

Summary
Sjögren's syndrome (SS) is an autoimmune disease and the second most common chronic systemic rheumatic disorder. Prevalence of primary SS in the general population has been estimated to be approximately 1–3%, whereas secondary SS has been observed in 10–20% of patients with rheumatoid arthritis, systemic lupus erythematosus (SLE) and scleroderma. Despite this, its exact aetiology and pathogenesis are largely unexplored. Nuclear factor‐kappa B (NF‐κB) signalling mechanisms provide central controls in SS, but how these pathways intersect the pathological features of this disease is unclear. The ubiquitin‐editing enzyme A20 (tumour necrosis factor‐α‐induced protein 3, TNFAIP3) serves as a critical inhibitor on NF‐κB signalling. In humans, polymorphisms in the A20 gene or a deregulated expression of A20 are often associated with several inflammatory disorders, including SS. Because A20 controls the ectodysplasin‐A1 (EDA‐A1)/ectodysplasin receptor (EDAR) signalling negatively, and the deletion of A20 results in excessive EDA1‐induced NF‐κB signalling, this work investigates the expression levels of EDA‐A1 and EDAR in SS human salivary glands epithelial cells (SGEC) and evaluates the hypothesis that SS SGEC‐specific deregulation of A20 results in excessive EDA1‐induced NF‐κB signalling in SS. Our approach, which combines the use of siRNA‐mediated gene silencing and quantitative pathway analysis, was used to elucidate the role of the A20 target gene in intracellular EDA‐A1/EDAR/NF‐κB pathway in SS SGEC, holding significant promise for compound selection in drug discovery.
Keywords: A20, EDA‐A1, EDAR, NF‐κB, Sjögren's syndrome
Introduction
Sjögren's syndrome (SS) is an autoimmune disorder and the second most common chronic systemic rheumatic disease characterized by extraglandular manifestations, variable severity of salivary gland hypofunction, infiltration of lymphocytes and plasmacytoid dendritic cells and decreased salivary secretion 1. The transcription factor nuclear factor‐κB (NF‐κB) represents a central regulator of signals that lead to inflammatory reaction, cell adhesion, cell growth, differentiation, apoptosis and regulation of immune responses 2, 3, and plays a prominent role in SS affecting the chronic inflammation characterizing this disease 4, 5, 6, 7, 8, 9, 10, 11. The NF‐κB activation was regulated negatively through the ubiquitin‐editing enzyme A20 (tumour necrosis factor‐α‐induced protein 3, TNFAIP3) 12, 13, 14, 15 that was deregulated in human salivary gland epithelial cells (SGEC) from SS patients 11. Recent findings demonstrated that A20 plays a role in controlling the activity of three genes, mutated in hypohidrotic/anhidrotic ectodermal dysplasia (HED/EDA), which has been involved in NF‐κB activation: ectodysplasin (EDA‐A1), EDA‐receptor (EDAR) and EDAR‐associated death domain (EDARADD) 16, 17, 18, 19. Like the TNF/TNFR system, EDA‐A1/EDAR signalling is thought to be primarily through the canonical NF‐κB pathway 20 that induces nuclear translocation of the NF‐κB 21. On the basis of these premises, in the present work we investigate the expression levels of EDA‐A1 and EDAR genes and proteins in SS salivary gland, aiming to demonstrate that SS SGEC‐specific deregulation of A20 results in excessive EDA‐A1/EDAR‐induced NF‐κB activation.
Materials and methods
Patient population
Twenty‐four primary SS (pSS) patients, confirmed to have definite disease according to the revised 2002 American–European Consensus Group, and displaying a salivary gland focus score of at least 3, were enrolled into the study 22. All patients had the clinical symptoms of dry eyes and mouth, the Schirmer's test (less than 5 mm wetting of a strip of filter paper per 5 min) performed was positive, and had Rose Bengal staining (increased uptake of Rose Bengal dye in devitalized areas in the conjunctiva and cornea. The presence of at least one of the following autoantibodies, anti‐SS‐related antigen A (Ro/SSA), anti‐SS‐related antigen La/SSB, anti‐nuclear antibodies and rheumatoid factor, was detected. None of the patients studied had received glucocorticoid and/or immunosuppressive drug treatment and/or biological therapy at the time of the lip biopsy. Healthy labial salivary gland biopsy (LSG) biopsies were derived from 15 healthy individuals awaiting removal of salivary mucoceles from the lower lip. Informed consent from the subjects and approval by the local ethics committee were obtained. The healthy subjects had no complaints of oral dryness, any autoimmune disease and normal salivary function. Labial minor glands were harvested from the lower lip under local anaesthesia through normal mucosa, according to the explants outgrowth technique 23.
SGEC culture
pSS and healthy SGEC were isolated from the minor salivary glands (MSG) by microdissection and collagenase (Millipore, Freehold, NJ, USA) digestion and resuspended in McCoy's 5a modified medium [10% fetal bovine serum (FBS), 1% antibiotic solution, 2 mM L‐glutamine, 50 ng/ml epidermal growth factor (EGF; Promega, Madison, WI, USA), 0·5 μg/ml insulin (Novo, Bagsvaerd, Denmark)] and incubated at 37°C, 5% CO2 in air. Ethylenediamine tetraacetic acid (EDTA) (0·02%) was used to remove contaminating fibroblasts. The epithelial origin of cultured cells was confirmed routinely by staining with monoclonal antibodies against epithelial‐specific markers, including the various cytokeratins and epithelial membrane antigens and the absence of myoepithelial, fibroblastoid and lymphoid markers, using immunocytochemistry as described previously 24.
Reverse transcription–polymerase chain reaction (RT–PCR)
Total RNA was isolated from variously treated healthy and pSS SGEC using TRIzol reagent, following the manufacturer's protocol (Invitrogen, Carlsbad, CA, USA). RNA quality was checked by gel electrophoresis to confirm the integrity of the RNA preparations. Two μg RNA were treated with DNase I (Gibco, Life Technologies, Carlsbad, CA, USA) prior to reverse transcription with Moloney murine leukaemia virus reverse transcriptase (Gibco) in the presence of RNaseOUT (Gibco) and 1/10 of the cDNA preparation for each polymerase chain reaction (PCR) was used. After initial denaturation at 94°C for 5 min, 35 cycles were performed. Equal amounts of PCR products were run on a 1·5% agarose gel containing ethidium bromide. The PCR primers used were: for EDA‐A1 forward 5′‐TGAACGGCTGAGGCAGACG‐3′, reverse 5′‐TCCGAGCGCAACTCTAGGTA‐3′; for EDAR forward 5′‐ACCAGGAGATGGAAAA‐3′, reverse 5′‐GCTGGATGAGTGTGCTGA‐3′; and for nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor alpha (IκBα) the primers used were: forward 5′‐AACCTGCAGCAGACTCCACT‐3′, reverse 5′‐ACACCAGGTCAGGATTTTGC‐3′. Densitometric analysis was performed by gel image software (Bio‐Profil Bio‐1D; ltf Labortechnik GmbH, Wasserburg, Germany) to quantify mRNA expression levels relative to glyceraldehyde‐3‐phosphate dehydrogenase (GADPH) (internal control for lane loading). Sequencing confirmed the identity of each PCR product.
Quantitative reverse transcription–polymerase chain reaction (qRT–PCR)
Total RNA was extracted from healthy and pSS SGEC, reverse‐transcribed and cDNA preparation was used for qRT–PCR. qRT–PCR was performed in a 96‐well microtitre plate with an ABI PRISM 7700 (Applied Biosystems, Foster City, CA, USA). Each reaction contained 5 μl of cDNA template, 2·5 μl of 20 × probes and primers mixture, 12·5 μl of TaqMan Universal PCR Master Mix, No AmpErase UNG (Applied Biosystems), in a total volume of 25 μl. Reactions were amplified for 40 cycles. The threshold was determined as 10 times the standard deviation (s.d.) of the baseline fluorescence signal. The cycle number at the threshold was used as the threshold cycle (Ct). Primers for real‐time PCR were purchased from SABiosciences [Frederick, MD, USA; catalogue numbers PPH10986A (EDA) and PPH09589B (EDAR)].
