

Summary
Anti‐phospholipid antibody syndrome (APS) is a systemic autoimmune disease characterized clinically by arterial and/or venous thromboses, recurrent abortions or fetal loss and serologically by the presence of ‘anti‐phospholipid antibodies’ (aPL). The main target antigen of the antibodies is β2glycoprotein I (β2GPI). Post‐translational oxidative modifications of the protein have been widely described. In this study we aimed to analyse sera reactivity to glucose‐modified β2GPI (G‐β2GPI). Sera collected from 43 patients with APS [15 primary APS (PAPS) and 28 APS associated with systemic lupus erythematosus (SLE) (SAPS)], 30 with SLE, 30 with rheumatoid arthritis (RA) and 40 healthy subjects were analysed by an enzyme‐linked immunosorbent assay (ELISA) using a G‐β2GPI. Nine of 15 consecutive PAPS out‐patients (60%) and 16 of 28 SAPS (57.1%) showed serum antibodies [immunoglobulin (Ig)G class] against G‐β2GPI (anti‐G‐β2GPI) by ELISA. The occurrence of anti‐G‐β2GPI was significantly higher in APS patients compared to patients suffering from SLE. No RA patients or control healthy subjects resulted positive for anti‐G‐β2GPI. Of note, aG‐β2GPI prompted to identify some APS patients (four PAPS and seven SAPS), who were negative in the classical anti‐β2GPI test. Moreover, in APS patients, anti‐G‐β2GPI titre was associated significantly with venous thrombosis and seizure in APS patients. This study demonstrates that G‐β2GPI is a target antigen of humoral immune response in patients with APS, suggesting that β2GPI glycation products may contain additional epitopes for anti‐β2GPI reactivity. Searching for these antibodies may be useful for evaluating the risk of clinical manifestations.
Keywords: β2glycoprotein I, anti‐phospholipid antibody syndrome, cryptic or neoepitopes, glycation, systemic lupus erythematosus
Introduction
Anti‐phospholipid antibody syndrome (APS) is a systemic autoimmune disease characterized clinically by arterial and/or venous thromboses, recurrent abortions or fetal loss and serologically by the presence of anti‐phospholipid antibodies (aPL) 1, 2. Diagnosis of APS requires the combination of at least one clinical and one laboratory criterion 3. Anti‐cardiolipin antibodies (aCL) and anti‐β2‐glycoprotein I antibodies (aβ2GPI) detected by enzyme linked immunosorbent assay (ELISA), and lupus anti‐coagulant (LA) detected by clotting assays, are the recommended tests for detection of aPL 4, 5. However, aPL represent a heterogeneous family of antibodies reacting with phospholipid‐binding proteins, including not only β2GPI 6, 7, but also different anionic phospholipids, proteins or phospholipid–protein complexes, such as prothrombin 8, protein S 9, 10, protein C 11, annexin V 12, annexin II 13, vimentin 14, oxidized low‐density lipoproteins, lyso‐bis‐phosphatidic acid (LBPA) and sulphatides, etc. 15, 16, 17. β2GPI, an abundant plasma glycoprotein involved in clotting mechanisms and lipid pathways 18, is the most common target for aPL associated frequently with vascular cell dysfunction 19, thrombotic events and proatherogenic mechanisms 20, 21, 22. The aPL action mechanism forms the basis of APS pathophysiology. Moreover, only autoimmune‐type aCL are usually dependent upon the presence of β2GPI and anti‐β2GPI have been involved in the expression of LA activity, an in‐vitro phenomenon associated with an increased risk of arterial and/or venous thromboembolic events. Understanding the mechanisms of how this abundant self‐plasma protein becomes a target of pathogenic autoantibodies will improve the knowledge of APS pathophysiology.
The molecular structure and location of the major epitopic region(s) of the β2GPI molecule are controversial, although they were recently characterized fully 23. Several studies have investigated whether the immune response is directed to native β2GPI 22, 23, 24, 25 or to cryptic or neoepitopes 26, 27. Decisive events generating cryptic or neoepitopes include β2GPI binding to anionic surfaces, such as phospholipids, and oxidative modifications that alter phospholipid binding 27, 28, 29, 30. Many mechanisms may be responsible for generating neoepitopes, and multiple mechanisms receive support from the heterogeneous antigenic specificities in β2GPI‐specific antibodies. One candidate mechanism is non‐enzymatic glycosylation (glycation), a process that leads to the formation of early, intermediate and advanced glycation end products (AGEs), able to modify self‐molecule structures and functions. Even though glycation is present physiologically and is modulated by several factors, diet, ageing as well as disorders of glucose metabolism and systemic autoimmune diseases associated with inflammation and oxidative stress may favour the formation and accumulation of these products 31, 32, 33. AGEs represent new epitopes and contain new antigenic structures, thereby possibly contributing to the generation of autoimmune responses. Recently, we showed that several potential glycation sites are present within the β2GPI primary structure and that, after in‐vitro exposure to glucose, β2GPI was sugar‐modified and this modification probably consisted of an AGE formation. Our results on the ability of AGE‐β2GPI to activate human monocyte‐derived immature dendritic cells suggest a possible role for glycation in the increase of β2GPI immunogenicity 34. Although there is accumulating evidence that AGEs are involved in the progression of inflammatory and immune‐mediated diseases 35, further investigations are needed to clarify the role of glycation in generating new antigenic epitopes in β2GPI possibly contributing to the heterogeneous specificity of aPL.
This study was designed to investigate whether β2GPI in vitro treated with glucose (G‐β2GPI) is a target of humoral response in APS patients and whether anti‐G‐β2GPI may be associated with clinical features.
