

Summary
Circulating immunoglobulin (Ig)A class anti‐neutrophil cytoplasm antibodies (ANCA) directed against proteinase 3 (PR3) have been reported in ANCA‐associated vasculitis (AAV) with mucosal involvement. However, secretory IgA (SIgA) PR3‐ANCA has not been reported previously. In this study we compared serum levels of SIgA PR3‐ANCA and IgA PR3‐ANCA with IgG PR3‐ANCA in relation to disease characteristics. Among 73 patients with AAV and PR3‐ANCA at diagnosis, 84% tested positive for IgG PR3‐ANCA, 47% for IgA‐ANCA and 36% for SIgA PR3‐ANCA at the time of sampling for the present study. IgA and IgG PR3‐ANCA were represented similarly among patients with different organ manifestations, i.e. upper airway, lung or kidney at time of sampling. However, SIgA PR3‐ANCA was significantly less represented among patients with upper airway involvement. During active disease, the proportions of IgA PR3‐ANCA and SIgA PR3‐ANCA‐positive patients were significantly higher compared to inactive disease. Eight patients were sampled prospectively during 24 months from onset of active disease. In these patients, IgA PR3‐ANCA and SIgA PR3‐ANCA turned negative more often after remission induction compared to IgG PR3‐ANCA. Our findings suggest that serum IgA PR3‐ANCA and SIgA PR3‐ANCA are related more closely to disease activity in AAV compared to IgG PR3‐ANCA. Further studies are required to reveal if this has implications for disease activity monitoring. The mean number of PR3‐ANCA isotypes increased along with disease activity, suggesting a global B cell activation during active disease.
Keywords: ANCA‐associated vasculitis, disease, IgA PR3‐ANCA, mucosal immunity activity
Introduction
Granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) are the two major forms of anti‐neutrophil cytoplasmic antibody (ANCA)‐associated vasculitis (AAV). AAV predominantly affects small and medium sized arteries, and can thus engage multiple organs, mainly the upper and lower respiratory tract and kidneys, but less frequently skin, joints and peripheral nerves 1, 2. Experimental animal models and in ‐ vitro set‐ups support the concept that ANCA is of pathogenic importance in AAV by targeting surface‐exposed myeloperoxidase (MPO) or proteinase 3 (PR3) either on cytokine‐primed neutrophils, vascular endothelial cells 3, 4, 5, 6 or on epithelial cells in glomeruli or lungs 7, 8. In experimental murine models, it has been demonstrated that ANCA‐stimulated neutrophils reacted by forming ‘neutrophil extracellular traps’ (NET) exposing PR3 and MPO 9, which may induce ANCA and subsequent autoimmunity 10.
Immunoglobulin (Ig)G‐class PR3‐ANCA as well as MPO‐ANCA can bind their target antigens exposed on the neutrophil surface (for instance, after cytokine‐priming), resulting in cross‐linking of Fc‐receptors, complement activation and neutrophil oxidative burst 3, 11, 12, 13, 14, 15, 16, 17, 18. ANCA of different isotypes have been described previously, including IgG, IgA and IgM‐ANCA, where IgG‐ANCA is the predominating circulating isotype in AAV, and is monitored frequently in GPA as a means to assess disease activity 19, 20, 21, although the clinical utility remains controversial 22, 23, 24. With regard to mucosal manifestations in GPA, and as secretory IgA (SIgA) is the dominating isotype at mucosal sites, it is of interest to study IgA‐ and SIgA‐class PR3‐ANCA in relation to organ manifestations and disease activity in AAV. Circulating IgA‐class PR3‐ANCA has been described previously in GPA 25, and IgA‐ANCAs have been observed in IgA vasculitis (formerly known as Henoch–Schönlein purpura) 26, IgA‐nephropathy 27, cutaneous vasculitis 28, liver cirrhosis 29 and inflammatory bowel diseases 30, 31. SIgA PR3‐ANCA, however, has not been described previously in AAV. The present study was undertaken to analyse the occurrence, levels and clinical correlates of circulating IgA and SIgA PR3‐ANCA in patients with IgG PR3‐AAV based on the hypothesis that IgA/SIgA PR3‐ANCAs correlate with mucosal disease manifestations (i.e. upper and/or lower respiratory tract) and disease activity.