Genomic DNA (gDNA) extraction from minor salivary gland tissues
gDNA was extracted from fresh SS and healthy salivary gland tissues using DNAzol (Molecular Research Center, Cincinnati, OH, USA), according to the manufacturer's instructions. Salivary glands were lysed in 10 mM Tris pH 7·4, 5 mM EDTA, 1% (v/v) Triton X‐100 for 20 min on ice. One ml of DNAzol was then added in the presence of proteinase K (Sigma, St Louis, MO, USA; 100 lg/ml) for 30 min at 37°C. After centrifugation at 11 000 g for 20 min at 10°C, the supernatant was collected and RNAse A (Sigma; 20 lg/ml) was added for 1 h at 37°C, in order to eliminate contaminating RNA. The DNA extracted was precipitated in 100% (v/v) ethanol and centrifuged at 11 000 g for 20 min. Purified DNA was dissolved in 8 mM NaOH, separated by electrophoresis (50 lg for well) in a 2% (w/v) agarose gel and visualized by ethidium bromide staining.
PCR on gDNA samples and exon sequencing of EDA gene
Selected exons and their flanking intronic sequences were amplified using the Accuprime kit (Life Technologies) according to the manufacturer's instructions and PCR assays were run by an Applied Biosystem Gene Amp PCR system 9600; PCR products were purified by the AMPure purification kit (Beckman Coulter, Brea, CA, USA); they were then sequenced in forward and reverse directions using the ABI prism Big Dye terminator version 3·1 ready reaction cycle sequencing kit and the ABI PRISM 3730xl Genetic Analyzer (Applied Biosystems). Exon‐specific primer pairs adopted for both PCR and sequencing were designed on the EDA gene reference sequence (NCBI Accession no. NG_009809.1) with respect to the eight exons containing the entire coding sequence, and comprised in the reference EDA‐A1 variant mRNA (NM_001399.4); details of selected oligonucleotide sequences are reported in Table 1.
Table 1.
Oligonucleotide sequences of exon‐specific primer pairs for polymerase chain reaction (PCR) and sequencing.
| Exon | Forward 5′ 3′ Sequence (Tm) | Reverse 5′ 3′ Sequence (Tm) | PCR size bp |
|---|---|---|---|
| 1 | TGAACGACAGCGCCAGTCA (59·5) | AACTTCTCCCTTGCTCGCCT (58) | 1000 |
| 2 | GTACAGTGGAGGGGAAGAT (53·3) | CACCATGCCCTACCAAGA (54·4) | 350 |
| 3 | TGACCCTTGGCTGTGAGACT (58·4) | AATAACAGACAGACAATGCTGA (52) | 270 |
| 4 | CGCCTGTAATCCCAGTTACT (54·2) | TACTGGCTTGTCCAATTCCTAGT (55·7) | 500 |
| 5 | CTCTGAGCCCTGGAGAATA (53·1) | GAAATAAAGCCTCAGGAAATCT (50·6) | 270 |
| 6 | GTAGTCAGTAACATCCCAAGACA (54) | AAAGAATGACTTCGTATGCCA (52) | 330 |
| 7 | GATAAAGACAGACAGGCAGA (51·6) | ATTGGATGGTCTTGGCTG (52·4) | 450 |
| 8 | AAGAACAATGCCTGTCACCT (54·5) | GGCATTTCTCTGCGGCTG (57) | 550 |
Nucleotide sequences and melting temperatures (tm)· are reported for primer pairs adopted for PCR and sequencing assays on exons 1–8 of the human ectodysplasin (EDA) gene. Each exon‐specific predicted PCR product size is reported· and comprises the flanking (upstream and downstream) intronic sequences included in the primer pair amplification region.
In‐silico analysis of exon sequences
Electropherograms from gDNA sequencing of the eight exonic regions within their flanking nucleotide traits were analysed and processed by Chromas Lite 2·1·1 chromatogram editor freeware (Technelysium Pty Ltd, South Brisbane, Australia). To check for nucleotide identity, forward and reverse sequences deriving from control and patient gDNA samples were aligned by the ClustalW2 multiple sequence alignment tool at http://www.ebi.ac.uk/Tools/msa/clustalw2/, and compared to the human reference mRNA sequence for the EDA gene (EDA A1 variant, RefSeq mRNA NCBI Accession no. NM_001399.4) by the CAP3 sequence assembly program at http://doua.prabi.fr/cgi-bin/run_cap3. Analysis of donor and acceptor splice sites at exon–intron–exon boundaries was conducted by comparing the exon‐specific multiple alignments to the mRNA‐to‐genomic alignment performed by the Spidey web‐based program (http://www.ncbi.nlm.nih.gov/spidey/) that was adopted to obtain feedback of the canonical splice sites and gene structure; specifically, Spidey was used to align the EDA gene reference sequence (Accession no. NG_009809.1) with seven mRNA reference sequences related to the splicing variants reported in the NCBI nucleotide database (namely, Accession nos NM_001399.4, BC144049.1, XM_006724630.1, NM_001005609.1, NM_001005612.2, NM_001005610.3, NM_001005613.3).
Immunohistochemistry
EDA‐A1 and EDAR proteins expression were assessed on paraffin‐embedded tissue samples derived from pSS patients and healthy subjects and were processed for immunohistochemical staining. The tissue samples were cut into 3‐μm‐thick sections by a microtome after deparaffinization and dehydration and the slides were washed in Tris‐buffered saline (TBS) (Sigma‐Aldrich, St Louis, MO, USA) containing 0·1% bovine serum albumin (BSA). Endogenous peroxidase activity was quenched by incubating the slides in a solution of 700 μl H2O2 (30%) in 70 ml methanol. The sections were pretreated with pepsin (0·4%) for 30 min at 37°C. Blocking was performed with normal serum. After incubation with primary antibody against EDA‐A1 (sc‐18927; dilution 1 : 50) and EDAR (sc‐15290; dilution 1 : 50) (both from Santa Cruz Biotechnology, Santa Cruz, CA, USA) at room temperature for 60 min, the slides were washed in TBS buffer, the horseradish peroxidase (HRP)‐conjugated secondary antibody (donkey anti‐goat HRP; Santa Cruz Biotechnology) was added and the slides were incubated at room temperature for 30 min. Afterwards, the slides were incubated with diaminobenzidine tetrahydrochloride (DAB) (Sigma) as substrate and counterstained with haematoxylin (Merck Eurolab, Dietikon, Switzerland). Sections with no primary antibody were used as a negative control. The slides were mounted and imaging acquisition was performed using a Nikon Eclipse inverted fluorescence microscope (Nikon, Inc., Melville, NY, USA) equipped with the camera system (Nikon).
Western blot analysis
Healthy and pSS labial salivary glands tissues were homogenized in a buffer (500 μl) containing 1% Triton X‐100, 50 mM Tris–HCl (pH 7·4), 1 mM phenylmethylsulphonyl fluoride (PMSF), 10 lg/ml soybean trypsin inhibitor and 1 mg/ml leupeptin. Bradford's protein assay was used to determine spectrophotometrically the protein concentration in the supernatant, and the protein lysates were subjected to sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE). Proteins (30 μg/lane) and prestained standards (BioRad Laboratories, Hercules, CA, USA) were loaded onto SDS‐polyacrylamide precast gels, and after electrophoresis proteins were transferred to nitrocellulose membranes. For gel and membrane saturation and blot, a blot buffer [20 mM Tris/150 mM glycine, pH 8, 20% (v/v) methanol] was used. Blots were blocked by phosphate‐buffered saline (PBS) pH 7·2 with 0·1% (v/v) Tween 20, 5% w/v non‐fat dried milk for 1 h and washed three times with 0·1% (v/v) Tween 20‐PBS 19 (T‐PBS). Membranes were incubated for 90 min with goat anti‐human EDA‐A1 polyclonal antibody (pAb) (sc‐18927; dilution 1 : 200), goat anti‐human EDAR pAb (sc‐15290; dilution 1 : 200), (both from Santa Cruz Biotechnology) and for 30 min with the relative secondary antibodies‐HRP conjugates (Santa Cruz Biotechnology). Proteins recognized by the antibodies were revealed using chemoluminescence luminal reagent (Santa Cruz Biotechnology) according to the protocol. The β‐actin (β‐actin protein) level was determined by Western blot and used as protein loading control.
