

Summary
Chronic mucocutaneous candidiasis (CMC) is characterized by recurrent and persistent superficial infections, with Candida albicans affecting the mucous membranes, skin and nails. It can be acquired or caused by primary immune deficiencies, particularly those that impair interleukin (IL)−17 and IL‐22 immunity. We describe a single kindred with CMC and the identification of a STAT1 GOF mutation by whole exome sequencing (WES). We show how detailed clinical and immunological phenotyping of this family in the context of WES has enabled revision of disease status and clinical management. Together with analysis of other CMC cases within our cohort of patients, we used knowledge arising from the characterization of this family to develop a rapid ex‐vivo screening assay for the detection of T helper type 17 (Th17) deficiency better suited to the routine diagnostic setting than established in‐vitro techniques, such as intracellular cytokine staining and enzyme‐linked immunosorbent assay (ELISA) using cell culture supernatants. We demonstrate that cell surface staining of unstimulated whole blood for CCR6+CXCR3–CCR4+CD161+ T helper cells generates results that correlate with intracellular cytokine staining for IL‐17A, and is able to discriminate between patients with molecularly defined CMC and healthy controls with 100% sensitivity and specificity within the cohort tested. Furthermore, removal of CCR4 and CD161 from the antibody staining panel did not affect assay performance, suggesting that the enumeration of CCR6+CXCR3–CD4+ T cells is sufficient for screening for Th17 deficiency in patients with CMC and could be used to guide further investigation aimed at identifying the underlying molecular cause.
Keywords: chemokine receptors, chronic mucocutaneous candidiasis, surface phenotyping, Th17
Introduction
Chronic mucocutaneous candidiasis (CMC) is characterized by recurrent and persistent superficial infections with Candida albicans affecting the mucous membranes, skin and nails. It can be acquired or caused by primary immune deficiencies (PIDs), particularly those that impair interleukin (IL)−17 and IL‐22 immunity 1, 2, 3. Examples of PIDs that cause CMC include hyper‐immunoglobulin (Ig)E syndrome due to dominant negative STAT3 mutations (HIGE) 4, 5, 6, 7 and autosomal recessive mutations in DOCK8 1, 8, 9, 10, 11, IL‐17F and IL‐17RA deficiency 12, IL‐12RB1 deficiency 13 and autoimmune polyglandular syndrome 1 (APS1) due to loss‐of‐function mutations in the autoimmune regulator (AIRE) and the production of neutralizing autoantibodies against IL‐17 and IL‐22 14, 15. More recently, gain‐of‐function (GOF) mutations in the coiled‐coil and DNA binding domains of STAT1 have been found to cause autosomal‐dominant CMC due to defective development and function of IL‐17‐producing T cells 16, 17, 18, 19.
The diagnosis of CMC associated with PID is based on clinical history, identification of differentiating clinical and immunological features associated with particular molecular causes, and genetic analysis. Approaches to identify the underlying molecular cause include single gene sequencing and next‐generation techniques such as targeted resequencing using gene panels, whole exome and genome sequencing. However, these can be costly and require specialized and often lengthy analysis. Functional studies can also be used to demonstrate T helper type 17 (Th17) dysfunction, for example IL‐17‐producing T cells can be measured using in‐vitro stimulation of peripheral blood mononuclear cells (PBMCs) followed by intracellular cytokine staining and analysis by flow cytometry or enzyme‐linked immunosorbent assay (ELISA) using cell culture supernatants. In addition, immunoblot or flow cytometry can be used to demonstrate increased signal transducer and activator of transcription‐1 (STAT‐1) phosphorylation in leucocytes after stimulation with interferon (IFN)‐γ 20. These techniques are labour‐intensive and time‐consuming and, in the context of CMC, the latter can only be used to demonstrate STAT‐1 GOF.
Surface phenotyping for chemokine receptors can also be used to identify T helper subsets. For example CXCR3, CXCR6 and CCR5 are expressed by Th1 cells and CCR3, CCR4 and CCR8 are expressed by Th2 cells 21. Co‐expression of CCR6 and CCR4, chemokine receptors that mediate homing to the skin and mucosae, has been shown to identify human memory T helper cells that produce IL‐17 and express RAR‐related orphan receptor gamma (RORγt), the key transcription factor that drives Th17 differentiation 21, 22. Th17 cells have also been shown to lack surface CXCR3, a marker which can be used to distinguish them from Th1 cells 21, 22. Microarray analysis has revealed that, in addition to IL‐17, IL‐23R, RORγt and CCR6, expression of CD161, a homologue of murine NK1·1, is up‐regulated in human Th17 clones compared to Th1 or Th2 clones 23. When human CD4+ T cells are sorted based on surface expression of CD161 and CCR6, production of IL‐17 after stimulation with phorbol myristate acetate (PMA) and ionomycin, as well as expression of IL‐23R and RORγt, is restricted almost completely to the CD161+CCR6+ double‐positive population 22.