Materials and methods
Patients and controls
Seventy‐three patients diagnosed previously with AAV (GPA, n = 61 or MPA, n = 12), according to the 2012 revised international Chapel Hill consensus nomenclature of vasculitis 32, participated in the study. Baseline patient characteristics are presented in Table 1. The majority were Swedish by ancestry (70 of 73; 96%). Disease activity was assessed according to the Birmingham Vasculitis Activity Score (BVAS) 3·.0 33. At the time of diagnosis, all patients tested positive for IgG PR3‐ANCA (performed at the accredited Clinical Immunology Laboratory at Linköping University Hospital, Linköping, Sweden). At the time of sampling, 22 of the 73 patients (30%) had active disease (BVAS > 0), and 51 were in remission (Table 1). In eight cases, we had access to serum samples collected longitudinally (every third month during 24 months). Plasma samples from 31 patients diagnosed previously with IgA nephropathy (n = 17) or IgA vasculitis (n = 14) and serum samples from 101 blood donors were used as controls. All samples were obtained with informed consent and the study was approved by the regional ethical review board in Linköping, Sweden. Serum samples were stored at −80°C until analysed.
Table 1.
Baseline characteristics of patients diagnosed previously with ANCA‐associated vasculitis (AAV) at the time of sampling.
| Cohort n = 73 | |
|---|---|
| Men/women | 42/31 |
| Mean age (years) (s.d.) | 62·41 (15·88) |
| GPA/MPA* diagnosis | 61/12 |
| Mean disease duration (years) (range) | 6·96 (0–21·3) |
| Active disease (%) | 30 |
| Mean prednisone dose (mg) (range) | 15·2 (0–80) |
| Other immune suppressive therapy (%)† | 63 |
*Granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA); s.d. = standard deviation.
†Other immune suppressive therapy besides prednisone were: methotrexate, mycophenolate mofetil, azathioprine, leflunomide, chlorambucil, and rituximab.
PR3‐ANCA analyses
Detection of circulating PR3‐ANCA was performed by high‐sensitivity anti‐PR3 enzyme‐linked immunosorbent assays (ELISAs) (Orgentec Diagnostika GmbH, Mainz, Germany). For IgG PR3‐ANCA antibodies, analyses and cut‐off procedures were performed according to the manufacturer's instructions. Regarding IgA PR3‐ANCA antibodies, samples were diluted 1 : 100 and the detection antibody was replaced by a horseradish peroxidase (HRP)‐conjugated rabbit anti‐human α‐chain antibody (Dako A/S, Glostrup, Denmark) diluted 1 : 3000. For the detection of serum SIgA PR3‐ANCA antibodies, samples were diluted 1 : 25, and an HRP‐conjugated goat detection antibody towards human secretory component (Nordic BioSite AB, Täby, Sweden) was applied. Incubation conditions for all assays were in accordance with the manufacturer's recommendations for the IgG assay. A serially diluted strongly positive reference serum served as standard curve in each IgA and SIgA experiment (Supporting information, Fig. S1). Cut‐off levels for the IgA and SIgA PR3‐ANCA antibody assays were set to > 99th percentile among 101 blood donors (IgA PR3‐ANCA cut‐off; 12·9 AU/ml, and SIgA PR3‐ANCA cut‐off; 18·0 AU/ml). Regarding IgG PR3‐ANCA, we used the cut‐off level suggested by the manufacturer (10 U/ml). Intra‐ and interassay variations were assessed by testing a medium‐level sample (mean SIgA PR3‐ANCA 119 AU/ml and mean IgA PR3‐ANCA 96 AU/ml) 10 and five times, respectively. SIgA PR3‐ANCA intra‐ and interassay variation was 7·2 and 11·1%, respectively. Regarding IgA PR3‐ANCA, the intra‐ and interassay variation was 10·0 and 10·3%, respectively.
Antibody affinity purification
IgA anti‐PR3 and IgG anti‐PR3 were purified from a highly double‐positive serum sample by affinity chromatography. A 3‐ml serum sample was added to a Protein G column (Pierce Biotechnology, Woburn, MA, USA) and IgG antibodies were eluted using IgG Elution buffer (Pierce Biotechnology). The flow‐through containing IgA‐class antibodies was applied to a peptide M column (Invivogen Inc., San Diego, CA, USA). IgA‐antibodies were eluted using 0·1 M sodium acetate (pH 4·0). Immediately after elution, both IgG and IgA class antibodies were neutralized with 1 M Tris‐HCl (pH 8·3). The IgG and IgA class antibodies were applied to an in‐house PR3 column (Euro Diagnostica AB, Malmö, Sweden). Bound antibodies were eluted with 0·1 M glycine (pH 2·7) and neutralized immediately with 1 M Tris‐HCl (pH 8·3). The purified antibodies were stored at −20°C until further use.