Flow cytometry analysis
For flow cytometric analysis, pSS and healthy SCEC were incubated with rabbit anti‐human EDA‐phycoerythrin monoclonal antibody (mAb) and rabbit anti‐human EDAR phycoerythrin mAb (both from Bioss Inc. Woburn, MA, USA, catalogue numbers bs‐12347R‐PE and bs‐13050R‐PE, respectively). To confirm the specificity of primary antibody binding and rule out non‐specific Fc receptor binding or other cellular protein interactions, an isotype control was employed and cells were Fc‐blocked by treatment with human immunoglobulin (Ig)G (R&D Systems, Minneapolis, MN, USA) prior to staining. In addition, indirect staining using only the secondary antibody was used as negative standard. The protein expressions were analysed by a Becton Dickinson (BD, Heidelberg, Germany) fluorescence activated cell sorter (FACS)Canto II flow cytometer and BD FACS Diva software. The data were expressed as mean fluorescence intensity (MFI).
NF‐κB activity assay
NF‐κB activity was measured in nuclear protein extracts by the TransAMTM NF‐kB p65 protein assay (Active Motif, Carlsbad, CA, USA), an enzyme‐linked immunosorbent assay (ELISA)‐based method designed specifically to detect and quantify NF‐κB p65 subunit activation, with high sensitivity and reproducibility. Briefly, nuclear fractions were harvested from cells using the nuclear extraction kit following the manufacturer's suggestions. Bradford's method was used to determine protein concentration in the nuclear extract. To each well containing a NF‐κB consensus binding site (5′‐GGGACTTTCC‐3′), 10 μg of nuclear extract in cell binding and cell lysis buffer were added in triplicate. We used 5 μg of nuclear extract of Raji cells (a Burkitt's lymphoma cell line) as positive control (Active Motif). To assess DNA binding specificity, excess wild‐type and mutant competitor oligonucleotides can be added into the binding reaction. The absorbance was measured at 450 nm by the VERSAmax microplate reader (Molecular Devices Corporation, Silicon Valley, CA, USA).
siRNA synthesis and transfection
The siRNA sequences used for targeted down‐regulation of human A20 were designed and synthesized by Ambion (Austin, TX, USA, pre‐designed siRNA). An irrelevant siRNA with random nucleotides and no known specificity was used to normalize relative gene inhibition of the target gene. Twenty‐four h before transfection, healthy SGEC cultured in McCoy's 5a modified medium were transferred onto a 24‐well plate (pre‐plating) to reach 50–80% confluence and were then transfected with siRNA for A20 using the siPORT NeoFX transfection agent (Ambion). The siRNAs were mixed and diluted in OPTI‐MEM1 medium to a final concentration of 30 nM/well. The siRNA/transfection agent was dispensed into culture plates, as directed by the manufacturer's manual. The highest efficiency in silencing the targeted gene was obtained by using mixtures of siRNA duplexes targeting different regions of the gene of interest. Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH, NM_204305.1) was used to design siRNA primers as positive controls (Ambion), and a negative control with no known sequence similarity to human genes was included. Silencing was observed by mRNA quantification by qRT–PCR.
Transient transfection with the dominant negative mutant form of IκBα
SS SGEC, 5 ×105, were seeded per well in a 24‐well tissue culture dish and transfected transiently using lipofectamine 2000 reagent (Invitrogen) with 10 μg/ml of dominant‐negative inhibitory κBα proteins (IκBα) vector (IκBαDN) and a plasmid DNA, including a NF‐κB luciferase reporter construct or the empty vector (pCMV; Clontech, San Diego, CA, USA), according to the manufacturer. The IκBαDN resulted mutated at the two inducible phosphorylation sites 32 and 36, and this disturbs phosphorylation and subsequent degradation. The IκBαDN was kindly gifted from Dr P. Bauerle, University of Munich and Dr M. Lienhard Schmitz, Deutsches Krebsforschungszentrum, Germany. The NF‐κB luciferase vector contains four tandem copies of the NF‐κB consensus sequence fused to a TATA‐like promoter region from the Herpes simplex virus thymidine kinase promoter. Luciferase activity was measured by the Luciferase Reporter Assay System (Promega, Madison, WI, USA) and detection of β‐galactosidase in the same sample was used to assay transfection efficiency.
Statistics
The data were analysed for normality using the Wilks–Shapiro test. Differences in means for paired observations were analysed by Student's t‐test, P < 0·05 being considered statistically significant.
Results
qRT–PCR measurement of EDA and EDAR mRNAs in SGEC from pSS patients and healthy subjects
We began by examining the expression of both EDA‐A1 and EDAR gene in SGEC from primary SS patients (n = 24; aged 2–76 years), compared with SGEC from healthy individuals (n = 15, aged 20–65 years) using quantitative RT–PCR analysis (Fig. 1). qRT–PCR was achieved successfully in all cases examined, as amplification was observed for both the EDA‐A1 (Fig. 1a) and EDAR gene markers (Fig. 1b). As shown in Fig. 1, the level of EDA‐A1 and EDAR mRNAs were expressed in higher amounts in pSS patients than in controls. The results monitored by real‐time PCR showed a significant increase (P < 0·01) of EDA‐A1 and EDAR mRNA expression in SS SGEC (3·1 ± 0·8 and 2·8 ± 0·9 times, respectively) when compared with healthy SGEC, revealing up‐regulation of EDA‐A1 and EDAR gene expression in SS.
Figure 1.
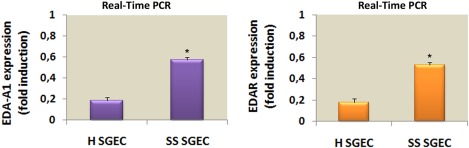
Quantitative real‐time polymerase chain reaction (PCR) for ectodysplasin (EDA)‐A1 and EDA‐receptor (EDAR) gene expression. EDA‐A1 (a) and EDAR (b) genes are up‐regulated in Sjögren's syndrome salivary gland epithelial cells (SS SGEC) derived from SS patients compared with human (H) SGEC. Representative histograms of mRNA levels expression of EDA‐A1 and EDAR in healthy (H) SGEC and SS SGEC. Data are presented as fold induction of EDA‐A1 and EDAR mRNA levels after normalizing to β‐2‐microglobulin expression [mean ± standard error (s.e.) of five independent experiments, *P < 0·01].
In‐silico analysis of sequenced EDA‐A1 exonic regions from control and patients’ gDNA
Mutations in mouse Eda or human EDA are associated with absent or hypoplastic sweat glands, sebaceous glands, lacrimal glands, salivary glands, mammary glands and/or nipples, and mucous glands of the bronchial, oesophageal and colonic mucosa 25. Patients with HED have been shown to have salivary gland hypoplasia and variably reduced salivary secretion 26, 27, 28, as also occurs in SS patients characterized by diminished salivary flow. Based on this evidence, in the current study DNA samples extracted from SS salivary glands were analysed to evaluate possible alterations of the EDA gene and to discover a possible correlation between the eventual mutations and clinical or morphological parameters of SS disease. Genomic DNA samples were extracted from four salivary gland biopsies derived from SS patients and from human healthy subjects, and exon‐specific primers were used for sequencing of exons 1–8, within their flanking non‐coding regions, along the EDA gene, in order to check for nucleotide identity in exon sequences from patient samples with respect to control. Oligonucleotide sequences of exon‐specific primer pairs for PCR and sequencing are reported in Table 1. By performing sequence alignments, exon regions were identified and consequent exon‐specific sequence analysis showed 100% nucleotide identity between control and patients for each exon analysed. Sequenced exons 1–8 also showed no difference with the corresponding nucleotide regions in the reference mRNA sequence (NCBI Accession no. NM_001399.4); the identified exonic regions comprised the entire predicted CDS, from the ATG initiation codon in exon 1 to the TAG termination codon in exon 8. Also, analysis of exon–intron and intron–exon junctions showed canonical consensus sequences of donor/acceptor splice sites, conserved between sequences from human control and SS patients (Fig. 2; for complete alignment details, please see Supporting information S1).