Materials and methods
Patients
This study included 73 consecutive out‐patients attending the Lupus Clinic of the Sapienza University of Rome. Forty‐three patients had APS, diagnosed according to the Sydney Classification Criteria 3, primary (PAPS, n = 15) or APS associated with systemic lupus erythematosus (SLE) (SAPS, n = 28); 30 patients had SLE fulfilling the ACR revised criteria for the classification of SLE 36. Sera were collected at several times and stored at −20°C until use. Finally, 30 patients with RA and 40 healthy subjects (normal blood donors) matched for age and sex were also studied as controls. This study was approved by the local ethic committees and participants gave written informed consent in accordance with the Declaration of Helsinki.
Bioinformatic analysis of β2GPI
The primary structure of β2GPI (Accession no. P02749) was analysed by means of tools available online at http://sysbio.unl.edu/SVMTriP/index.php, to predict the presence of flexible and functional sites, including antigenic epitopes 37, related closely or structurally to potential glycation sites 34. The most recommended epitope(s) within β2GPI sequence were calculated using 20 amino acids as the default epitope length search for putative epitopes on the web server, as reported 38.
Preparation of glucose‐modified β2GPI
Human native β2GPI was purchased from Calbiochem (La Jolla, CA, USA). Purity of β2GPI > 98% was checked by mass spectrometry, as reported by the manufacturer.
To prepare glucose‐modified β2GPI (G‐β2GPI), human β2GPI was dissolved in glycation buffer solution (GB) (0·144 g/l KH2PO4, 0·426 g/l Na2HPO4) pH 7·4, at 10 μg/ml final concentration and frozen immediately at −80°C under sterile conditions. Then, β2GPI aliquots were incubated in the presence of 250 mM glucose or mannitol (Sigma‐Aldrich, Milan, Italy) in the dark, at 37°C for increasing intervals of time (0, 12 h, 10 and 22 days) (in sealed vials), as described previously 34, 39. As glycation control, a highly purified preparation of human albumin (Sigma‐Aldrich) was treated with D‐glucose under the same conditions used for β2GPI.
Characterization of glucose‐modified β2GPI
Size exclusion chromatography was performed by fast protein liquid chromatography (FPLC; Pharmacia, Uppsala, Sweden) interfaced to an ultraviolet (UV) monochromator detector (ProteomeLab PF2D Protein Fractionation System; Beckman Coulter, Brea, CA). Twenty micrograms of native or G‐β2GPI resuspended into 50 μl of phosphate‐buffered saline (PBS) without calcium and magnesium (PBS−/−) were injected onto a Superose S12 Pharmacia column equilibrated in PBS−/−, pH 7·4. Elution was carried out with a 0·4 ml/min flow rate at 22°C, PBS−/− as elution buffer, and protein peaks were detected under UV recording (optical density at 214 nm). Purity of β2GPI and its molecular weight under denatured conditions was checked by electrophoretic analysis 34.
UV spectrophotometry
The UV absorption characteristics of G‐β2GPI and native‐β2GPI were recorded on a UV/visible spectrophotometer (dual beam Uvikon 860 Instrument; Kontron, Zurich, CH) between 200 and 400 nm, using 1 cm optical length UV quartz cuvettes, as described previously 40, with some modifications: G‐β2GPI was analysed onto the ‘sample’ quartz cuvette, while native‐β2GPI was analysed onto the ‘reference’ quartz cuvette. In this manner, the differential absorption spectrum indicates the spectral regions of β2GPI affected by glucose‐induced modifications.
Non‐tryptophan fluorescence studies
Fluorescence studies were carried out as reported previously 34, 39. Steady‐state fluorescence emission spectra of β2GPI preparations were collected with a FluoroMax‐2 spectrofluorometer (Jobin Yvon‐Spex, Edison, NJ, USA) using an excitation wavelength of 370 nm, equal bandwidths for excitation and emission (5/5). Emission fluorescence at 450 nm was measured by subtracting contributions of sugar solutions from the fluorescence of the sugar‐β2GPI mixture.
ELISA for aCL and anti‐β2GPI antibodies
aCL and anti‐β2GPI ELISA kits were obtained from Inova Diagnostics Inc. (San Diego, CA, USA). ELISA was performed for all the patients’ and healthy subjects’ sera according to the manufacturer's instructions. aβ2GPI were also tested by ELISA, as reported below.
LA assay
LA for all the patients’ and healthy subjects’ sera was studied in two coagulation systems, a dilute sensitized activated partial thromboplastin time (aPTT) and a dilute Russell's viper venom time (dRVVT), followed by confirmatory test, using reagents and instrumentation by Hemoliance Instrumentation Laboratory (Lexington, MA, USA).
ELISA for glucose‐modified β2GPI
Ninety‐six‐well polystyrene plates were coated and incubated overnight at 4°C with 1 µg/well of native‐β2GPI or G‐β2GPI in 0·05 μM NaHCO3 buffer, pH 9·5. Coated plates were incubated overnight at 4°C and then washed three times with PBS containing 0·05% Tween 20 (PBS‐T). Plates were blocked for 2 h at room temperature with 100 µl of 1% bovine serum albumin (BSA) in PBS. After washing three times with PBS‐T, the wells were incubated for 1 h at room temperature with 100 µl of patient sera diluted 1 : 100 in the blocking buffer. Each serum was analysed in triplicate. Goat polyclonal anti‐β2GPI (Affinity Biologicals Inc., Ancaster, ON, Canada) was used as positive control. After three washes with PBS‐T, the plates were incubated for 1 h at room temperature with horseradish peroxidase (HRP)‐conjugated antibodies, anti‐human immunoglobulin (Ig)G (Sigma‐Aldrich) or anti‐goat IgG (Dako Italia S.p.A., Cernusco sul Naviglio, Italy) diluted in the blocking buffer. The plates were washed three times with PBS‐T; the bound peroxidase was then revealed with 100 µl of O‐phenylenediamine dihydrochloride buffer and colour development was stopped with H2SO4 0·2 M for 5 min. Absorbance was measured at 492 nm in a microplate reader. Data were presented as the mean optical density (OD) corrected for background (wells without coated antigen). Forty sera from healthy subjects were also tested and a cut‐off value was established at the mean of OD ± 3 standard deviations (s.d.) of normal human sera. Parallel experiments were performed with an identical procedure but without coated G‐β2GPI.