Western blot
Purified antibodies were mixed with Laemmli buffer 1 : 1 (BioRad, Hercules, CA, USA) without 2‐beta mercaptoethanol for a native sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and applied to a precast Mini‐Protean TGX gel (BioRad), which was run for 45 min at 180 V (Bio‐Rad Power Pac HC). Precision plus protein all blue standards (BioRad) was used as a molecular weight marker and could be used for fluorescent blot detection at red wavelengths (635 nm laser). Bands on the SDS‐PAGE gel were transferred to a polyvinylidine fluoride (PVDF) membrane using a PVDF transfer pack (BioRad) and the Trans‐Blot Turbo Transfer system (BioRad) for 7 min at 25 V. The membrane was then blocked in 5% bovine serum albumin (BSA) in Tris‐buffered saline‐Tween 20 (TBST) with 0·1% Tween for 1 h. After blocking the membrane was incubated with a goat anti‐human secretory component peroxidase antibody (GAHU/SC/PO; NordicBiosite, Täby, Sweden) 1 : 5000 in TBS with 0·1% Tween for 1 h at room temperature (RT). Thereafter, the membrane was washed in TBST with 0·1% Tween, mounted and incubated with enhanced chemiluminescence (ECL) prime Western blotting detection reagent (GE Healthcare, Little Chalfont, UK) for 1 min. Bands on the membrane were detected using the ChemiDoc MP system (BioRad) and the molecular weight marker was detected with fluorescent Western blotting (Qdots 625).
Statistical analyses
Correlations were analysed by Spearman's correlation, categorical data by Fisher's exact test and ordinal data by Mann–Whitney U‐test. All the above‐mentioned analyses were performed with IBM spss statistics version 22. Statistical analyses for distribution of anti‐PR3 isotypes in organ involvement were performed using two‐way analysis of variance (anova) with GraphPad Prism version 6.
Results
PR3‐ANCA isotypes
PR3‐ANCA levels of different isotypes in patients and controls are shown in Fig. 1a–c. IgG PR3‐ANCA occurred in 84% (61 of 73) (Fig. 1a), whereas SIgA PR3‐ANCA occurred in 36% (26 of 73) of the patients (Fig. 1b), and IgA PR3‐ANCA in 34 of 73 (47%) of AAV patients (Fig. 1c). IgG PR3‐ANCA was significantly more common than the occurrence of IgA PR3‐ANCA (P < 0·0001) or SIgA anti‐PR3 (P < 0·0001). The occurrence of IgA PR3‐ANCA did not differ significantly from SIgA PR3‐ANCA (P = 0·24). Among IgG PR3‐ANCA‐ positive patients ever diagnosed with MPA, 17% (two of 12) were positive regarding IgA PR3‐ANCA, whereas among the GPA patients, 53% (32 of 61) were IgA PR3‐ANCA‐positive. Among AAV patients, all anti‐PR3 isotype levels correlated significantly; for IgG versus IgA PR3‐ANCA the correlation coefficient was 0·56 (P < 0·001), for IgG versus SIgA PR3‐ANCA 0·51 (P < 0·001), and for IgA versus SIgA PR3‐ANCA 0·53 (P < 0·001).
Figure 1.
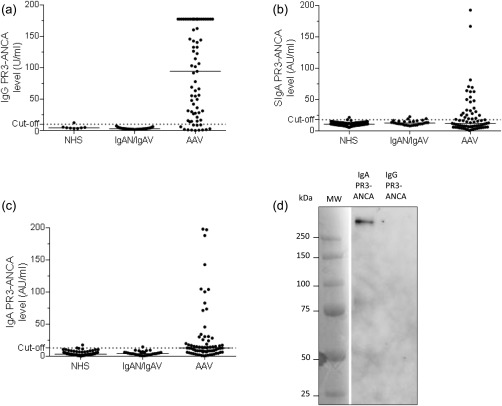
Occurrence and levels of immunoglobulin (Ig)G proteinase 3‐ anti‐neutrophil cytoplasm antibodies (PR3‐ANCA) (a), secretory IgA (SIgA) PR3‐ANCA (b), and IgA PR3‐ANCA (c) in sera from patients diagnosed with ANCA‐associated vasculitis (AAV). Western blot for secretory component (d). IgA and IgG were purified from a serum sample taken from a double‐positive AAV patient and were then subjected separately to affinity purification on a PR3‐column. Anti‐secretory component reactivity was detected only in the IgA fraction. NHS = normal healthy subjects; IgAN = IgA nephropathy; IgAV = IgA vasculitis.