Figure 2.
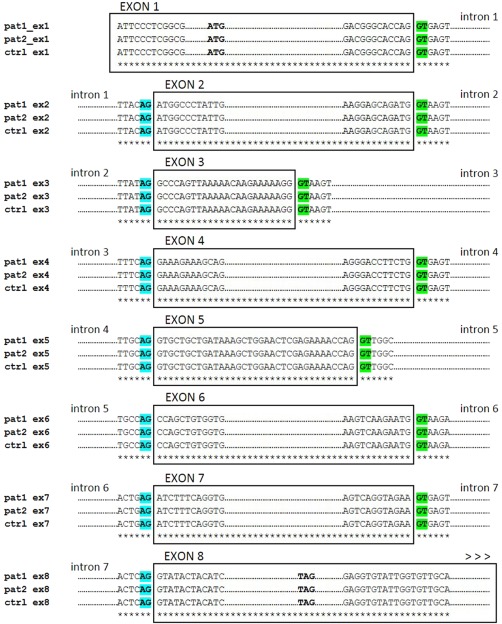
In‐silico analysis of sequenced ectodysplasin (EDA) exonic regions. Rectangles include representative alignments of the 8 exon sequences identified in control (ctrl) and two selected patients (pat1, pat2) gDNA samples. Underscoring asterisks indicate single nucleotide identity. The up‐ and down‐stream intron sequences are reported, showing the donor (GT, green) and acceptor (AG, blue) canonical splice sites, respectively. In exon 1 alignments, the conserved ATG initiation codon is reported; also, in exon 8 alignment the predicted TAG termination codon has been detected in each sequence analysed.
EDA‐A1 and EDAR proteins are over‐expressed in patients with Sjögren's syndrome
In order to validate the up‐regulation of EDA‐A1 and EDAR gene expression level in SS patients, we next analysed the cell‐specific distribution of these proteins in paraffin‐embedded sections from the salivary glands of normal subjects and patients with SS by immunohistochemical analysis (Fig. 3). The histological examination of the glandular epithelium of the normal salivary glands revealed moderate immune‐positive EDA‐A1 and EDAR proteins, both of which showed staining that was localized predominantly in the ductal epithelial cells (Fig. 3b,e), while negligible in the acinar cells (Fig. 3b,e). By contrast, we detected a remarkable positive staining for the EDA‐A1 (Fig. 3c) and its receptor EDAR (Fig. 3f) in the biopsy specimens from SS patients in respect of healthy control subjects, distributed in the cytoplasm and membrane of ductal cells. Acinar cells exhibited a very weak expression of EDA‐A1 and EDAR compared to ductal cells (Fig. 3c,f). Diffuse infiltrating mononuclear cells were found in the salivary glands of SS biopsies around the blood vessels (Fig. 3c,f), but not in those of normal subjects (Fig. 3b,e). Omission of the primary antibody resulted in the absence of specific labelling (Fig. 3a,d). Our immunohistochemical findings clearly revealed substantial differences in the levels of EDA‐A1 and EDAR protein expression between healthy controls and SS patients’ salivary gland biopsies.
Figure 3.
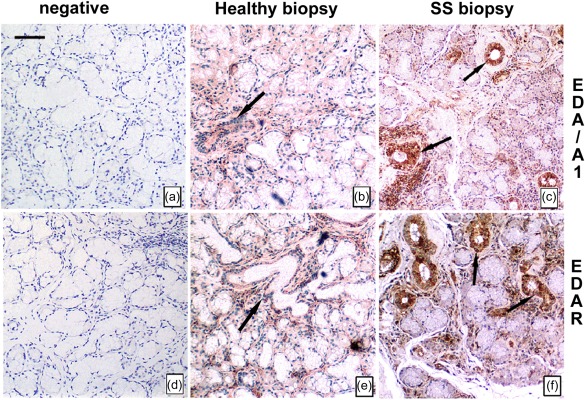
Increased immunoreactivity of ectodysplasin (EDA)‐A1 and EDA‐receptor (EDAR) in Sjögren's syndrome (SS) tissues in comparison to healthy control cells. Expression and localization of EDA‐A1 and EDAR proteins in sections from the salivary glands of normal subjects (b,e) and patients with SS (c,f) by immunohistochemistry. Selected healthy and SS biopsy tissues were processed as described in Materials and methods. Immunostaining of labial salivary gland sections shows a strong EDA‐A1 (c) and EDAR (e) reactivity on ductal epithelial cells in SS patients. Negative controls without primary antibody were included in each experiment to verify antibody specificity (a,d). Brown staining shows positive immune reaction; blue staining shows nuclei. Bar = 20 μm. Arrows show EDA‐A1 and EDAR distribution in the cytoplasm of ductal cells of healthy (b,e) and SS (c,f) salivary glands.
To support the notion that EDA‐A1 and EDAR protein are expressed strongly in SS tissues, we next assessed cultured SGEC cells for EDA‐A1 and EDAR protein expression by Western blot analyses. The protein levels of EDA‐A1 and EDAR in healthy and SS SGEC are shown in Fig. 4. Blotting for EDA‐A1 and EDAR detected two strong bands at 41 kDa and 49 kDa, respectively (Fig. 4c,d), based on the relative intensity of EDA‐A1 and its receptor (by comparing with β‐actin), resulting in over‐expressed SGEC derived from SS salivary gland tissues in comparison with healthy SGEC. Specificity was proved in controls without the primary antibody, which was deemed negative (not shown). As suggested by the results of the densitometric analysis (Fig. 4a,b), EDA‐A1 and EDAR was expressed more abundantly in the SS patients than in the healthy controls. The over‐expression of each protein was statistically significant (P < 0·01). To confirm immunohistochemistry and Western blot results, one detailed analysis of EDA‐A1 and EDAR expression was performed with flow cytometry of SGEC derived from Sjögren's syndrome biopsies and healthy donors and reported in Fig. 4e–h, which shows an example of flow cytometric images from one representative experiment. The expression of EDA‐A1 protein (e) was increased from 61·8% ± 1·9 of the healthy SCEC value to 97·4% ± 1·3 (P < 0·01) of SS SGEC. The expression of EDAR (f) followed a similar trend (59% ± 1·5 for healthy control SGEC versus 96% ± 1·2 for SGEC derived from SS patients). A significant increase in the levels of EDA‐A1 expression (Fig. 4g), as measured by MFI, was observed on the SS SGEC (median MFI = 13 589 ± 220) compared with that of healthy subjects (median MFI = 3312 ± 183, P < 0·01). A similar result was also observed for EDAR protein (Fig. 4h) (MFI = 3500 ± 220 for healthy SGEC versus MFI = 12 600 ± 250 for SS SGEC, P < 0·01), providing strong evidence that EDA‐A1 and EDAR proteins were over‐expressed in SS SGEC.
Figure 4.