Absorption tests
Sera of patients with APS, positive for both aβ2GPI and anti‐G‐β2GPI, and of healthy subjects, were diluted 1 : 100 in the blocking buffer and incubated with native β2GPI (100 μg/ml) for 1 h at 37°C and then overnight at 4°C. The mixture was centrifuged for 15 min at 27 000 g at 4°C, according to Alessandri et al. 16; the supernatant fraction was kept as absorbed serum and tested for anti‐G‐β2GPI and anti‐native‐β2GPI antibodies by ELISA, as reported above.
Statistical analysis
All the statistical procedures were performed by GraphPad Prism software Inc. (San Diego, CA, USA). Normally distributed variables were summarized using the mean ± s.d. and non‐normally distributed variables by the median and range. Differences between numerical variables were tested with the Wilcoxon test. Correlation was tested with Spearman's rank order or Pearson's correlation coefficient. For comparison of categorical variables or percentages, Fisher's exact and χ2 tests were used when appropriate. P‐values less than 0·05 were considered significant.
Results
Clinical and serological characteristics of APS and SLE patients
All patients enrolled into this study were Caucasian. APS patients were 39 females and four males with a median age of 45 years (range = 17–75), and a median disease duration of 7 years (range = 0·5–24). SLE patients were 28 females and two males, with a median age of 42 years (range = 25–64) and a median disease duration of 6 years (range = 1–22). The clinical characteristics of APS and SLE patients are reported in Table 1. All patients were screened for aCL, anti‐β2GPI (tested by both ELISA methods with overlapping results) and LA; the prevalence of the antibodies is reported in Table 2.
Table 1.
Clinical characteristics of patients.
| Characteristics n (%) | PAPS n = 15 | SAPS n = 28 | SLE n = 30 |
|---|---|---|---|
| Arterial thrombosis | 9 (60) | 11 (39·3) | 0 |
| Venous thrombosis | 4 (26·7) | 16 (57·1) | 0 |
| Pregnancy morbidity | 4 (26·7) | 5 (17·9) | 3 (10) |
| Livedo reticularis | 6 (40) | 12 (42·9) | 5 (16·6) |
| Thrombocytopenia | 4 (26·7) | 8 (28·6) | 1 (3·3) |
| Migraine | 3 (20) | 2 (7·1) | 2 (6·6) |
| Seizures | 2 (13·3) | 2 (7·1) | 1 (3·3) |
PAPS = primary anti‐phospholipid antibody syndrome; SLE = systemic lupus erythematosus; SAPS = APS associated with SLE.
Table 2.
Prevalence of antibodies of patients.
| Autoantibodies n (%) | PAPS n = 15 | SAPS n = 28 | SLE n = 30 |
|---|---|---|---|
| aCL | 12 (80) | 22 (78·6) | 3 (10) |
| aβ2GPI | 5 (33·3) | 9 (32·1) | 1 (3·3) |
| LA | 8 (53·3) | 16 (57·1) | 1 (3·3) |
| aG‐β2GPI | 9 (60) | 16 (57·1) | 8 (26·6) |
PAPS = primary anti‐phospholipid antibody syndrome; SLE = systemic lupus erythematosus; SAPS = APS associated with SLE; aCL = anti‐cardiolipin antibodies; LA = lupus anti‐coagulant.
Bioinformatic analysis of β2GPI
Analysis of functional and immunological sites within β2GPI sequence showed that the most recommended epitope corresponds to the residues 51–70. Of note, this region contains the residue with the highest potentiality to be glycated (#63: score = 0.967, as calculated by Netglycate‐1·0 prediction), as published previously 34 (Supporting information, Table S1).
Characterization of glucose‐modified β2GPI
Purified β2GPI preparation was incubated for increasing times in the presence of D‐ glucose and characterized as described in the Methods section.
In order to verify whether G‐β2GPI might undergo a significant molecular size modification, a size exclusion chromatographic separation was carried out. UV recording at 214 nm of eluate showed the formation of a molecular size increase due probably to the formation of glucose‐β2GPI adducts (G‐β2GPI) (see arrow in Fig. 1a). Analysis of the chromatographic profile showed a molecular size increase in the 22‐day glycated β2GPI corresponding to approximately 18% of total protein.
Figure 1.
