 in the nucleus and exhibit activation of the β-catenin/Tcf4-responsive LacZ-reporter consistent with a role for Wnt signaling in mPanIN development (12). To our knowledge, N-cadherin protein expression has not been examined in PanIN, although its expression has been reported in primary and metastatic PDA (13).
Canonical Wnt signaling was shown recently to be required for mPanIN formation (14). Three complementary approaches were used to inhibit Wnt/β-catenin signaling in the K-rasG12D mouse model. First, genetic mosaicism of β-catenin in the K-rasG12D pancreas demonstrated that β-catenin-null cells did not contribute to mPanIN lesions. Second, induction of the secreted Wnt inhibitor Dkk1 in the K-rasG12D pancreas inhibited mPanIN development and progression. Third, treatment of K-rasG12D mice with a monoclonal antibody that binds to multiple Frizzled receptors and blocks their activity resulted in fewer mPanINs compared to nontreated K-rasG12D animals. Taken together, these data demonstrate a need for canonical Wnt signaling in K-rasG12D-induced mPanIN development.
Herein we report that N-cadherin is induced in human and murine PanINs, and that deletion of N-cadherin (Cdh2) gene in ductal cells leads to activation of Wnt/β-catenin target genes and accelerated mPanIN formation in the K-rasG12D mouse model. These in vivo results demonstrate for the first time that N-cadherin functions as a growth suppressor in the context of oncogenic K-ras.
Results and Discussion
N-cadherin is expressed in PanIN lesions
N-cadherin expression is associated primarily with poorly differentiated tumors, but no information is available concerning N-cadherin expression in PanIN, precursor lesions to PDA. Therefore, we examined N-cadherin expression in both human and murine PanIN specimens. In human pancreas, E-cadherin was located primarily at the lateral borders between the pancreatic ductal epithelial cells (PDEC) in normal tissue, whereas N-cadherin was never observed in normal ducts (Fig. 1A,B). By contrast, in human PanIN lesions N-cadherin was found to be expressed in ductal cells where it localized to regions of cell-cell contact together with E-cadherin (Fig. 1C,D and Suppl. Fig. 1). N-cadherin staining was heterogeneous and often less intense compared to E-cadherin in PanINs. The human data were confirmed in LSL-K-rasG12D; Pdx1/Cre mice (referred to as KC mice) that develop mPanIN lesions similar to humans (15). Co-expression of N-cadherin and E-cadherin was observed in mPanIN, along with N-cadherin-positive ductal cells with weak or little E-cadherin expression (Fig. 1E,F). Next, we took advantage of a N-cadherin reporter mouse line (NcadlacZ/+) where the lacZ gene is under the transcriptional regulation of the endogenous N-cadherin promoter (16). The NcadlacZ/+ reporter allele was introduced into the KC and KPC (17) (i.e., LSL-K-rasG12D; LSL-Trp53R172H; Pdx1/Cre) mouse models. In addition to islets of Langerhans and nerve bundles where N-cadherin is expressed normally, β-galactosidase activity was observed in PDA of the KPC mice confirming the utility of the NcadlacZ/+ reporter (Suppl. Fig. 2). Importantly, the NcadlacZ/+ reporter was active in mPanIN lesions of the KC mice, thus confirming expression of N-cadherin in these early precursor lesions (Fig. 1H). Collectively, these in vivo data demonstrate that ectopic N-cadherin expression occurs initially at the PanIN stage of the disease prior to EMT.
Figure 1. E- and N-cadherin expression in human and murine PanIN.

Double immunofluorescence analysis of E-cadherin (antibody from Santa Cruz) and N-cadherin (antibody from Invitrogen) in human (A–D) and murine (E, F) pancreas specimens. Pancreas samples were obtained from patients at Johns Hopkins Medical Center (JHMC) following approval by JHMC IRB. E-cadherin was present both in normal pancreatic duct (A) and PanIN (C). By contrast, N-cadherin was absent from normal ducts (B), but found upregulated in a subset of PanIN lesions (D). Arrowheads point to areas shown in merged image (insets). E-cadherin (E) and N-cadherin (F) expression in mPanIN from K-rasG12D; Pdx1/Cre (KC) mice. Note ductal cells with decreased E-cadherin and increased N-cadherin. Histological analysis of β-galactosidase-stained mPanIN from Ncad+/+ (G) and NcadlacZ/+ (H) KC mice. N-cadherin LacZ reporter showed a positive signal (blue) in ductal cells and islets of Langerhans (Is) in the KC NcadlacZ/+ mice (H) whereas KC mice lacking the LacZ reporter served as a negative control (G).