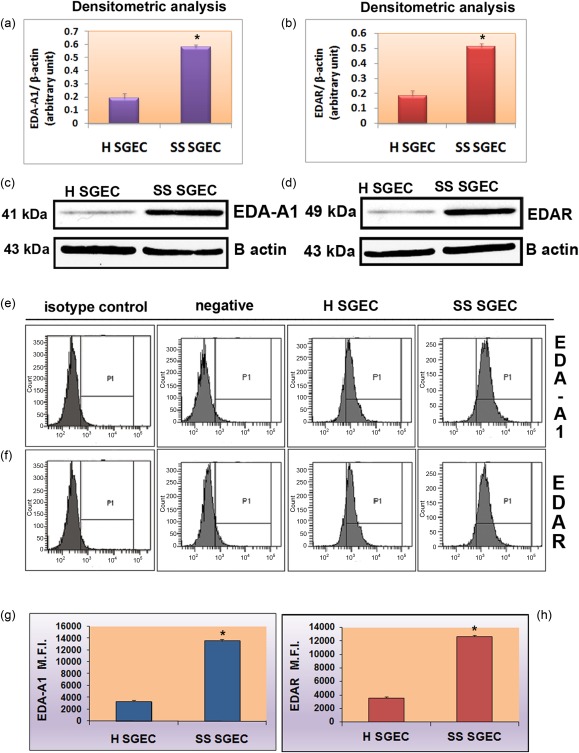
Over‐expression of ectodysplasin (EDA)‐A1 and EDA‐receptor (EDAR) protein in Sjögren's syndrome salivary gland epithelial cells (SS SGEC). Densitometric analysis (a,b) and Western blot analysis (c,d) of EDA‐A1 and EDAR. Immunoblotting gave rise to bands of the expected size (41 kDa for EDA‐A1 and 49 kDa for EDAR). The EDA‐A1 and EDAR protein expression levels were increased significantly in Sjögren's syndrome (SS) patients with active SS cytometric analysis of EDA‐A1 and EDAR proteins. (e,f) Examples of flow cytometric images from one representative experiment. Negative controls represent cells stained only with secondary antibody. Isotype controls represent isotype‐specific control antibody staining. (g,h) Results obtained by flow cytometry were expressed as the relative mean fluorescence intensity (MFI). Data represent the mean ± standard error (s.e.) of MFI from five independent experiments. *P < 0·01.
A20 acts as a regulator of the EDA‐A1 pathway to NF‐κB activation in pSS SGEC
As well as its role in controlling inflammation and preventing apoptosis, NF‐κB is also involved in the development of epidermal derivatives, such as hair, nails and sweat glands 29, 30, in which signalling is initiated by the binding of EDA‐A1 to the epidermal‐specific TNF‐receptor family member EDAR. A20 is required for terminating NF‐κB signalling and the in‐vivo importance of the A20 anti‐inflammatory activities is illustrated by severe multi‐organ inflammation 11, 14, 15. Based on this evidence we hypothesized that A20 might be a negative regulator of the EDA‐A1 pathway in pSS SGEC. Indeed, by using an ELISA‐based method designed to specifically detect and quantify NF‐κB p65 subunit activation, we demonstrated that the down‐regulated expression of A20 could be responsible for the observed increased expression of the EDA‐A1/EDAR system in SS SGEC that leads to an increased nuclear translocation and activation of NF‐κB in SS SGEC (Fig. 5). To verify this approach, we investigated the regulatory effect of A20 on the EDA‐A1/EDAR/NF‐κB system by silencing its expression in human healthy control SGEC that have high A20 expression in comparison with SS SGEC 11. By transiently silencing the expression of A20 with siRNA in healthy SGEC, we found increased expression of the EDA‐A1/EDAR system compared with untreated healthy SGEC and scrambled siRNA‐treated healthy SGEC. EDA‐A1 and EDAR expression was measured by flow cytometry (Fig. 5a,b) and EDA‐A1/EDAR expression, determined and expressed as mean fluorescence intensity (MFI), increased after A20 gene knock‐down (P < 0·01) (Fig. 5a,b). Concurrently, in the same transfected cells, increased NF‐κB nuclear translocation and activation were observed, as revealed by the TransAMTM NF‐κB p65 protein assay application (Fig. 5c). To confirm these results, RT–PCR and real‐time PCR was then used to quantify accurately differences in the expression levels of the NF‐κB‐target gene Iκβα, which is a known as a NF‐κB target gene in cells with activated NF‐κB p50/p65 complexes, after treatment with siRNA directed against A20. Figure 5d,e shows the relative amount of mRNA from the Iκβα gene. The results indicate that the expression level for the gene tested was increased significantly by A20 siRNA transfection, confirming the increased activity of NF‐κB. As EDA‐A1 signals through its receptor and instigates downstream activation of NF‐κB 29, the results obtained through the gene silencing experimental procedure indicate that, in SGEC, A20 is an NF‐κB response gene in the EDA‐A1/EDAR pathway acting in negative feedback regulation of EDA‐A1/EDAR‐induced NF‐κB signalling and then, when A20 is under‐expressed, as occurs in SS SGEC, the negative feedback regulation of the EDA/EDAR‐induced NF‐κB pathway was compromised and this leads to a sustained NF‐κB activation.
Figure 5.
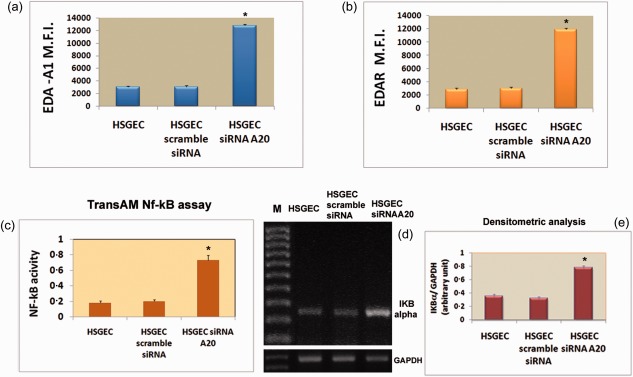
Effects of A20 gene knock‐down on ectodysplasin (EDA)‐A1 and EDA‐receptor (EDAR) protein expression in human healthy salivary gland epithelial cells (SS SGEC). (a,b) Human healthy SGEC were transfected with either A20 siRNA or scramble siRNA and then analysed by flow cytometry. Values are the mean ± standard error (s.e.) of mean fluorescence intensity (MFI) of triplicate cultures of three independent experiments. (c) Measurement of nuclear factor kappa B (NF‐κB) activity by TransAM NF‐κB assay in the nuclear extracts from healthy SGEC transfected with A20 siRNA and scramble siRNA. Activity of p65 family member of NF‐κB was up‐regulated significantly as a result of A20 gene silencing. Data represent mean ± s.e. of three independent experiments. *P < 0·01. (d) Levels of nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor alpha (IκBα) expression were analysed by reverse transcription–polymerase chain reaction (RT–PCR) on mRNA extracted from SS SGEC and SS SGEC transfected with siRNA against A20 gene (M = marker). (e) Band intensities were analysed by densitometry (data represent the mean ± s.e. of three independent experiments).*P < 0·01.
NF‐κB activity in SS SGEC is induced downstream of EDA‐A1/EDAR signalling
Having demonstrated the activation of the EDA‐A1/EDAR‐induced NF‐κB signalling in SGEC, we used the dominant negative super‐repressor inhibitory protein IκBα (IκBαDN) as the NF‐κB‐DNA binding inhibitor to clarify whether, in SGEC, NF‐κB acts down‐ or upstream of the EDA‐A1/EDAR system, given that the expression of several members of the TNF ligand and receptor multi‐gene families [e.g. TNF‐α, lymphotoxin beta (LT‐β), LT‐ β receptor, CD40, etc.] are under the control of NF‐κB. As shown from the literature, NF‐κB in unstimulated cells is sequestered in the cytoplasm by inhibitory IκB proteins. Upon stimuli such as EDA, IκBα is phosphorylated by the I‐κB kinase (IKK) and, as a result, IκBα is degraded and NF‐κB translocates into the nucleus and regulates expression of target genes. EDA signalling is, therefore, mediated in part by IκBα 21, 29. EDA phenotypes, along with immunodeficiency, were observed in a mouse strain in which IkBα truncated at its N‐terminus was inserted into a β‐catenin gene 30. The mutant form acts as a ‘super‐repressor’ of NF‐κB, maintaining NF‐κB in the cytoplasm 21, 30. Our results, obtained using semiquantitative and qRT–PCR, confirmed data collected from the literature; as expected, in SS SGEC transfected with IκBαDN, EDA‐A1 and EDAR mRNAs expression was still present and the levels were indistinguishable from those in untransfected SS SGEC, demonstrating that EDA‐A1 and EDAR expression do not require NF‐κB activation (Fig. 6). This study produced results which corroborate the findings that the EDA‐A1/EDAR‐NF‐κB signalling pathway is active in the SS SGEC, apparently working through the IkBα‐dependent canonical NF‐κB cascade.