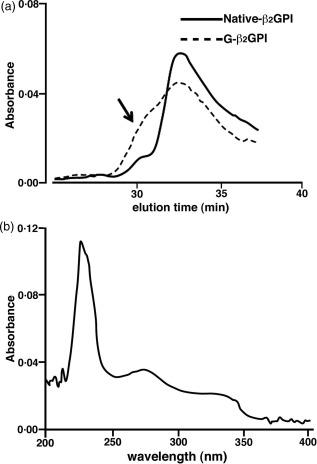
(a) Chromatographic elution of native and glucose‐modified β2glycoprotein I (G‐β2GPI). Twenty micrograms of highly purified β2GPI incubated previously with sugar were injected onto Superose S12 fast protein liquid chromatography (FPLC) column as described in the Methods section and protein peaks were detected under ultraviolet (UV) recording (optical density at 214 nm). The dashed line indicates that G‐β2GPI elutes as a complex protein population involving higher molecular weight complexes (highlighted by the arrow). A semiquantitative evaluation of such polymeric G‐β2GPI, corresponding to the left‐side shoulder of the dashed line, indicates that under these non‐denaturating chromatographic conditions, such complexes may represent the 15–20% of total G‐β2GPI proteins; (b) differential absorption spectrum between native β2GPI and G‐β2GPI. Using a dual‐beam UV spectrophotometer, absorbance of G‐β2GPI and native‐β2GPI were analysed. The absorption spectrum shows the different spectral regions of β2GPI affected by glucose‐induced modification. The analysis indicates that three main regions are affected: the change of absorbance observed at region corresponding to 310–340 nm suggests the formation of non‐enzymatic glycosylation adducts 34, whereas the changes of absorbance observed at 260–280 and 215–240 suggest structural modifications occurring on the β2GPI protein folding.
UV absorption spectrum indicated that three main regions of β2GPI were affected by glucose treatment. The change of absorbance observed at region corresponding to 310–340 nm suggests the formation of non‐enzymatic glycosylation adducts, as reported by published studies 40, whereas the changes of absorbance observed at 260–280 and 215–240 nm suggest structural modifications occurring on β2GPI protein folding (Fig. 1b).
Fluorescence at 450 nm, specific for AGE formation, was measured and confirmed the creation of time‐dependent AGE products (Supporting information, Table S2).
Detection of antibodies to glucose modified‐β2GPI by ELISA
Nine of 15 consecutive PAPS out‐patients (60%) and 16 of 28 SAPS (57·1%) showed serum antibodies (IgG class) against G‐β2GPI (anti‐G‐β2GPI). No significant difference was found between PAPS and SAPS patients (Table 2). Of note, four sera from patients with PAPS and seven with SAPS were positive for anti‐G‐β2GPI but negative for anti‐native‐β2GPI (Fig. 2, arrows).
Figure 2.
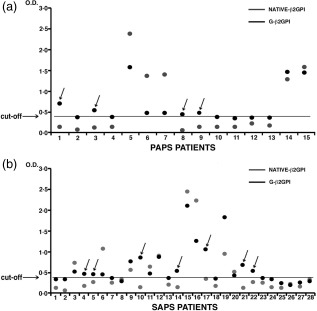
Relationship between anti‐native β2glycoprotein I (β2GPI) and anti‐glucose‐modified GPI (G‐β2GPI) antibodies in primary anti‐phospholipid antibody syndrome (PAPS) and APS associated with systemic lupus erythematosus (SLE) (SAPS) patients. Sera from patients with PAPS and SAPS were analysed by enzyme‐linked immunosorbent assay (ELISA) for the detection of anti‐native‐β2GPI and anti‐G‐β2GPI immunoglobulin (Ig)G. The arrows indicate the sera from patients (PAPS or SAPS) positive for anti‐G‐β2GPI but negative for anti‐native‐β2GPI. The horizontal line shows the cut‐off level calculated as mean optical density (OD) value in control healthy subjects’ sera ± 3 standard deviations (s.d.).
The occurrence of anti‐G‐β2GPI was significantly higher in APS patients (25 of 43, 58·1%) compared to patients suffering from SLE (26·6%) (P = 0·037). No RA patients or control healthy subjects resulted positive for aG‐β2GPI (Fig. 3).
Figure 3.
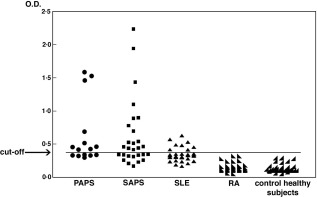
Anti‐glucose‐modified β2glycoprotein I (G‐β2GPI) antibodies in patients and healthy subjects. Sera from patients with anti‐phospholipid antibody syndrome (PAPS) and APS associated with systemic lupus erythematosus (SLE) (SAPS), SLE, rheumatoid arthritis (RA) and of healthy subjects were analysed by enzyme‐linked immunosorbent assay (ELISA) for the detection of anti‐G‐β2GPI immunoglobulin (Ig)G. All the sera from RA patients and healthy subjects were negative for anti‐G‐β2GPI IgG antibodies. The horizontal line shows the cut‐off level calculated as mean optical density (OD) value in control healthy subjects’ sera ± 3 standard deviations (s.d.).
Sera of patients with APS, positive for both anti‐β2GPI and anti‐G‐β2GPI, were absorbed with native β2GPI and then tested for anti‐G‐β2GPI antibodies, or alternatively for anti‐native‐β2GPI, by ELISA. Reactivity with G‐β2GPI showed significant inhibition (63% inhibition, P < 0·001) by first absorbing sera with native β2GPI (Fig. 4a). As expected, reactivity with native‐β2GPI showed almost complete inhibition (83·8% inhibition, P < 0·001) (Fig. 4b). These findings suggest that anti‐G‐β2GPI recognize specific glycation‐related epitopes.
Figure 4.