None of the 31 sera from patients with IgA‐nephropathy or IgA vasculitis tested positive for IgG PR3‐ANCA (Fig. 1a). IgA PR3‐ANCA occurred in one patient (7%) diagnosed previously with IgA vasculitis (Fig. 1c), while SIgA PR3‐ANCA occurred at low levels in two cases diagnosed previously with IgA vasculitis (14%), and in one patient diagnosed previously with IgA‐nephropathy (6%) (Fig. 1b).
A shown by Western blot in Fig. 1d, the anti‐human secretory component antibody used in the high‐sensitivity anti‐PR3 ELISA detected a > 250 kDa band (compatible with 385 kDa SIgA) in the IgA PR3‐ANCA eluate, but not in the IgG PR3‐ANCA fraction.
PR3‐ANCA isotypes and disease activity
In patients with active disease (BVAS > 0) at the time of sampling (n = 22), the frequencies of IgA PR3‐ANCA‐ and SIgA PR3‐ANCA‐positive patients were significantly higher (P = 0·0001 and P = 0·035, respectively) than in patients with inactive disease (BVAS = 0) (Fig. 2a). There was no significant difference regarding the occurrence of IgG PR3‐ANCA (P = 0·092). At the time of sampling, IgA and IgG PR3‐ANCA were represented similarly among different types of organ involvement, i.e. upper airway, lung or kidney (Fig. 2b).The occurrence of serum SIgA PR3‐ANCA, however, was significantly lower among patients with upper airway involvement (P = 0·013). Three of 12 patients testing positive for SIgA PR3‐ANCA antibodies had neither upper airway nor lung involvement. We found statistically significant, although moderate, correlations between PR3‐ANCA isotype levels and BVAS: 0·43 for IgA PR3‐ANCA (P < 0·001), 0·37 for SIgA PR3‐ANCA (P = 0·001) and 0·33 for IgG PR3‐ANCA (P = 0·005). In the total cohort, mean BVAS was associated with the number of PR3‐ANCA isotypes present in serum (Fig. 3).
Figure 2.
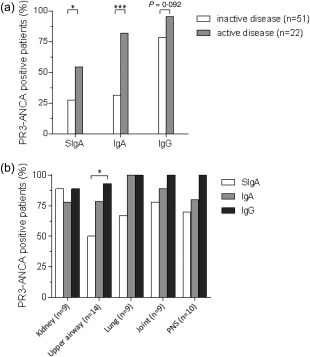
Proteinase 3‐anti‐neutrophil cytoplasm antibodies (PR3‐ANCA) levels in relation to disease activity (a) and in relation to organ involvement in patients with active disease at sampling (n = 22) (b). PNS = peripheral nervous system.
Figure 3.
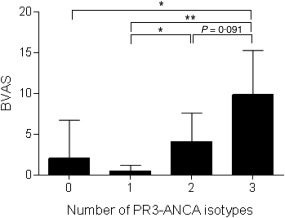
Number of serum proteinase 3‐ anti‐neutrophil cytoplasm antibodies (PR3‐ANCA) isotypes in relation to disease activity in ANCA‐associated vasculitis (n = 73). Bars represent mean Birmingham Vasculitis score with 95% confidence intervals depicted with error bars.
Serial monitoring of PR3‐ANCA isotype levels in patients with disease flare at baseline showed that, in four of eight patients, the IgG PR3‐ANCA antibody levels remained stable during 24 months, while remission (BVAS = 0) was achieved from 3 months and onwards. By contrast, in six of eight patients IgA PR3‐ANCA antibody levels declined below the cut‐off level during the same period (Fig. 4). Regarding SIgA PR3‐ANCA antibodies, only two patients tested positive for SIgA PR3‐ANCA at baseline, and in both cases the SIgA antibody levels declined 3 months after disease flare onset (Fig. 4). None of the eight patients were in active disease flare during the 24‐month follow‐up or the following 6 months after the last samples were collected. Furthermore, these longitudinally collected samples displayed a significantly higher frequency of IgA PR3‐ANCA and SIgA PR3‐ANCA positive patients in active disease (at 0 months) compared to inactive disease (3 months to 24 months) (P = 0·022, and P = 0·014, respectively). This could not be demonstrated regarding IgG PR3‐ANCA (P = 0·58) (Fig. 5).