Loss of N-cadherin in the context of oncogenic K-ras leads to increased mPanIN incidence and progression
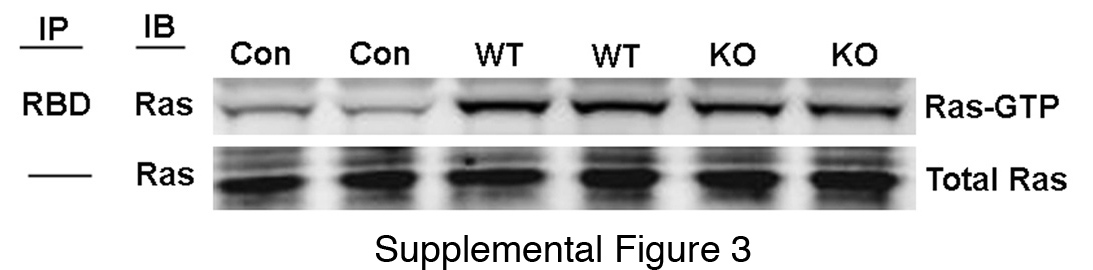
To determine the consequences of interfering with N-cadherin function during mPanIN development, the N-cadherin floxed allele (18) was introduced into the KC model to generate LSL-K-rasG12D; N-cadfl/fl; Pdx1/Cre mice referred to as Ncad knockout (KO). KC mice not containing the N-cadherin floxed alleles are referred to as Ncad wild-type (WT). LSL-K-rasG12D; N-cadfl/fl mice lacking the Cre transgene served as negative controls. Pancreas development and morphology are normal in N-cadfl/fl; Pdx1/Cre mice lacking the K-ras mutation (19). Surprisingly, the Ncad KO pancreata were significantly larger compared to Ncad WT (Fig. 2A,B). In comparison to Ncad WT, the Ncad KO exhibited extensive mPanINs accompanied by periductal desmoplasia (Fig. 2C). Significant collagen deposition was associated with the desmoplastic reaction in the Ncad KO as depicted by Masson Trichrome stain (Fig. 2D). Alcian blue staining identified abundant mucin in the Ncad KO mPanIN lesions (Fig. 2E). The changes in Ncad KO mice were not seen in Ncad heterozygous mice (LSL-K-rasG12D; N-cadlacZ/+; Pdx1/Cre) (data not shown). The proliferative index of mPanINs was assessed by PCNA immunohistochemistry. There was a two-fold increase in PCNA-positive ductal cells in N-cad KO compared to N-cad WT mPanIN lesions at comparative mPanIN grades (Fig. 2F,G). To determine if loss of N-cadherin affected K-ras activity, we examined levels of activated, or GTP-bound, Ras. There was no change in Ras-GTP levels between Ncad KO and WT pancreata (Suppl. Fig. 3). Together, these data indicate that loss of N-cadherin caused an increase in ductal cell proliferation leading to excessive mPanIN development in the context of K-rasG12D.
Figure 2. Enlarged pancreata, increased mPanIN lesions and desmoplasia in Ncad KO KC mice.

(A) Whole mount images of pancreata from 5- to 6-month old control (Cre minus), Ncad WT, and Ncad KO mice and (B) comparison of pancreas mass (gram) in 6- and 9- to 12-month old mice. *, p<0.05. Transgenic mice were in a mixed genetic background. All mouse experiments were performed under the approval of the Thomas Jefferson University IACUC. Representative pancreas sections from 5- to 6-month old mice stained with H&E (C), Masson trichrome (D), and Alcian blue (E). Representative images of immunohistochemical analysis of PCNA (antibody from Invitrogen) in pancreata from 6-month old control, Ncad WT, and Ncad KO mice (F). (G) Quantification of PCNA nuclear staining. n, number of independent animals examined by immunohistochemistry; **, p<0.01.