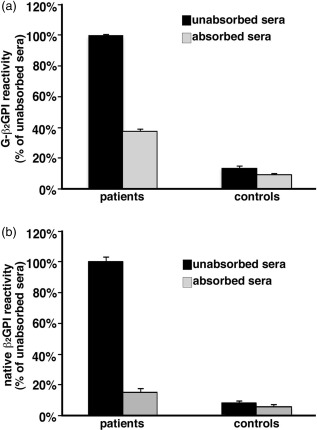
Absorption tests for detection of anti‐glucose‐modified β2glycoprotein I (G‐β2GPI) specificity. Sera of patients with anti‐phospholipid antibody syndrome (APS), positive for both anti‐native β2GPI and anti‐G‐β2GPI, and of healthy subjects (controls), unabsorbed or absorbed with native β2GPI, were analysed by enzyme‐linked immunosorbent assay (ELISA) for the detection of: (a) anti‐G‐β2GPI IgG, mean absorbance of unabsorbed sera [optical density (OD) 2·264] was set to 100%. Absorbed patients’ sera versus unabsorbed patients’ sera: P < 0·001; absorbed controls’ sera versus unabsorbed controls’ sera: P > 0·05. (b) anti‐β2GPI IgG, mean absorbance of unabsorbed sera (OD 1·116) was set to 100%. Absorbed patients’ sera versus unabsorbed patients’ sera: P < 0·001; absorbed controls’ sera versus unabsorbed controls’ sera: P > 0·05.
Associations of antibodies to glucose‐modified β2GPI with classical aPL and clinical features
Analysis of the correlations between anti‐G‐β2GPI OD and classical aPL in APS patients revealed a significant association with aCL (P = 0·013) and anti‐native β2GPI (P = 0·009). Furthermore, a significant correlation was found between anti‐G‐β2GPI with venous thrombosis (P = 0·017) and seizure (P = 0·027) in these patients.
Discussion
This study provides new findings showing that G‐β2GPI is a target antigen of humoral immune response in patients with APS and suggests a possible usefulness of anti‐G‐β2GPI to improve the diagnosis of this disease.
Several studies have suggested that post‐translational oxidative modifications of β2GPI may affect antigenic properties of the molecule. Among these, glycosylation processes may play a relevant role 41. In this study we analysed the presence of antibodies to glucose‐modified β2GPI by glycation (non‐enzymatic glycosylation). Glycation is the non‐enzymatic addition or insertion of saccharide derivatives to proteins, lipids or nucleic acids, leading to the formation of intermediary Schiff bases and Amadori products and, finally, to irreversible AGEs 42. Glycation reaction, similarly to other decisive events, including β2GPI binding to anionic surfaces, such as phospholipids, and oxidative modifications 29, 30, 43, 44, may induce a significant misfolding effect on the β2GPI structure, contributing to the expression of cryptic or neoepitopes recognized by the immune system. In a previous investigation we demonstrated, by bioinformatic analyses of the β2GPI primary structure, that several potential glycation sites are present within the molecule 34. The intriguing co‐localization of high glycation sites with potential epitopes found in the present study suggests that sugar‐induced modifications occurring on β2GPI may be able to affect its antigenic behaviour.
In our previous investigation we also showed that a 10‐day β2GPI treatment with glucose induces a protein modification probably consisting of AGE‐β2GPI formation, and that this glucose‐modified protein is able to activate human monocyte‐derived immature dendritic cells 34. In the present study, we investigated the effects of a longer incubation (22 days) with glucose on β2GPI structure. G‐β2GPI was studied under non‐denaturing conditions, in order to evaluate the formation of glycation end products and/or adducts with sugar characterized by non‐covalent bonds. The chromatographic analysis of G‐β2GPI indicated the presence of a protein population with shorter elution time, i.e. with larger molecular size. Peak integration of the chromatogram suggested that this molecular size shift could involve a significant portion of β2GPI protein.
Therefore, we verified that glucose treatment may induce a significant misfolding effect on the β2GPI structure, probably leading to expression of cryptic or neoepitopes recognized by the immune system. Upon modification with glucose, increase in UV absorbance was recorded. This finding provided a useful insight into the structural perturbation of β2GPI protein probably inducing the formation of glycation adducts. The fluorescence spectrophotometric analysis of G‐β2GPI confirmed the creation of time‐dependent advanced glycation end products (AGEs).
In this study we observed that anti‐G‐β2GPI were present in a significant percentage of patients with APS. Interestingly, ELISA for anti‐G‐β2GPI prompted to identify some APS patients (four PAPS and seven SAPS), who were negative in the ‘classical’ anti‐β2GPI test.
The mechanisms by which G‐β2GPI accumulates and may trigger a humoral immune response in APS patients are not completely known. A possible explanation is that G‐β2GPI accumulation occurs in patients with APS driven by oxidative stress and inflammation, thereby initiating a local autoimmune process and becoming the target of autoimmune responses. The role of oxidative post‐translational modification of β2GPI has been described widely 45, 46. In particular, it can produce new antigens that are not represented in the thymus, so that autoreactive T cells can escape negative selection and move to the periphery. Alternatively, the way by which antigen‐presenting cells process oxidized β2GPI may be different from that processing the reduced form. In both cases an autoimmune response can be triggered. Thus, as epitope dominance is influenced by protein structure, glycation events may change the molecular context of β2GPI epitopes (by altering secondary or tertiary structure), thus permitting the efficient presentation of cryptic and neodeterminants 45. According to our previous results 34, 44, we can hypothesize that a pro‐oxidant and proinflammatory microenvironment predisposes local β2GPI to glycation and/or oxidation, thereby initiating a local autoimmune process.