Figure 6.
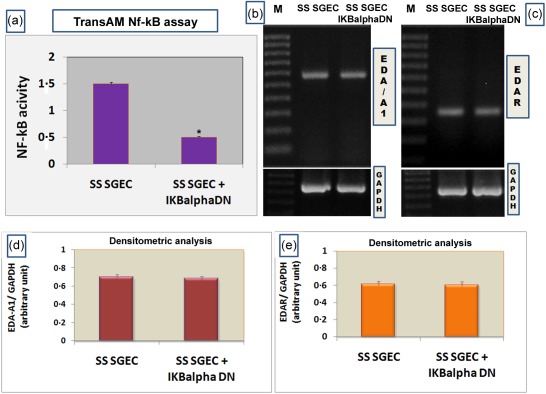
Effect of inhibition by nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor alpha (IκBα) dominant negative (DΝ) on Sjögren's syndrome salivary gland epithelial cells (SS SGEC). (a) Measurement of nuclear factor kappa B (NF‐κB) activity by TransAM NF‐κB assay in the nuclear extracts from SS SGEC and SS SGEC transfected transiently with the dominant negative super‐repressor inhibitory protein IKBα DΝ. Cell lysate was prepared and assayed for binding of p65 to DNA using the Trans‐Am NF‐κB p65 transcription factor assay kit as described in Materials and methods. Activity of p65 family member of NF‐κB was under‐regulated significantly in transfected SS SGEC as a result of inhibition by IKBα DΝ. Data represent mean ± standard error (s.e.) of three independent experiments. *P < 0·01. (b,c) Levels of ectodysplasin (EDA)‐A1 and EDA‐receptor (EDAR) expression were analysed by reverse transcription–polymerase chain reaction (RT–PCR) on mRNA extracted from SS SGEC and SS SGEC transfected with IKBα DΝ. M = marker; glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) served as an internal control for amplification. Results presented are from one experiment but representative of three independent experiments. (d,e) EDA‐A1 and EDAR gene expression levels were quantified by densitometric analysis (n = 3). *P < 0·01.
Discussion
During recent years, substantial progress has been made in explaining the mechanisms that control the dynamics of NF‐κB activation, and several autoregulatory feedback loops terminating the NF‐κB response have been identified 31. As NF‐κB signalling is involved in the regulation of cellular processes and, at each stage, the different family members must be regulated tightly for each function, several NF‐κB signalling protein factors have been identified as crucial steps in the control of the NF‐κB pathway 32. This well‐orchestrated biological process is controlled tightly, and distortions in the sequence of events can result in different disorders such as inflammation, cancer and autoimmune diseases 33, 34, 35. In this context, several recent studies identified new molecular targets involved in the control mechanism governing the inflammatory function of the NF‐κB cascade. For example, Tracey and colleagues described an alternative mechanism for cytokine production during the inflammatory response based on the cholinergic modulation of cytokines synthesis 36; in addition, the involvement of muscarinic acetylcholine receptors in the activation of the NF‐κB cascade in SS was also demonstrated, as suggested from the published studies of Gilboa‐Geffen and colleagues 37. In the last year there has been great progress in understanding the regulation of NF‐κB transcription in SS. A20 has been proposed as a key player in the termination of NF‐κB signalling, as A20 is capable of inhibiting TNF‐α‐induced NF‐κB activation by acting on IκB‐α phosphorylation in response to TNF‐α 38. Given its key function in the fine‐tuning of NF‐κB signalling, it was to be expected that defects in A20 expression or function could lead to chronic inflammation and tissue damage. Confirming this hypothesis, recent genetic studies demonstrate an association between the human ubiquitin‐editing enzyme A20 gene and autoimmune pathologies, classifying it as a potential target for the treatment of multiple inflammatory disorders 15. The importance of A20 in limiting inflammation is underscored by the association of polymorphisms in the A20 genomic region with multiple human autoimmune and inflammatory diseases, including rheumatoid arthritis 39, psoriasis 40, systemic lupus erythematosus 41, 42 and type 1 diabetes 43. Thus, A20 has been proposed as a critical anti‐inflammatory molecule acting to control prolonged inflammation. A putative association of A20 polymorphism with SS has been described recently 42. In addition, recent works reported decreased levels of A20 in SS SGEC, thus contributing to the chronic invasive immune processes in these patients resulting from an initial aberrant inflammation 11. Recently, experimental research works have reported that NF‐κB signalling plays a critical role during embryonic salivary gland development initiated by the binding of EDA‐A1 to the receptor EDAR 44, 45. The epidermis‐specific A20 deletion leads to multiple abnormalities in ectodermal organs, similar to those observed during the over‐activation of the EDA pathway, and these findings identify A20 as an EDA‐A1‐induced protein acting as an inhibitor of EDAR‐dependent canonical NF‐κB pathway signalling 29, 46, 47, 48, 49, 50, 51, 52, 53, 54. A schematic representation of the EDA‐A1/EDAR/NF‐κB pathway is reported in Fig. 7. Salivary gland hypofunction or xerostomia, as an inevitable consequence of SS, compromises significantly the quality of life of millions patients through associated poor oral health 55. A large number of studies in vitro on the SMG branching morphogenesis suggested that EDA‐A1/EDAR signalling regulates ontogeny largely through the canonical NF‐κB pathway 44, but the expression levels of the molecular factors involved in the EDA‐A1/EDAR/NF‐κB and the regulation of this pathway by A20 have not been investigated in SS salivary glands. To our knowledge, EDA‐A1 and EDAR protein expression levels have never been evaluated previously in salivary glands derived from patients affected by primary SS, and in the experiments presented here we have sought to bridge the altered expression of A20 in SS SGEC, already demonstrated 11, with a possible deregulation of the EDA‐A1/EDAR/NF‐κB signalling in SS patients. Interestingly, our study revealed, first, a statistically significant higher expression of EDA‐A1 and EDAR in SS biopsies compared with healthy controls both at gene and protein levels. Immunohistochemical analysis of inflamed salivary gland tissues of SS patients has indicated that epithelial ductal cells display strong staining for EDA‐A1 and its receptors EDAR compared with healthy biopsies. These results were in agreement with those obtained employing several experimental procedures, as reported in the Results section. Secondly, we show that specific A20 deletion in healthy SCEC leads to over‐activation of the EDA‐A1/EDAR expression. We used the siRNA strategy to silence A20 and to investigate the cellular effect of A20 knock‐down on the EDA‐A1/EDAR/NF‐κB pathway. By transiently silencing the expression of A20 with siRNA mixtures in healthy SGEC, we documented, in vitro, a significantly higher expression of the EDA‐A1/EDAR system compared with untransfected healthy SGEC. Concurrently, A20 gene knock‐down determined an increased NF‐κB nuclear translocation and activation. This accords with A20 being a NF‐κB response gene in the EDA‐A1/EDAR pathway, acting as a negative feedback regulator of EDA‐A1/EDAR‐induced NF‐κB signalling and of the inducible NF‐κB‐dependent gene transcription. Interestingly, we demonstrate for the first time that in SS SGEC NF‐κB is activated downstream of EDA‐A1 and EDAR signalling. Transfecting SS SGEC with the mutated form of the regulatory protein IκBα, our studies demonstrate that the EDA‐A1/EDAR‐NFκB signalling pathway in salivary gland apparently works through the IκBα‐dependent canonical NF‐κB cascade. Because translating basic research into clinical practice is fundamental to uncover and understand the deregulated cellular processes driving autoimmune initiation and progression, data obtained from genetic analysis are producing an explosion in the content and complexity of autoimmune mutation databases. Based on these premises, and given the demonstration of over‐expression of the EDA‐A1/EDAR/NF‐κB pathway in the SS salivary gland tissues, our efforts are focused upon the identification of eventual genetic variants of EDA‐A1 gene in SS patients, performed by sequencing genetic analysis. Genomic DNA samples, extracted from salivary gland biopsies derived from SS patients and from human healthy subjects, were subjected to sequencing of exons 1–8 using exon‐specific primers in order to check for nucleotide identity in exon sequences from patient samples with respect to controls. Although mutations were not detected in all the coding exons of EDA gene in SS SGEC in comparison with healthy subjects, genomic analysis requires further investigation and, in particular, detailed analysis of intronic sequences of EDA gene could be carried out in future. As the sequencing costs often set limits to the amount of sequences that can be generated and, consequently, the biological outcomes that can be achieved from an experimental design, however, the result obtained suggests utility to expand the genetic analysis for improving the molecular diagnosis of the SS disease.