Figure 4.
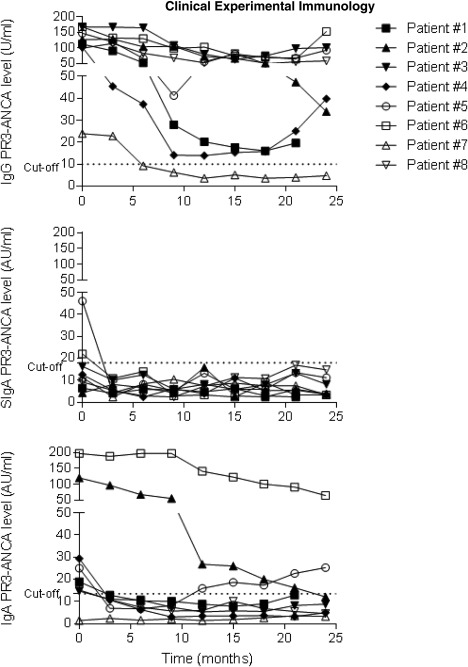
Proteinase 3‐anti‐neutrophil cytoplasm antibodies (PR3‐ANCA) levels in serum samples collected longitudinally from patients with disease flare at baseline and in remission from month 3 and onwards.
Figure 5.
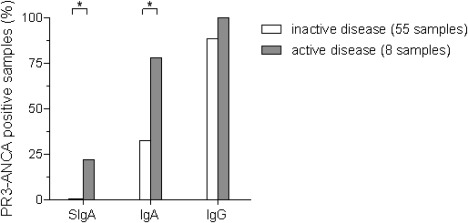
Proteinase 3‐anti‐neutrophil cytoplasm antibodies (PR3‐ANCA) isotype occurrence according to disease activity in longitudinally collected serum samples from eight patients.
Discussion
In this study, based on IgG PR3‐ANCA‐positive AAV patients, we found that serum IgA PR3‐ANCA correlated more clearly with disease activity than did IgG PR3‐ANCA, regardless of clinically apparent mucosal involvement of the disease. To our knowledge, this is the first report on circulating SIgA‐ANCA in vasculitis.
Kelley et al. have described previously that IgA PR3‐ANCA antibodies in GPA occurred predominantly among patients with upper airway involvement and without renal disease 25. With this in mind, and given the mucosal connection of IgA antibodies in general, we hypothesized that IgA PR3‐ANCA, and SIgA in particular, would be better markers of ‘mucosal disease activity’ in AAV compared to IgG PR3‐ANCA. Indeed, we found IgA PR3‐ANCA to be less prevalent in inactive disease. However, in contrast to the study by Kelley et al., we found no clear difference across different organ manifestations. Longitudinal sampling over 24 months revealed that IgG PR3‐ANCA levels were high during disease flare and remained positive in remission. Conversely, IgA PR3‐ANCA and SIgA PR3‐ANCA tended to decline more obviously during remission. Accordingly, both the cross‐sectional and longitudinal serum sample sets showed that the proportion of IgA‐ and SIgA PR3‐ANCA‐positive samples were significantly higher among patients with active disease compared to those in remission, while this was not seen regarding IgG PR3‐ANCA. Moreover, among the tested isotypes, IgA PR3‐ANCA had the highest correlation with BVAS. Taken together, this implies that analysis of IgA PR3‐ANCA may be superior in monitoring disease activity.
To our surprise, serum SIgA PR3‐ANCA was not more common among patients with upper or lower airway involvement. In fact, upper airway involvement was the only manifestation where SIgA PR3‐ANCA was significantly less prevalent compared with IgA and IgG PR3‐ANCA (Fig. 2b). The fact that 25% of the patients testing positive for SIgA PR3‐ANCA had neither upper airway nor lung involvement shows that circulating SIgA PR3‐ANCA can be present without clinically apparent AAV‐related mucosal manifestations. It remains to be elucidated whether this reflects subclinical upper airway or lung involvement or, instead, originates from other mucosal compartments, e.g. gastrointestinal or urogenital mucosa. The mechanism by which SIgA PR3‐ANCA may be redirected to the circulation in AAV remains to be determined. Leakage across epithelial surfaces disrupted by inflammation is one obvious possibility, but there are also reports on active apical‐to‐basal retransportation mechanisms of SIgA from the gut lumen to submucosa 34, 35, 36.