Another interesting finding of this study is the observation that anti‐G‐β2GPI were correlated significantly with several clinical manifestations of APS, including venous thrombosis and seizure. To date, the most commonly investigated antigenic target in APS patients is β2GPI, and anti‐β2GPI antibodies represent a highly specific test for diagnosis of the syndrome. In particular, IgG anti‐β2GPI antibodies comprise a family of antibodies which recognize different epitopes of the protein. In recent years, several studies showed that antibodies to domain I (DI), that specifically recognize the glycine40‐arginine43 epitope, showed a good correlation with thrombosis and pregnancy morbidity 47, 48. Moreover, anti‐β2GPI antibodies with DI specificity were found in the majority of APS patients and were associated significantly with LA and venous thrombosis 49. Our findings extend these data, strongly suggesting a relationship between the presence of anti‐G‐β2GPI and thrombosis. This observation is not surprising. Indeed, since 1985, Vlassara et al. showed that AGE‐modified proteins can be bound to a special receptor receptor for advanced glycation end‐product (RAGE) 50, and the same binding has been shown for AGE‐β2GPI 34. Engagement of RAGE results in intracellular signalling, which leads to activation of the proinflammatory transcription factor nuclear factor kappa B (NF‐κB), with consequent production of cytokines, adhesion molecules, prothrombotic and vasoconstrictive gene products 34, 51. For example, the endothelial surface is changed in a way that coagulation events are favoured; indeed, AGE‐bound RAGE on the endothelium may result in alteration of the cell surface structure, inducing a procoagulant endothelium, via reduced thrombomodulin activity 52 concomitant with increased tissue factor expression 53.
Taken together, our data demonstrate the existence of anti‐G‐β2GPI in APS patients, suggesting that β2GPI glycation products may represent additional epitopes for anti‐β2GPI reactivity, possibly contributing to the heterogeneous specificity of aPL 54. Searching for these antibodies may be useful for evaluating the risk of clinical manifestations in APS patients.
Disclosure
The authors declare that they have no disclosures.
Author contributions
M.S. designed the study and drafted the manuscript, B.B. designed the study and carried out the experiments, A.C. carried out the experiments, E.P. designed the study and carried out the experiments, F.F. carried out the experiments (preparation of G‐β2GPI and chromatographic analyses), C.A. and S.T. collected the sera samples, characterized the patients clinically and performed statistical analysis, R.M. drafted the manuscript and revised the manuscript critically, F.C. and G.V. revised the manuscript critically, R.R. designed the study and drafted the manuscript.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Table S1. Bioinformatic analysis of functional and immunological sites within β2GPI sequence.
{kind=link}
Table S2. Time‐dependent non‐tryptophan fluorescence of sugar‐treated β2GPI.
{kind=link}
Acknowledgements
The technological support from the Facility for Complex Protein Mixture (CPM) Analysis at ISS (Rome) is kindly acknowledged. All authors read and approved the final manuscript.
References
- 1. Hughes GR, Harris NN, Gharavi AE. The anticardiolipin syndrome. J Rheumatol 1986; 13:486–9. [PubMed] [Google Scholar]
- 2. Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–63. [DOI] [PubMed] [Google Scholar]
- 3. Miyakis S, Lockshin MD, Atsumi T et al International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306. [DOI] [PubMed] [Google Scholar]
- 4. Tincani A, Balestrieri G, Spatola L, Cinquini M, Meroni PL, Roubey RA. Anticardiolipin and anti‐beta 2glycoprotein I immunoassays in the diagnosis of antiphospholipid syndrome. Clin Exp Rheumatol 1998; 16:396–402. [PubMed] [Google Scholar]
- 5. Bertolaccini ML, Gomez S, Pareja JF et al Antiphospholipid antibody tests: spreading the net. Ann Rheum Dis 2005; 64:1639–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Galli M, Barbui T, Zwaal RF, Comfurius P, Bevers EM. Antiphospholipid antibodies: involvement of protein cofactors. Haematologica 1993; 78:1–4. [PubMed] [Google Scholar]
- 7. Bas de Laat H, Derksen RH, de Groot PG. Beta2‐glycoprotein I, the playmaker of the antiphospholipid syndrome. Clin Immunol 2004; 112:161–8. [DOI] [PubMed] [Google Scholar]
- 8. Arvieux J, Darnige L, Caron C, Reber G, Bensa JC, Colomb MG. Development of an ELISA for autoantibodies to prothrombin showing their prevalence in patients with lupus anticoagulants. Thromb Haemost 1995; 74:1120–5. [PubMed] [Google Scholar]
- 9. Sorice M, Griggi T, Circella A et al Protein S antibodies in acquired protein S deficiencies. Blood 1994; 83:2383–4. [PubMed] [Google Scholar]
- 10. Sorice M, Arcieri P, Griggi T et al Inhibition of protein S by autoantibodies in patients with acquired protein S deficiency. Thromb Haemost 1996; 75:555–9. [PubMed] [Google Scholar]
- 11. Oosting JD, Derksen RH, Bobbink IW, Hackeng TM, Bouma BN, de Groot PG. Antiphospholipid antibodies directed against a combination of phospholipids with prothrombin, protein C, or protein S: an explanation for their pathogenic mechanism? Blood 1993; 81:2618–25. [PubMed] [Google Scholar]