Similar to human disease, the KC mice recapitulate the progression from mPanIN-1 – PanIN-3 eventually developing PDA after 1 year of age (15). We examined sections from the pancreata of each transgenic model to score the presence of different grades of mPanIN or PDA, which were defined by the consensus criteria of the pancreatic mouse modeling community (20). Histological examination showed evidence of accelerated mPanIN development in Ncad KO compared to Ncad WT KC mice. Between 8 and 12 months of age, the majority of mPanIN lesions in Ncad WT mice were classified as mPanIN-2 whereas Ncad KO displayed less mPanIN-2 and more advanced mPanIN-3 lesions as well as invasive carcinoma (Ncad KO, 6/26 vs Ncad WT 1/18, p < 0.05) (Fig. 3A). PET imaging detected a strong 18F-FDG signal in the abdominal region of an 8-month-old Ncad KO (Fig. 3B). The pancreas was removed and a solid mass identified (Fig. 3C), and histological examination confirmed the carcinoma pathology (Fig. 3D). Taken together, we conclude that mPanIN progression and tumorigenesis were accelerated in Ncad KO compared to Ncad WT KC mice.
Figure 3. Accelerated mPanIN and tumor development in Ncad KO KC mice.

(A) Quantification of mPanIN and PDA in 8- to 12-month old Ncad KO (n=26) and WT (n=18) mice. Percent (%) total indicates the ratio of mice that developed mPanIN (highest grade counted) or PDA to the total number of mice. An experienced pancreatic pathologist (CS) reviewed tissue specimens in a blinded fashion. Chi-square analysis showed a significant difference between Ncad KO and Ncad WT KC mice (p < 0.05). (B) Whole-body PET scan using 18F-FDG (Fludeoxyglucose (18F)) on 8-month old control, Ncad WT, and Ncad KO mice. Strong uptake of 18F-FDG was observed in the pancreatic region of Ncad KO mouse (arrow) and absent from control and Ncad WT mice. (C) Whole mount images of the pancreata removed from the PET imaged mice (B). A solid mass was found in the pancreas (arrow) of Ncad KO mouse consistent with PET image. (D) Histological analysis of the pancreata confirmed carcinoma in the Ncad KO and mPanIN in the Ncad WT.
E-cadherin and β-catenin expression in N-cadherin KO mPanIN lesions
E-cadherin was expressed strongly at the lateral borders of the Ncad WT ductal epithelial cells (Suppl. Fig. 4B). In comparison, E-cadherin expression was weaker in the Ncad KO ducts (Suppl. Fig. 4C). Western analysis was performed on total pancreata lysates (Suppl. Fig. 4D). Quantification of E-cadherin relative to the ductal marker cytokeratin 19 (CK19) indicated a reduction in E-cadherin levels in Ncad KO compared to Ncad WT KC mice (Suppl. Fig. 4E).
Changes in E-cadherin levels can modulate β-catenin activity and growth of neoplastic cells (21). Therefore, we examined β-catenin expression in pancreata from 6-month old Ncad KO and WT KC mice. β-catenin was increased in the cytoplasm and nuclei of the Ncad KO compared to WT ductal cells (Fig. 4A). Moreover, in adjacent Ncad KO sections we observed an increase in nuclear localization of the β-catenin binding partner, Tcf4 (Fig. 4B,C). Next, we examined the expression of representative β-catenin/Tcf4 target genes in these pancreata from 6-month-old mice. Loss of N-cadherin was associated with increased expression of axin2 and MMP-7 transcripts in Ncad KO pancreata compared to Ncad WT (Fig. 4D). To further examine the cellular distribution of β-catenin, primary PDEC lines were derived from Ncad KO and WT pancreata as previously described (22). As predicted from the genotypes, N-cadherin was absent from the N-cad KO PDEC lines (Fig. 4E). Cellular fractionation demonstrated that β-catenin was increased in the nuclear fraction of the Ncad KO PDECs (Fig. 4F). Moreover, expression of cyclin D1, a Wnt/β-catenin target, was increased in the Ncad KO PDECs (Fig. 4E). Taken together, these data suggest that an overall decrease in cadherin expression leads to increased nuclear β-catenin and expression of Wnt/β-catenin target genes, thus contributing to the acceleration of mPanIN development in the Ncad KO KC mice.
Figure 4. Enhanced β-catenin/Tcf4 activity Ncad KO KC.