The PR3‐ANCA isotype repertoire appears to increase with global disease activity, resembling the situation reported for IgG subclasses of ANCA, where all patients with active disease had more than one IgG ANCA subclass while a single subclass was common in patients in remission 37. Taken together, this indicates that active vasculitis is associated with a generalized B cell activation, promoted by, for instance, elevated B cell activating factor/B lymphocyte stimulator (BAFF/BLyS) levels 38, decreased numbers of peripheral blood regulatory B cells 39 and defective regulatory T cell (Treg) function 40.
The generalized B cell activation in active vasculitis most probably involves B1 cells (shown to produce IgA in lungs and intestinal lamina propria 41) and marginal zone B lymphocytes (MZ cells), as well as B2 cells activated in germinal centres. The possibility that MZ cells and B1 cells are more prone to give rise to short‐lived IgA‐producing plasma cells, while B2 cells are more prone to give rise to long‐lived IgG producing plasma cells, may provide an explanation to why IgA/SIgA PR3‐ANCA diminishes quicker after onset of treatment and correlates more effectively with disease activity. Upon successful treatment, the resolution of inflammation would then lead hypothetically to degeneration of short‐lived plasma cell survival niches, and thereby decreased IgA production. It is also possible that the shorter half‐life of IgA compared with IgG may contribute to a faster decline in IgA autoantibody levels. Although any type of activated B cell can become a plasma cell precursor (plasmablast), not all plasma cells have the capacity to become long‐lived plasma cells. This may explain transient peak levels of circulating immunoglobins. For instance, this was illustrated in systemic lupus erythematosus (SLE), where large numbers of plasmablasts were also found during disease flare 42. Moreover, following acute viral infections, the number of short‐lived class‐switched IgA‐producing plasmablasts was found to be increased 43.
A weakness of the current study is the relatively small number of patients with active disease, especially in subgroups according to organ involvement. However, the patients are well‐characterized, and we believe that they constitute a representative ‘real‐world’ PR3‐ANCA‐positive AAV patient sample.
In conclusion, this study describes the occurrence of circulating SIgA PR3‐ANCA in AAV, and provides evidence that IgA PR3‐ANCA antibody analyses may improve serological monitoring of AAV patients.
Disclosure
The authors declare no financial disclosures.
Author contributions
A. K. conceived the study and C. S. performed the experiments. M. S. and P. E. were responsible for patient recruitment and characterization. C. S. and A. K. analysed the data. C. S., P. E., M. S., T. S. and A. K. were involved in the design of the study and manuscript preparation.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Representative standard curves for the detection of secretory immunoglobulin (Ig)A (SIgA) proteinase 3‐anti‐neutrophil cytoplasm antibodies (PR3‐ANCA) (a) and IgA PR3‐ANCA (b).
Acknowledgements
We thank Marianne Peterson at the Rheumatology Clinic, and Christina Sandell at the Clinical Immunology Laboratory for preparation of research samples. This study was supported by grants from the Swedish Society of Medicine, the Reinhold Sund foundation for Rheumatology Research, the Swedish Association against Rheumatism and the Östergötland County Council.
References
- 1. Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody‐mediated disease. Nat Rev Rheumatol 2014; 10:463–73. [DOI] [PubMed] [Google Scholar]
- 2. Kelley JM, Edberg JC, Kimberly RP. Wegener's granulomatosis: a model of auto‐antibodies in mucosal autoimmunity. Clin Immunol 2010; 134:104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti‐neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro . Proc Natl Acad Sci USA 1990; 87:4115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xiao H, Heeringa P, Liu Z et al The role of neutrophils in the induction of glomerulonephritis by anti‐myeloperoxidase antibodies. Am J Pathol 2005; 167:39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Primo VC, Marusic S, Franklin CC et al Anti‐PR3 immune responses induce segmental and necrotizing glomerulonephritis. Clin Exp Immunol 2010; 159:327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Little MA, Al‐Ani B, Ren S et al Anti‐proteinase 3 anti‐neutrophil cytoplasm autoantibodies recapitulate systemic vasculitis in mice with a humanized immune system. PLOS ONE 2012; 7:e28626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schwarting A, Hagen D, Odenthal M et al Proteinase‐3 mRNA expressed by glomerular epithelial cells correlates with crescent formation in Wegener's granulomatosis. Kidney Int 2000; 57:2412–22. [DOI] [PubMed] [Google Scholar]