- 12. Kaburaki J, Kuwana M, Yamamoto M, Kawai S, Ikeda Y. Clinical significance of anti‐annexin V antibodies in patients with systemic lupus erythematosus. Am J Hematol 1997; 54:209–13. [DOI] [PubMed] [Google Scholar]
- 13. Salle V, Maziere JC, Smail A et al Anti‐annexin II antibodies in systemic autoimmune diseases and antiphospholipid syndrome. J Clin Immunol 2008; 28:291–7. [DOI] [PubMed] [Google Scholar]
- 14. Ortona E, Capozzi A, Colasanti T et al Vimentin/cardiolipin complex as a new antigenic target of the antiphospholipid syndrome. Blood 2010; 116:2960–7. [DOI] [PubMed] [Google Scholar]
- 15. Kobayashi T, Stang E, Fang KS, de Moerloose P, Parton RG, Gruenberg J. A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature 1998; 392:193–7. [DOI] [PubMed] [Google Scholar]
- 16. Alessandri C, Bombardieri M, Di Prospero L, Conigliaro P, Conti F, Labbadia G. Anti‐lysobisphosphatidic acid antibodies in patients with antiphospholipid syndrome and systemic lupus erythematosus. Clin Exp Immunol 2005; 140:173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Valesini G, Alessandri C. New facet of antiphospholipid antibodies. Ann NY Acad Sci 2005; 1051:487–97. [DOI] [PubMed] [Google Scholar]
- 18. Muller G, Biering A, Lux E, Richter V. Beta 2 glycoprotein I analysis in patients with hyperlipoproteinemia, arteriosclerotic occlusive disease and diabetes mellitus. Z Gesamte Inn Med 1983; 38:282–4. [PubMed] [Google Scholar]
- 19. Hattori N, Kuwana M, Kaburaki J, Mimori T, Ikeda Y, Kawakami Y. T cells that are autoreactive to beta2‐glycoprotein I in patients with antiphospholipid syndrome and healthy individuals. Arthritis Rheum 2000; 43:65–75. [DOI] [PubMed] [Google Scholar]
- 20. Ferro D, Pittoni V, Quintarelli C et al Coexistence of anti‐phospholipid antibodies and endothelial perturbation in systemic lupus erythematosus patients with ongoing prothrombotic state. Circulation 1997; 95:1425–32. [DOI] [PubMed] [Google Scholar]
- 21. George J, Shoenfeld Y, Harats D. The involvement of beta2‐glycoprotein I (beta2‐GPI) in human and murine atherosclerosis. J Autoimmun 1999; 13:57–60. [DOI] [PubMed] [Google Scholar]
- 22. Garcia CO, Kanbour‐Shakir A, Tang H, Molina JF, Espinoza LR, Gharavi AE. Induction of experimental antiphospholipid antibody syndrome in PL/J mice following immunization with beta 2 GPI. Am J Reprod Immunol 1997; 37:118–24. [DOI] [PubMed] [Google Scholar]
- 23. Zager U, Kveder T, Cučnik S, Božič B, Lunder M. Anti‐β2‐glycoprotein I paratopes and β2‐glycoprotein I epitopes characterization using random peptide libraries. Autoimmunity 2014; 47:438–44. [DOI] [PubMed] [Google Scholar]
- 24. Tincani A, Spatola L, Prati E et al The anti‐beta2‐glycoprotein I activity in human anti‐phospholipid syndrome sera is due to monoreactive low‐affinity autoantibodies directed to epitopes located on native beta2‐glycoprotein I and preserved during species’ evolution. J Immunol 1996; 157:5732–8. [PubMed] [Google Scholar]
- 25. Roubey RA, Eisenberg RA, Harper MF, Winfield JB. ‘Anticardiolipin’ autoantibodies recognize beta 2‐glycoprotein I in the absence of phospholipid. Importance of Ag density and bivalent binding. J Immunol 1995; 154:954–60. [PubMed] [Google Scholar]
- 26. Aron AL, Cuellar ML, Brey RL et al Early onset of autoimmunity in MRL/++ mice following immunization with beta 2 glycoprotein I. Clin Exp Immunol 1995; 101:78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Artenjak A, Locatelli I, Brelih H et al Immunoreactivity and avidity of IgG anti‐β2‐glycoprotein I antibodies from patients with autoimmune diseases to different peptide clusters of β2‐glycoprotein I. Immunol Res 2015; 61:35–44. [DOI] [PubMed] [Google Scholar]
- 28. Wang MX, Kandiah DA, Ichikawa K et al Epitope specificity of monoclonal anti‐beta 2‐glycoprotein I antibodies derived from patients with the antiphospholipid syndrome. J Immunol 1995; 155:1629–36. [PubMed] [Google Scholar]
- 29. Horkko S, Miller E, Branch DW, Palinski W, Witztum JL. The epitopes for some antiphospholipid antibodies are adducts of oxidized phospholipid and beta2 glycoprotein 1 (and other proteins). Proc Natl Acad Sci USA 1997; 94:10356–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arvieux J, Regnault V, Hachulla E, Darnige L, Berthou F, Youinou P. Oxidation of beta2‐glycoprotein I (beta2GPI) by the hydroxyl radical alters phospholipid binding and modulates recognition by anti‐beta2GPI autoantibodies. Thromb Haemost 2001; 86:1070–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ramasamy R, Yan SF, Schmidt AM. RAGE: therapeutic target and biomarker of the inflammatory response‐the evidence mounts. J Leukoc Biol 2009; 86:505–12. [DOI] [PubMed] [Google Scholar]
- 32. Shanmugam N, Kim YS, Lanting L, Natarajan R. Regulation of cyclooxygenase‐2 expression in monocytes by ligation of the receptor for advanced glycation end products. J Biol Chem 2003; 278:34834–44. [DOI] [PubMed] [Google Scholar]