Immunohistochemical analysis of β-catenin (antibody from Invitrogen) (A) and Tcf4 (antibody from Millipore) (B) expression on adjacent sections of 6-month old control, Ncad WT, and Ncad KO pancreata. Note the corresponding increase in both β-catenin and Tcf4 in cells from adjacent sections in Ncad KO. (C) Quantification of Tcf4-positive nuclei in mPanIN lesions. (D) qPCR of β-catenin/Tcf4 target genes axin2 and MMP7 in pancreata from control (n=3), Ncad WT (n=8), and Ncad KO (n=6) mice. Total RNA was isolated from pancreas tissue disassociated into single cells using RNeasy Mini Kit (Qiagen). Expression of the target gene was compared with the expression level of GAPDH. Primer sequences used were Axin2 forward: 5´ AGCCGCCATAGTC 3´, Axin2 reverse: 5´ GGTCCTCTTCATAGC 3´; MMP7 forward: 5' GCAAGGAGAGATCATGGAGACAGCTT 3', MMP7 reverse: 5' AAGTTCACTCCTGCGTCCTCACCAT 3'; GAPDH forward: 5’ CCACTCTTCCACCTTCGATG 3’, GAPDH reverse: 5’ TCCACCACCCTGTTGCTGTA 3’. (E) Immunoblot analysis of N-cadherin and cyclin D1 (antibody from Santa Cruz) in total protein lysates of PDEC isolated from Ncad WT and Ncad KO mice. (F) Immunoblot analysis of β-catenin in total lysate, cytoplasmic, and nuclear fractions prepared from PDEC isolated from Ncad WT and Ncad KO mice. Lamin B1 (antibody from Abcam) served as a control for enrichment of the nuclear fraction.
Perspective
Contact inhibition of cell proliferation is often invoked in the context of epithelial cells where E-cadherin-mediated adhesion restrains cell growth and division (23). By contrast, N-cadherin expression is generally associated with EMT and the acquisition of mesenchymal morphology and migratory capabilities (6). In this study, we found N-cadherin expression at an earlier stage of pancreatic cancer than previously appreciated. Subpopulations of ductal cells in human and murine PanIN lesions were positive for N-cadherin. Surprisingly, genetic ablation of N-cadherin caused ductal cell hyperproliferation and accelerated mPanIN progression in the K-rasG12D mice. This result indicates that N-cadherin regulates the proliferative potential of a unique subset of ductal cells in mPanIN lesions.
It has been proposed that oncogenic K-ras regulates N-cadherin expression in pancreatic cancer, however its relevance to disease progression is poorly understood. In PDEC grown in culture, activated K-ras was shown to induce N-cadherin expression (24, 25). In comparison to these in vitro results, here we show that only a subset of ductal cells express N-cadherin in K-rasG12D-induced mPanIN lesions. However, when PDEC isolated from N-cad WT K-rasG12D mice were grown in vitro, N-cadherin induction was observed in all the cells. Interestingly, N-cadherin expression in pancreatic cancer cell lines depends on signals from the extracellular matrix. Cells grown on collagen I activate N-cadherin whereas fibronectin or laminin substrates had no effect on N-cadherin expression (9). The difference between these in vitro and in vivo findings suggest the PanIN microenvironment, e.g., basement membrane, may regulate N-cadherin activation in vivo. In the future, it will be of interest to examine N-cadherin expression in PanIN lesions with respect to changes in basement membrane integrity.
Studies indicate that the ability of N-cadherin to regulate cell proliferation depends on the cellular context and the health of the tissue. N-cadherin-mediated adhesion was shown to promote cell cycle arrest of CHO cells (26) and C2C12 myoblasts (27, 28) in culture. Moreover, we reported recently that interfering with α-catenins, which link N-cadherin to the actin cytoskeleton, results in aberrant N-cadherin expression and hyperproliferation of cardiomyocytes (29). By contrast, in another study it was shown by use of N-cadherin conditional knockout mice that N-cadherin is a positive regulator of smooth muscle cell proliferation following vascular injury (30). Moreover, we previously reported that reduced N-cadherin levels in the context of both K-rasG12D and p53R172H mutations (i.e. KPC mice) led to a decrease in proliferation of pancreatic cancer cells consistent with a growth-promoting role for N-cadherin in later stage cancer (10). The different affects of N-cadherin on PDEC growth in the KPC (10) and KC (this study) mouse models depends on N-cadherin gene dosage (summarized in Suppl. Table I). Taken together, these data show that N-cadherin can either inhibit or promote cell proliferation depending on the cellular context and disease state.