- 8. Brockmann H, Schwarting A, Kriegsmann J et al Proteinase‐3 as the major autoantigen of c‐ANCA is strongly expressed in lung tissue of patients with Wegener's granulomatosis. Arthritis Res 2002; 4:220–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kessenbrock K, Krumbholz M, Schonermarck U et al Netting neutrophils in autoimmune small‐vessel vasculitis. Nat Med 2009; 15:623–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sangaletti S, Tripodo C, Chiodoni C et al Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood 2012; 120:3007–18. [DOI] [PubMed] [Google Scholar]
- 11. Nolle B, Specks U, Ludemann J, Rohrbach MS, DeRemee RA, Gross WL. Anticytoplasmic autoantibodies: their immunodiagnostic value in Wegener granulomatosis. Ann Intern Med 1989; 111:28–40. [DOI] [PubMed] [Google Scholar]
- 12. Kettritz R, Jennette JC, Falk RJ. Crosslinking of ANCA‐antigens stimulates superoxide release by human neutrophils. J Am Soc Nephrol 1997; 8:386–94. [DOI] [PubMed] [Google Scholar]
- 13. Kallenberg CG. Pathogenesis of PR3‐ANCA associated vasculitis. J Autoimmun 2008; 30:29–36. [DOI] [PubMed] [Google Scholar]
- 14. Porges AJ, Redecha PB, Kimberly WT, Csernok E, Gross WL, Kimberly RP. Anti‐neutrophil cytoplasmic antibodies engage and activate human neutrophils via Fc gamma RIIa. J Immunol 1994; 153:1271–80. [PubMed] [Google Scholar]
- 15. Kocher M, Edberg JC, Fleit HB, Kimberly RP. Antineutrophil cytoplasmic antibodies preferentially engage Fc gammaRIIIb on human neutrophils. J Immunol 1998; 161:6909–14. [PubMed] [Google Scholar]
- 16. Ben‐Smith A, Dove SK, Martin A, Wakelam MJ, Savage CO. Antineutrophil cytoplasm autoantibodies from patients with systemic vasculitis activate neutrophils through distinct signaling cascades: comparison with conventional Fcgamma receptor ligation. Blood 2001; 98:1448–55. [DOI] [PubMed] [Google Scholar]
- 17. Skogh T, Dahlgren C, Holmgren K, Peen E, Stendahl O. Anti‐granulocyte antibodies (C‐ANCA, P‐ANCA, GS‐ANA) studied by confocal scanning laser fluorescence microscopy, ELISA, and chemiluminescence techniques. Scand J Immunol 1991; 34:137–45. [DOI] [PubMed] [Google Scholar]
- 18. Ohlsson SM, Ohlsson S, Soderberg D et al Neutrophils from vasculitis patients exhibit an increased propensity for activation by anti‐neutrophil cytoplasmic antibodies. Clin Exp Immunol 2014; 176:363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Finkielman JD, Lee AS, Hummel AM et al ANCA are detectable in nearly all patients with active severe Wegener's granulomatosis. Am J Med 2007; 120:643.e9–14. [DOI] [PubMed] [Google Scholar]
- 20. Specks U, Wheatley CL, McDonald TJ, Rohrbach MS, DeRemee RA. Anticytoplasmic autoantibodies in the diagnosis and follow‐up of Wegener's granulomatosis. Mayo Clin Proc 1989; 64:28–36. [DOI] [PubMed] [Google Scholar]
- 21. Segelmark M, Phillips BD, Hogan SL, Falk RJ, Jennette JC. Monitoring proteinase 3 antineutrophil cytoplasmic antibodies for detection of relapses in small vessel vasculitis. Clin Diagn Lab Immunol 2003; 10:769–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davenport A, Lock RJ, Wallington T. Clinical significance of the serial measurement of autoantibodies to neutrophil cytoplasm using a standard indirect immunofluorescence test. Am J Nephrol 1995; 15:201–7. [DOI] [PubMed] [Google Scholar]
- 23. Kerr GS, Fleisher TA, Hallahan CW, Leavitt RY, Fauci AS, Hoffman GS. Limited prognostic value of changes in antineutrophil cytoplasmic antibody titer in patients with Wegener's granulomatosis. Arthritis Rheum 1993; 36:365–71. [DOI] [PubMed] [Google Scholar]