- 33. Bierhaus A, Hofmann MA, Ziegler R, Nawroth PP. AGEs and their interaction with AGE‐receptors in vascular disease and diabetes mellitus. I. The AGE concept. Cardiovasc Res 1998; 37:586–600. [DOI] [PubMed] [Google Scholar]
- 34. Buttari B, Profumo E, Capozzi A et al Advanced glycation end products of human beta(2) glycoprotein I modulate the maturation and function of DCs. Blood 2011; 117:6152–61. [DOI] [PubMed] [Google Scholar]
- 35. Nienhuis HL, Westra J, Smit AJ, Limburg PC, Kallenberg CG, Bijl M. AGE and their receptor RAGE in systemic autoimmune diseases: an inflammation propagating factor contributing to accelerated atherosclerosis. Autoimmunity 2009; 42:302–4. [DOI] [PubMed] [Google Scholar]
- 36. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997; 40:1725. [DOI] [PubMed] [Google Scholar]
- 37. Facchiano AM, Facchiano A, Facchiano F. Active Sequences Collection (ASC) database: a new tool to assign functions to protein sequences. Nucleic Acids Res 2003; 31:379–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yao B, Zhang L, Liang S, Zhang C. SVMTriP: a method to predict antigenic epitopes using support vector machine to integrate tri‐peptide similarity and propensity. PLOS ONE 2012; 7:e45152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Facchiano F, D'Arcangelo D, Russo K et al Glycated fibroblast growth factor‐2 is quickly produced in vitro upon low‐millimolar glucose treatment and detected in vivo in diabetic mice. Mol Endocrinol 2006; 20:2806–18. [DOI] [PubMed] [Google Scholar]
- 40. Ansari NA, Dash D. Biochemical studies on methylglyoxal‐mediated glycated histones: implications for presence of serum antibodies against the glycated histones in patients with type 1 diabetes mellitus. ISRN Biochem 2013; 7:198065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kondo A, Miyamoto T, Yonekawa O, Giessing AM, Osterlund EC, Jensen ON. Glycopeptide profiling of beta‐2‐glycoprotein I by mass spectrometry reveals attenuated sialylation in patients with antiphospholipid syndrome. J Proteomics 2009; 73:123–33. [DOI] [PubMed] [Google Scholar]
- 42. Smit AJ, Lutgers HL. The clinical relevance of advanced glycation endproducts (AGE) and recent developments in pharmaceutics to reduce AGE accumulation. Curr Med Chem 2004; 11:2767–84. [DOI] [PubMed] [Google Scholar]
- 43. Matsuura E, Igarashi Y, Fujimoto M et al Heterogeneity of anticardiolipin antibodies defined by the anticardiolipin cofactor. J Immunol 1992; 148:3885–91. [PubMed] [Google Scholar]
- 44. Buttari B, Profumo E, Mattei V et al Oxidized beta2‐glycoprotein I induces human dendritic cell maturation and promotes a T helper type 1 response. Blood 2005; 106:3880–7. [DOI] [PubMed] [Google Scholar]
- 45. Passam FH, Giannakopoulos B, Mirarabshahi P, Krilis SA. Molecular pathophysiology of the antiphospholipid syndrome: the role of oxidative post‐translational modification of beta 2 glycoprotein I. J Thromb Haemost 2011; 9:275–82. [DOI] [PubMed] [Google Scholar]
- 46. Mirarabshahi P, Abdelatti M, Krilis S. Post‐translational oxidative modification of beta2‐glycoprotein I and its role in the pathophysiology of the antiphospholipid syndrome. Autoimmun Rev 2012; 11:779–80. [DOI] [PubMed] [Google Scholar]
- 47. Sebire NJ, Fox H, Backos M, Rai R, Paterson C, Regan L. Defective endovascular trophoblast invasion in primary antiphospholipid antibody syndrome‐associated early pregnancy failure. Hum Reprod 2002; 17:1067–71. [DOI] [PubMed] [Google Scholar]
- 48. Iverson GM, Victoria EJ, Marquis DM. Anti‐beta2 glycoprotein I (beta2GPI) autoantibodies recognize an epitope on the first domain of beta2GPI. Proc Natl Acad Sci USA 1998; 95:15542–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. de Laat B, de Groot. PG. Autoantibodies directed against domain I of beta2‐glycoprotein I. Curr Rheumatol Rep 2011; 13:70–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vlassara H, Brownlee M, Cerami A. High‐affinity‐receptor‐mediated uptake and degradation of glucose‐modified proteins: a potential mechanism for the removal of senescent macromolecules. Proc Natl Acad Sci USA 1985; 82:5588–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bierhaus A, Humpert PM, Morcos M et al Understanding RAGE, the receptor for advanced glycation end products. J Mol Med (Berl) 2005; 83:876–86. [DOI] [PubMed] [Google Scholar]
- 52. Esposito C, Gerlach H, Brett J, Stern D, Vlassara H. Endothelial receptor‐mediated binding of glucose‐modified albumin is associated with increased monolayer permeability and modulation of cell surface coagulant properties. J Exp Med 1989; 170:1387–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bierhaus A, Illmer T, Kasper M et al Advanced glycation end product (AGE)‐mediated induction of tissue factor in cultured endothelial cells is dependent on RAGE. Circulation 1997; 96:2262–71. [DOI] [PubMed] [Google Scholar]
- 54. Gharavi AE, Sammaritano LR, Wen J, Elkon KB. Induction of antiphospholipid autoantibodies by immunization with beta 2 glycoprotein I (apolipoprotein H). J Clin Invest 1992; 90:1105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Table S1. Bioinformatic analysis of functional and immunological sites within β2GPI sequence.
Table S2. Time‐dependent non‐tryptophan fluorescence of sugar‐treated β2GPI.