It is well established that activation of Wnt signaling plays a critical role in mPanIN development (14). In the present study, we demonstrate an increase in nuclear localization of β-catenin and Tcf4, and in expression of target genes in the N-cad KO KC mice. These data support the idea that decreased cadherin expression in N-cad KO KC mice alters the cellular distribution of β-catenin (membrane vs. cytoplasmic) and facilitates its accumulation in the nucleus where it activates genes involved in mPanIN formation. Additional signaling pathways including the Hippo pathway may work in concert with N-cadherin-mediated adhesion to regulate proliferation in the K-rasG12D mice (31). Interestingly, it was shown recently that E-cadherin mediates contact inhibition of proliferation in mammary epithelial cells via Hippo signaling pathway components and the regulation of the subcellular localization of Yes-associated protein (Yap) (32). Yap is a transcriptional co-activator that interacts with members of the TEAD transcription factor family to regulate genes involved in cell proliferation and survival. Importantly, it was shown that Yap is required in β-catenin-dependent cancers (33). Yap and the transcription factor Tbx5 were found to form a complex with β-catenin. Whether loss of N-cadherin modulates Yap nuclear translocation in mPanIN lesions is under investigation.
Previous studies indicated that N-cadherin was capable of promoting pancreatic tumor cell growth (9, 10). By contrast, here we report for the first time that N-cadherin functions as a growth suppressor in the context of K-ras-induced mPanIN development. Loss of N-cadherin causes activation of the Wnt/β-catenin signaling pathway and ductal cell hyperproliferation in K-rasG12D mice. Our unexpected results provide new insight into N-cadherin function in early pancreatic cancer.
Supplementary Material
Double immunofluorescence analysis of E-cadherin (antibody from Santa Cruz) and N-cadherin (antibody from Invitrogen) in human PanIN specimen. Note the co-expression of E-cadherin (A) and N-cadherin (B) shown in the merged image (C).
{kind=link}
Histological analysis of β-galactosidase-stained KPC N-cadherinlacZ/+ pancreas. LacZ expression was detected in (A) Islets of Langerhans (Is), (B) neural plexus (N), (C) mPanIN lesions, and (D) PDA in LSL-K-rasG12D; LSL-Trp53R172H; Pdx1/Cre; NcadlacZ/+ mice. N-cadherin lacZ expression (blue) was observed in a subpopulation of ductal cells (arrows) in mPanIN lesions (C) and in poorly differentiated tumor cells (D). The latter cells represent tumor cells that have undergone epithelial-mesenchymal transition (EMT).
{kind=link}
Immunoblot analysis of Ras and RAS-GTP in total protein lysates from 6-month old Ncad WT and Ncad KO mice. Ras activation assay was performed using Ras activation assay kit (Upstate).
{kind=link}
Immunohistochemical detection of E-cadherin (antibody from Santa Cruz) in pancreata from 6-month old control (A), Ncad WT (B), and Ncad KO (C) mice. Both membrane and cytoplasmic E-cadherin staining were observed in mPanINs. Note the weaker E-cadherin signal in Ncad KO (C) compared to Ncad WT (B). (D) Immunoblot analysis of E-cadherin expression in pancreata from control (con), Ncad WT, and Ncad KO mice. Cytokeratin 19 (CK19) (antibody from Developmental Studies Hybridoma Bank, University of Iowa) was used to account for the different proportion of epithelium between different samples. (E) Quantification of the protein levels revealed a reduction of E-cadherin in Ncad KO compared to Ncad WT pancreata. Densitometry of E-cadherin protein level was normalized to ductal marker CK19. *, p<0.05.
{kind=link}
Acknowledgements
We are grateful to Frans van Roy, Anil Rustgi, and Jennifer Wilson for comments. We thank David Tuveson for the LSL-K-rasG12D mice, Andrew Lowy for the Pdx1/Cre mice, and Michael Goggins for the PanIN microarrays. We are grateful to Mathew Thakur for assistance with PET imaging, and Han Du, Craig Riley, and David Kurz for technical assistance. Research in this study includes work carried out by the Jefferson Kimmel Cancer Center Small Animal Imaging Facility, which is supported in part by NCI Cancer Center Support Grant P30 CA56036. This work was supported by NIH R21 CA176097 (G.R.). This study was also supported by the SPORE grant CA62924 (R.H.)