Figure 7.
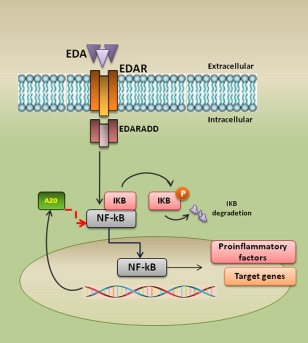
A simplified overview of the ectodysplasin/EDA‐receptor/nuclear factor kappa B (EDA/EDAR/NF‐κB) pathway. Trimeric receptor EDAR is activated by trimeric ligand EDA leading to the recruitment of the adaptor EDAR‐associated death (EDARADD) and to the formation of a complex containing EDARADD which ubiquitinates nuclear factor of kappa light chain gene enhancer in B cells (IkB) triggering to the release of NF‐κB transcription factor. Nuclear factor kappa B (NF‐κB) is then translocated into the nucleus to activate target genes. Strongly induced NF‐κB target genes include those that encode the negative regulators IkB alpha (IκBα) and A20.
In conclusion, our results suggest that the pathways in ectoderm development and inflammation may not be fundamentally different, and that the specificity of target gene activation may be determined largely by the general context of a given cell type and/or by a specific pathological condition. Recent findings have demonstrated that, as well as its role in controlling inflammation and preventing apoptosis, NF‐κB is also involved in the development of epidermal derivatives, such as hair, nails and sweat glands 56, in which signalling is initiated by the binding of EDA‐A1 to EDAR. The implication of the NF‐κB pathway in development was a surprising discovery, because it is involved primarily in inflammation and immunity, acting downstream of the activation of TNF immune receptors 16. By contrast, the activation of NF‐κB by the EDA pathway does not seem to be obviously linked to inflammation. This raises the recurrent question of how cells can differentiate between the types of messages transduced by the NF‐κB pathway and how specific target genes are activated for developmental or inflammatory responses. Interestingly, recent data suggest that the EDA pathway induces the transcription of chemokines known as key players of inflammation, which also have a developmental role 21. Furthermore, the findings reported here suggest that there is need of a more profound understanding of the role of A20 over‐expression on EDA gene transcription, which may play an instrumental role in the intricate inflammatory pathways. Thus, current knowledge for gene transcription regulation by the EDA‐A1/EDAR/NF‐κB pathway does not provide a satisfactory mechanistic framework to explain how an incoming activating signal provided by EDA‐A1 would be enabled to activate the transcription of genes involved in the exacerbation of inflammation observed in autoimmune conditions but, conversely, may provide new insights into treating or preventing autoimmunity and inflammation‐based disease.
Disclosure
The authors declare that there are no disclosures.
Supporting information
Supporting Information
Acknowledgements
We are grateful to M.V.C. Pragnell BA for critical reading of the manuscript. We thank Dr P. Bauerle, University of Munich and Dr M. Lienhard Schmitz, Deutsches Krebsforschungs zentrum, Germany.
References
- 1. Fox RI. Sjogren's syndrome. Lancet 2005; 366:321–31. [DOI] [PubMed] [Google Scholar]
- 2. Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol 2001; 11:372–7. [DOI] [PubMed] [Google Scholar]
- 3. Makarov SS. NF‐kappaB as a therapeutic target in chronic inflammation: recent advances. Mol Med Today 2000; 6:441–8. [DOI] [PubMed] [Google Scholar]
- 4. Peng B, Ling J, Lee AJ et al Defective feedback regulation of NF‐kappaB underlies Sjogren's syndrome in mice with mutated kappaB enhancers of the IkappaBalpha promoter. Proc Natl Acad Sci USA 2010; 107:15193–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sisto M, Lisi S, D'Amore M, Lofrumento DD. Rituximab‐mediated Raf kinase inhibitor protein induction modulates NF‐κB in Sjögren syndrome. Immunology 2014; 143:42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sisto M, Lisi S, Lofrumento DD, Ingravallo G, De Lucro R, D'Amore M. Salivary gland expression level of IκBα regulatory protein in Sjögren's syndrome. J Mol Histol 2013; 44:447–54. [DOI] [PubMed] [Google Scholar]
- 7. Lisi S, Sisto M, D'Amore M, Lofrumento DD, Ribatti D. Emerging avenues linking inflammation, angiogenesis and Sjögren's syndrome. Cytokine 2013; 61:693–703. [DOI] [PubMed] [Google Scholar]
- 8. Lisi S, Sisto M, Lofrumento DD, D'Amore M. Altered IκBα expression promotes NF‐κB activation in monocytes from primary Sjögren's syndrome patients. Pathology 2012; 44:557–61. [DOI] [PubMed] [Google Scholar]
- 9. Sisto M, Lisi S, Lofrumento DD, D'Amore M, Frassanito MA, Ribatti D. Sjögren's syndrome pathological neovascularization is regulated by VEGF‐A‐stimulated TACE‐dependent crosstalk between VEGFR2 and NF‐κB. Genes Immun 2012; 13:411–20. [DOI] [PubMed] [Google Scholar]
- 10. Lisi S, Sisto M, Lofrumento DD, D'Amore M. Sjögren's syndrome autoantibodies provoke changes in gene expression profiles of inflammatory cytokines triggering a pathway involving TACE/NF‐κB. Lab Invest 2012; 92:615–24. [DOI] [PubMed] [Google Scholar]
- 11. Sisto M, Lisi S, Lofrumento DD, Ingravallo G, Maiorano E, D'Amore M. A failure of TNFAIP3 negative regulation maintains sustained NF‐κB activation in Sjögren's syndrome. Histochem Cell Biol 2011; 135:615–25. [DOI] [PubMed] [Google Scholar]
- 12. Pujari R, Hunte R, Khan WN, Shembade N. A20‐mediated negative regulation of canonical NF‐κB signaling pathway. Immunol Res 2013; 57:166–71. [DOI] [PubMed] [Google Scholar]
- 13. Catrysse L, Vereecke L, Beyaert R, van Loo G. A20 in inflammation and autoimmunity. Trends Immunol 2014; 35:22–31. [DOI] [PubMed] [Google Scholar]
- 14. Lee EG, Boone DL, Chai S et al Failure to regulate TNF induced NF‐kappaB and cell death responses in A20‐deficient mice. Science 2000; 289:2350–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vereecke L, Beyaert R, van Loo G. The ubiquitin‐editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol 2009; 30:383–91. [DOI] [PubMed] [Google Scholar]
- 16. Lippens S, Lefebvre S, Gilbert B et al Keratinocyte‐specific ablation of the NF‐κB regulatory protein A20 (TNFAIP3) reveals a role in the control of epidermal homeostasis. Cell Death Differ 2011; 18:1845–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ferguson BM, Brockdorff N, Formstone E, Ngyuen T, Kronmiller JE, Zonana J. Cloning of Tabby, the murine homolog of the human EDA gene: evidence for a membrane‐associated protein with a short collagenous domain. Hum Mol Genet 1997; 6:1589–94. [DOI] [PubMed] [Google Scholar]
- 18. Srivastava AK, Pispa J, Hartung AJ et al The Tabby phenotype is caused by mutation in a mouse homologue of the EDA gene that reveals novel mouse and human exons and encodes a protein (ectodysplasin‐A) with collagenous domains. Proc Natl Acad Sci USA 1997; 94:13069–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ezer S, Bayés M, Elomaa O, Schlessinger D, Kere J. Ectodysplasin is a collagenous trimeric type II membrane protein with a tumor necrosis factor‐like domain and co‐localizes with cytoskeletal structures at lateral and apical surfaces of cells. Hum Mol Genet 1999; 8:2079–86. [DOI] [PubMed] [Google Scholar]