- 24. Tomasson G, Grayson PC, Mahr AD, Lavalley M, Merkel PA. Value of ANCA measurements during remission to predict a relapse of ANCA‐associated vasculitis – a meta‐analysis. Rheumatology 2012; 51:100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kelley JM, Monach PA, Ji C et al IgA and IgG antineutrophil cytoplasmic antibody engagement of Fc receptor genetic variants influences granulomatosis with polyangiitis. Proc Natl Acad Sci USA 2011; 108:20736–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ronda N, Esnault VL, Layward L et al Antineutrophil cytoplasm antibodies (ANCA) of IgA isotype in adult Henoch–Schonlein purpura. Clin Exp Immunol 1994; 95:49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martin SJ, Audrain MA, Baranger T et al Recurrence of immunoglobulin A nephropathy with immunoglobulin A antineutrophil cytoplasmic antibodies following renal transplantation. Am J Kidney Dis 1997; 29:125–31. [DOI] [PubMed] [Google Scholar]
- 28. Rovel‐Guitera P, Diemert MC, Charuel JL et al IgA antineutrophil cytoplasmic antibodies in cutaneous vasculitis. Br J Dermatol 2000; 143:99–103. [DOI] [PubMed] [Google Scholar]
- 29. Papp M, Sipeki N, Vitalis Z et al High prevalence of IgA class anti‐neutrophil cytoplasmic antibodies (ANCA) is associated with increased risk of bacterial infection in patients with cirrhosis. J Hepatol 2013; 59:457–66. [DOI] [PubMed] [Google Scholar]
- 30. Peen E, Almer S, Bodemar G et al Anti‐lactoferrin antibodies and other types of ANCA in ulcerative colitis, primary sclerosing cholangitis, and Crohn's disease. Gut 1993; 34:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Arias‐Loste MT, Bonilla G, Moraleja I et al Presence of anti‐proteinase 3 antineutrophil cytoplasmic antibodies (anti‐PR3 ANCA) as serologic markers in inflammatory bowel disease. Clin Rev Allergy Immunol 2013; 45:109–16. [DOI] [PubMed] [Google Scholar]
- 32. Jennette JC, Falk RJ, Bacon PA et al 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013; 65:1–11. [DOI] [PubMed] [Google Scholar]
- 33. Flossmann O, Bacon P, de Groot K et al Development of comprehensive disease assessment in systemic vasculitis. Ann Rheum Dis 2007; 66:283–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Matysiak‐Budnik T, Moura IC, Arcos‐Fajardo M et al Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. The J Exp Med 2008; 205:143–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lebreton C, Menard S, Abed J et al Interactions among secretory immunoglobulin A, CD71, and transglutaminase‐2 affect permeability of intestinal epithelial cells to gliadin peptides. Gastroenterology 2012; 143:698–707.e1–4. [DOI] [PubMed] [Google Scholar]
- 36. Rey J, Garin N, Spertini F, Corthesy B. Targeting of secretory IgA to Peyer's patch dendritic and T cells after transport by intestinal M cells. J Immunol 2004; 172:3026–33. [DOI] [PubMed] [Google Scholar]
- 37. Segelmark M, Wieslander J. IgG subclasses of antineutrophil cytoplasm autoantibodies (ANCA). Nephrol Dial Transplant 1993; 8:696–702. [DOI] [PubMed] [Google Scholar]
- 38. Krumbholz M, Specks U, Wick M, Kalled SL, Jenne D, Meinl E. BAFF is elevated in serum of patients with Wegener's granulomatosis. J Autoimmun 2005; 25:298–302. [DOI] [PubMed] [Google Scholar]
- 39. Bunch DO, McGregor JG, Khandoobhai NB et al Decreased CD5(+) B cells in active ANCA vasculitis and relapse after rituximab. Clin J Am Soc Nephrol 2013; 8:382–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Free ME, Bunch DO, McGregor JA et al Patients with antineutrophil cytoplasmic antibody‐associated vasculitis have defective Treg cell function exacerbated by the presence of a suppression‐resistant effector cell population. Arthritis Rheum 2013; 65:1922–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baumgarth N. The double life of a B‐1 cell: self‐reactivity selects for protective effector functions. Nat Rev Immunol 2011; 11:34–46. [DOI] [PubMed] [Google Scholar]
- 42. Radbruch A, Muehlinghaus G, Luger EO et al Competence and competition: the challenge of becoming a long‐lived plasma cell. Nat Rev Immunol 2006; 6:741–50. [DOI] [PubMed] [Google Scholar]
- 43. Fink K. Origin and function of circulating plasmablasts during acute viral infections. Front Immunol 2012; 3:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Representative standard curves for the detection of secretory immunoglobulin (Ig)A (SIgA) proteinase 3‐anti‐neutrophil cytoplasm antibodies (PR3‐ANCA) (a) and IgA PR3‐ANCA (b).