Footnotes
Conflict of interest
The authors declare no conflicts of interest.
References
- 1.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. The New England journal of medicine. 2014 Sep 11;371(11):1039–1049. doi: 10.1056/NEJMra1404198. PubMed PMID: 25207767. [DOI] [PubMed] [Google Scholar]
- 2.Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010 Oct 28;467(7319):1109–1113. doi: 10.1038/nature09460. PubMed PMID: 20981101. Pubmed Central PMCID: 3137369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008 Sep 26;321(5897):1801–1806. doi: 10.1126/science.1164368. PubMed PMID: 18772397. Pubmed Central PMCID: 2848990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Roy F. Beyond E-cadherin: roles of other cadherin superfamily members in cancer. Nature reviews Cancer. 2014 Feb;14(2):121–134. doi: 10.1038/nrc3647. PubMed PMID: 24442140. [DOI] [PubMed] [Google Scholar]
- 5.Hotz B, Arndt M, Dullat S, Bhargava S, Buhr HJ, Hotz HG. Epithelial to mesenchymal transition: expression of the regulators snail, slug, and twist in pancreatic cancer. Clin Cancer Res. 2007 Aug 15;13(16):4769–4776. doi: 10.1158/1078-0432.CCR-06-2926. PubMed PMID: 17699854. [DOI] [PubMed] [Google Scholar]
- 6.Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y, Johnson KR. Cadherin switching. J Cell Sci. 2008 Mar 15;121(Pt 6):727–735. doi: 10.1242/jcs.000455. PubMed PMID: 18322269. Epub 2008/03/07. eng. [DOI] [PubMed] [Google Scholar]
- 7.Maeda M, Johnson KR, Wheelock MJ. Cadherin switching: essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J Cell Sci. 2005 Mar 1;118(Pt 5):873–887. doi: 10.1242/jcs.01634. PubMed PMID: 15713751. [DOI] [PubMed] [Google Scholar]
- 8.Qian X, Anzovino A, Kim S, Suyama K, Yao J, Hulit J, et al. N-cadherin/FGFR promotes metastasis through epithelial-to-mesenchymal transition and stem/progenitor cell-like properties. Oncogene. 2014 Jun 26;33(26):3411–3421. doi: 10.1038/onc.2013.310. PubMed PMID: 23975425. Pubmed Central PMCID: 4051865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shintani Y, Hollingsworth MA, Wheelock MJ, Johnson KR. Collagen I promotes metastasis in pancreatic cancer by activating c-Jun NH(2)-terminal kinase 1 and up-regulating N-cadherin expression. Cancer Res. 2006 Dec 15;66(24):11745–11753. doi: 10.1158/0008-5472.CAN-06-2322. PubMed PMID: 17178870. Epub 2006/12/21. eng. [DOI] [PubMed] [Google Scholar]
- 10.Su Y, Li J, Witkiewicz AK, Brennan D, Neill T, Talarico J, et al. N-cadherin haploinsufficiency increases survival in a mouse model of pancreatic cancer. Oncogene. 2012 Oct 11;31(41):4484–4489. doi: 10.1038/onc.2011.574. PubMed PMID: 22158044. Pubmed Central PMCID: 3714178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Al-Aynati MM, Radulovich N, Riddell RH, Tsao MS. Epithelial-cadherin and beta-catenin expression changes in pancreatic intraepithelial neoplasia. Clin Cancer Res. 2004 Feb 15;10(4):1235–1240. doi: 10.1158/1078-0432.ccr-03-0087. PubMed PMID: 14977820. Epub 2004/02/24. eng. [DOI] [PubMed] [Google Scholar]
- 12.Pasca di Magliano M, Biankin AV, Heiser PW, Cano DA, Gutierrez PJ, Deramaudt T, et al. Common activation of canonical Wnt signaling in pancreatic adenocarcinoma. PloS one. 2007;2(11):e1155. doi: 10.1371/journal.pone.0001155. PubMed PMID: 17982507. Pubmed Central PMCID: 2048934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakajima S, Doi R, Toyoda E, Tsuji S, Wada M, Koizumi M, et al. N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma. Clin Cancer Res. 2004 Jun 15;10(12 Pt 1):4125–4133. doi: 10.1158/1078-0432.CCR-0578-03. PubMed PMID: 15217949. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Morris JPt, Yan W, Schofield HK, Gurney A, Simeone DM, et al. Canonical wnt signaling is required for pancreatic carcinogenesis. Cancer Res. 2013 Aug 1;73(15):4909–4922. doi: 10.1158/0008-5472.CAN-12-4384. PubMed PMID: 23761328. Pubmed Central PMCID: 3763696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003 Dec;4(6):437–450. doi: 10.1016/s1535-6108(03)00309-x. PubMed PMID: 14706336. Epub 2004/01/07. eng. [DOI] [PubMed] [Google Scholar]