- 20. Courtney JM, Blackburn J, Sharpe PT. The Ectodysplasin and NFkappaB signalling pathways in odontogenesis. Arch Oral Biol 2005; 50:159–63. [DOI] [PubMed] [Google Scholar]
- 21. Smahi A, Courtois G, Rabia SH et al The NF‐κB signalling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune‐deficiency syndromes. Hum Mol Genet 2002; 11:2371–5. [DOI] [PubMed] [Google Scholar]
- 22. Vitali C, Bootsma H, Bowman SJ et al Classification criteria for Sjogren's syndrome: we actually need to definitively resolve the long debate on the issue. Ann Rheum Dis 2013; 72:476–8. [DOI] [PubMed] [Google Scholar]
- 23. Sens DA, Hintz DS, Rudisill MT, Sens MA, Spicer SS. Explant culture of human submandibular gland epithelial cells: evidence for ductal origin. Lab Invest 1985; 52:559–67. [PubMed] [Google Scholar]
- 24. Kapsogeorgou EK, Dimitriou ID, Abu‐Helu RF, Moutsopoulos HM, Manoussakis MN. Activation of epithelial and myoepithelial cells in the salivary glands of patients with Sjogren's syndrome: high expression of intercellular adhesion molecule‐1 (ICAM.1) in biopsy specimens and cultured cells. Clin Exp Immunol 2001; 124:126–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Melnick M, Phair RD, Lapidot SA, Jaskoll T. Salivary gland branching morphogenesis: a quantitative systems analysis of the Eda/Edar/NFkappaB paradigm. BMC Dev Biol 2009; 6:9–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nordgarden H, Jensen JL, Storhaug K. Oligodontia is associated with extra‐oral ectodermal symptoms and low whole salivary flow rates. Oral Dis 2001; 7:226–32. [PubMed] [Google Scholar]
- 27. Nordgarden H, Storhaug K, Lyngstadaas SP, Jensen JL. Salivary gland function in persons with ectodermal dysplasias. Eur J Oral Sci 2003; 111:371–6. [DOI] [PubMed] [Google Scholar]
- 28. Lexner MO, Bardow A, Hertz JM, Almer L, Nauntofte B, Kreiborg S. Whole saliva in X‐linked hypohidrotic ectodermal dysplasia. Int J Paediatr Dent 2007; 17:155–62. [DOI] [PubMed] [Google Scholar]
- 29. Kumar A, Eby MT, Sinha S, Jasmin A, Chaudhary PM. The ectodermal dysplasia receptor activates the nuclear factor‐kappaB, JNK, and cell death pathways and binds to ectodysplasin A. J Biol Chem 2001; 276:2668–77. [DOI] [PubMed] [Google Scholar]
- 30. Schmidt‐Ullrich R, Aebischer T, Hulsken J, Birchmeier W, Klemm U, Scheidereit C. Requirement of NF‐kappaB/Rel for the development of hair follicles and other epidermal appendices. Development 2001; 128:3843–53. [DOI] [PubMed] [Google Scholar]
- 31. Renner F, Schmitz ML. Autoregulatory feedback loops terminating the NF‐kappaB response. Trends Biochem Sci 2009; 34:128–35. [DOI] [PubMed] [Google Scholar]
- 32. Skaug B, Jiang X, Chen ZJ. The role of ubiquitin in NF‐kappaB regulatory pathways. Annu Rev Biochem 2009; 78:769–96. [DOI] [PubMed] [Google Scholar]
- 33. Christman JW, Sadikot RT, Blackwell TS. The role of nuclear factor‐kappa B in pulmonary diseases. Chest 2000; 117:1482–7. [DOI] [PubMed] [Google Scholar]
- 34. Yamamoto Y, Gaynor RB. Therapeutic potential of inhibition of the NF‐kappaB pathway in the treatment of inflammation and cancer. J Clin Invest 2001; 107:135–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Karin M. The IkB kinase: a bridge between inflammation and cancer. Cell Res 2008; 18:334–42. [DOI] [PubMed] [Google Scholar]
- 36. Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest 2007; 117:289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gilboa‐Geffen A, Wolf Y, Hanin G et al Activation of the alternative NFκB pathway improves disease symptoms in a model of Sjogren's syndrome. PLOS ONE 2011; 6:e28727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang SQ, Kovalenko A, Cantarella G, Wallach D. Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity 2000; 12:301–11. [DOI] [PubMed] [Google Scholar]
- 39. Plenge RM, Cotsapas C, Davies L et al Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet 2007; 39:1477–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nair RP, Duffin KC, Helms C et al Genome‐wide scan reveals association of psoriasis with IL‐23 and NF‐kappa B pathways. Nat Genet 2009; 41:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Graham RR, Cotsapas C, Davies L et al Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet 2008; 40:1059–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Musone SL, Taylor KE, Lu TT et al Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet 2008; 40:1062–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fung EY, Smyth DJ, Howson JM et al Analysis of 17 autoimmune disease associated variants in type 1 diabetes identifies 6q23/TNFAIP3 as a susceptibility. Genes Immun 2009; 10:188–91. [DOI] [PubMed] [Google Scholar]
- 44. Melnick M, Jaskoll T. Mouse submandibular gland morphogenesis: a paradigm for embryonic signal processing. Crit Rev Oral Biol Med 2000; 11:199–215. [DOI] [PubMed] [Google Scholar]
- 45. Häärä O, Fujimori S, Schmidt‐Ullrich R, Hartmann C, Thesleff I, Mikkola ML. Ectodysplasin and Wnt pathways are required for salivary gland branching morphogenesis. Development 2011; 138:2681–91. [DOI] [PubMed] [Google Scholar]
- 46. Mustonen T, Pispa J, Mikkola ML et al Stimulation of ectodermal organ development by ectodysplasin‐A1. Dev Biol 2003; 259:123–36. [DOI] [PubMed] [Google Scholar]
- 47. Tucker AS, Headon DJ, Courtney JM, Overbeek P, Sharpe PT. The activation level of the TNF family receptor, Edar, determines cusp number and tooth number during tooth development. Dev Biol 2004; 268:185–94. [DOI] [PubMed] [Google Scholar]
- 48. Pispa J, Mustonen T, Mikkola ML et al Tooth patterning and enamel formation can be manipulated by misexpression of TNF receptor Edar. Dev Dyn 2004; 231:432–40. [DOI] [PubMed] [Google Scholar]
- 49. Chang SH, Jobling S, Brennan K, Headon DJ. Enhanced Edar signalling has pleiotropic effects on craniofacial and cutaneous glands. PLOS ONE 2009; 4:e7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cui CY, Schlessinger D. EDA signaling and skin appendage development. Cell Cycle 2006; 5:2477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thesleff I, Mikkola ML. Death receptor signaling giving life to ectodermal organs. Sci STKE 2002; 131:pe22. [DOI] [PubMed] [Google Scholar]
- 52. Yan M, Wang LC, Hymowitz SG et al Two‐amino acid molecular switch in an epithelial morphogen that regulates binding to two distinct receptors. Science 2000; 290:523–7. [DOI] [PubMed] [Google Scholar]
- 53. Döffinger R, Smahi A, Bessia C et al X‐linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF‐kappaB signaling. Nat Genet 2001; 27:277–85. [DOI] [PubMed] [Google Scholar]
- 54. Headon DJ, Emmal SA, Ferguson BM et al Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001; 414:913–6. [DOI] [PubMed] [Google Scholar]
- 55. Nederfors T. Xerostomia and hyposalivation. Adv Dent Res 2000; 14:48–56. [DOI] [PubMed] [Google Scholar]
- 56. Pispa J, Pummila M, Barker PA, Thesleff I, Mikkola ML. Edar and Troy signalling pathways act redundantly to regulate initiation of hair follicle development. Hum Mol Genet 2008; 17:3380–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information