- 16.Luo Y, Kostetskii I, Radice GL. N-cadherin is not essential for limb mesenchymal chondrogenesis. Dev Dyn. 2005 Feb;232(2):336–344. doi: 10.1002/dvdy.20241. PubMed PMID: 15614770. Epub 2004/12/23. eng. [DOI] [PubMed] [Google Scholar]
- 17.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005 May;7(5):469–483. doi: 10.1016/j.ccr.2005.04.023. PubMed PMID: 15894267. [DOI] [PubMed] [Google Scholar]
- 18.Kostetskii I, Li J, Xiong Y, Zhou R, Ferrari VA, Patel VV, et al. Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ Res. 2005 Feb 18;96(3):346–354. doi: 10.1161/01.RES.0000156274.72390.2c. PubMed PMID: 15662031. Epub 2005/01/22. eng. [DOI] [PubMed] [Google Scholar]
- 19.Johansson JK, Voss U, Kesavan G, Kostetskii I, Wierup N, Radice GL, et al. N-cadherin is dispensable for pancreas development but required for beta-cell granule turnover. Genesis. 2010 Jun;48(6):374–381. doi: 10.1002/dvg.20628. PubMed PMID: 20533404. Pubmed Central PMCID: 2921608. Epub 2010/06/10. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hruban RH, Adsay NV, Albores-Saavedra J, Anver MR, Biankin AV, Boivin GP, et al. Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Cancer Res. 2006 Jan 1;66(1):95–106. doi: 10.1158/0008-5472.CAN-05-2168. PubMed PMID: 16397221. [DOI] [PubMed] [Google Scholar]
- 21.Gottardi CJ, Wong E, Gumbiner BM. E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion-independent manner. J Cell Biol. 2001 May 28;153(5):1049–1060. doi: 10.1083/jcb.153.5.1049. PubMed PMID: 11381089. Pubmed Central PMCID: 2174337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schreiber FS, Deramaudt TB, Brunner TB, Boretti MI, Gooch KJ, Stoffers DA, et al. Successful growth and characterization of mouse pancreatic ductal cells: functional properties of the Ki-RAS(G12V) oncogene. Gastroenterology. 2004 Jul;127(1):250–260. doi: 10.1053/j.gastro.2004.03.058. PubMed PMID: 15236190. Epub 2004/07/06. eng. [DOI] [PubMed] [Google Scholar]
- 23.McClatchey AI, Yap AS. Contact inhibition (of proliferation) redux. Current opinion in cell biology. 2012 Oct;24(5):685–694. doi: 10.1016/j.ceb.2012.06.009. PubMed PMID: 22835462. [DOI] [PubMed] [Google Scholar]
- 24.Agbunag C, Bar-Sagi D. Oncogenic K-ras drives cell cycle progression and phenotypic conversion of primary pancreatic duct epithelial cells. Cancer Res. 2004 Aug 15;64(16):5659–5663. doi: 10.1158/0008-5472.CAN-04-0807. PubMed PMID: 15313904. Epub 2004/08/18. eng. [DOI] [PubMed] [Google Scholar]
- 25.Deramaudt TB, Takaoka M, Upadhyay R, Bowser MJ, Porter J, Lee A, et al. N-cadherin and keratinocyte growth factor receptor mediate the functional interplay between Ki-RASG12V and p53V143A in promoting pancreatic cell migration, invasion, and tissue architecture disruption. Mol Cell Biol. 2006 Jun;26(11):4185–4200. doi: 10.1128/MCB.01055-05. PubMed PMID: 16705170. Pubmed Central PMCID: 1489079. Epub 2006/05/18. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levenberg S, Yarden A, Kam Z, Geiger B. p27 is involved in N-cadherin-mediated contact inhibition of cell growth and S-phase entry. Oncogene. 1999 Jan 28;18(4):869–876. doi: 10.1038/sj.onc.1202396. PubMed PMID: 10023662. [DOI] [PubMed] [Google Scholar]
- 27.Charrasse S, Meriane M, Comunale F, Blangy A, Gauthier-Rouviere C. N-cadherin-dependent cell-cell contact regulates Rho GTPases and beta-catenin localization in mouse C2C12 myoblasts. J Cell Biol. 2002 Sep 2;158(5):953–965. doi: 10.1083/jcb.200202034. PubMed PMID: 12213839. Pubmed Central PMCID: 2173149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gavard J, Marthiens V, Monnet C, Lambert M, Mege RM. N-cadherin activation substitutes for the cell contact control in cell cycle arrest and myogenic differentiation: involvement of p120 and beta-catenin. The Journal of biological chemistry. 2004 Aug 27;279(35):36795–36802. doi: 10.1074/jbc.M401705200. PubMed PMID: 15194693. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Gao E, Vite A, Yi R, Gomez L, Goossens S, et al. Alpha-catenins control cardiomyocyte proliferation by regulating Yap activity. Circ Res. 2015 Jan 2;116(1):70–79. doi: 10.1161/CIRCRESAHA.116.304472. PubMed PMID: 25305307. Pubmed Central PMCID: 4282606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mui KL, Bae YH, Gao L, Liu SL, Xu T, Radice GL, et al. N-Cadherin Induction by ECM Stiffness and FAK Overrides the Spreading Requirement for Proliferation of Vascular Smooth Muscle Cells. Cell reports. 2015 Mar 4; doi: 10.1016/j.celrep.2015.02.023. PubMed PMID: 25753414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gumbiner BM, Kim NG. The Hippo-YAP signaling pathway and contact inhibition of growth. J Cell Sci. 2014 Feb 15;127(Pt 4):709–717. doi: 10.1242/jcs.140103. PubMed PMID: 24532814. Pubmed Central PMCID: 3924201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim NG, Koh E, Chen X, Gumbiner BM. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proceedings of the National Academy of Sciences of the United States of America. 2011 Jul 19;108(29):11930–11935. doi: 10.1073/pnas.1103345108. PubMed PMID: 21730131. Pubmed Central PMCID: 3141988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012 Dec 21;151(7):1457–1473. doi: 10.1016/j.cell.2012.11.026. PubMed PMID: 23245941. Pubmed Central PMCID: 3530160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Double immunofluorescence analysis of E-cadherin (antibody from Santa Cruz) and N-cadherin (antibody from Invitrogen) in human PanIN specimen. Note the co-expression of E-cadherin (A) and N-cadherin (B) shown in the merged image (C).
Histological analysis of β-galactosidase-stained KPC N-cadherinlacZ/+ pancreas. LacZ expression was detected in (A) Islets of Langerhans (Is), (B) neural plexus (N), (C) mPanIN lesions, and (D) PDA in LSL-K-rasG12D; LSL-Trp53R172H; Pdx1/Cre; NcadlacZ/+ mice. N-cadherin lacZ expression (blue) was observed in a subpopulation of ductal cells (arrows) in mPanIN lesions (C) and in poorly differentiated tumor cells (D). The latter cells represent tumor cells that have undergone epithelial-mesenchymal transition (EMT).
Immunoblot analysis of Ras and RAS-GTP in total protein lysates from 6-month old Ncad WT and Ncad KO mice. Ras activation assay was performed using Ras activation assay kit (Upstate).
Immunohistochemical detection of E-cadherin (antibody from Santa Cruz) in pancreata from 6-month old control (A), Ncad WT (B), and Ncad KO (C) mice. Both membrane and cytoplasmic E-cadherin staining were observed in mPanINs. Note the weaker E-cadherin signal in Ncad KO (C) compared to Ncad WT (B). (D) Immunoblot analysis of E-cadherin expression in pancreata from control (con), Ncad WT, and Ncad KO mice. Cytokeratin 19 (CK19) (antibody from Developmental Studies Hybridoma Bank, University of Iowa) was used to account for the different proportion of epithelium between different samples. (E) Quantification of the protein levels revealed a reduction of E-cadherin in Ncad KO compared to Ncad WT pancreata. Densitometry of E-cadherin protein level was normalized to ductal marker CK19. *, p<0.05.