Here we describe the clinical and immunological phenotypes of a single kindred found to have a STAT1 GOF mutation underlying CMC. Using members from this family, along with other patients with CMC and characterized genetic mutations known to cause Th17 dysregulation, we sought to assess whether surface staining for CCR6, CCR4, CD161 and CXCR3 21, 22, 23, 24 could be used as a surrogate measure of IL‐17‐producing T cells in peripheral blood without the need for T cell activation and intracellular staining, with the aim of creating a rapid screening assay for Th17 dysfunction in patients with CMC.
Materials and methods
Patient samples for whole exome and Sanger sequencing
Ethylenediamine tetraacetic acid (EDTA) whole blood was obtained from seven individuals in the family (II.7, III.1, III.2, III.5, III.6, III.7 and III.8) for whole exome and Sanger sequencing; additionally, stored DNA from another affected member (II.6) was used for Sanger sequencing (see Table 1 for patient characteristics).
Table 1.
Clinical and immunological features of a single kindred found to have a STAT1 gain‐of‐function (GOF) mutation underlying chronic mucocutaneous candidiasis (CMC). T helper type 17 (Th17) cells were measured using in‐vitro stimulation of peripheral blood mononuclear cells (PBMCs) with phorbol myristate acetate (PMA) and ionomycin for 6 h followed by intracellular cytokine staining for interleukin (IL)−17A and analysis by flow cytometry (as detailed in the main text).
| Patient | Chronic mucocutaneous candidiasis | Other clinical features | Immunological phenotype | STAT1 Genetics | |||||
|---|---|---|---|---|---|---|---|---|---|
| Onset | Extent of disease | Treatment | Fungal cultures | T/B/NK | Igs | Th17 | |||
| II.3 | – | Reported to have suffered with CMC | – | – | Oropharyngeal carcinoma (died 40 y/o) | – | – | – | – |
| II.6 | Neonatal |
• Childhood:discreet episodes of mild disease affecting nails & oral mucosa • Adulthood: progressively more severe & persistent with oral ulceration & oesophageal involvement |
• Azoles for treatment of acute episodes • Azole prophylaxis started in adulthood |
• Candida albicans
• Progressive azole resistance |
Oesophageal strictures, severe dental caries, iron deficiency, oral squamous cell carcinoma (died 43 y/o) | n | n | – | WT/R274W |
| II.7 | 5 y/o |
• Childhood:discreet episodes of mild disease affecting nails & oral mucosa • Adulthood: progressively more frequent & severe with oesophageal & genital involvement. Persistent oropharyngeal CMC |
• Childhood: topical/oral azoles for acute episodes • Daily azole prophylaxis started at 25 y/o, stopped at 48 y/o • Pulsed i.v. ambisome for acute episodes from 48 y/o |
• C. albicans
• Progressive azole resistance • Pan‐azole resistance 48 y/o |
Hyperthyroidism, severe dental caries, anxiety, depression, iron deficiency, venous thromboembolism, allergic rhinitis, asthma, bronchiectasis | Mild T cell lymphocytosis | Polyclonal increase | ↓ | WT/R274W |
| III.1 | 2 y/o |
• Childhood:discreet episodes of mild disease affecting nails & oral mucosa • Adulthood: progressively more frequent & severe with oesophageal involvement |
• Childhood: topical/oral azoles for acute episodes • Daily azole prophylaxis started at 18 y/o, stopped at 30 y/o • Pulsed i.v. ambisome for acute episodes from 30 y/o |
• C. albicans
• Progressive azole resistance • Pan‐azole resistance 30 y/o |
Severe dental caries, frequent respiratory tract infections, psoriasis, depression, personality disorder | n | n | ↓ | WT/R274W |
| III.2 | Neonatal |
• Childhood:discreet episodes of mild disease affecting nails & oral mucosa • Adulthood:intermittent oesophageal infection |
• Childhood: topical/oral azoles for acute episodes • Daily azole prophylaxis started at 5 y/o • Pulsed i.v. caspofungin for acute episodes from 22 y/o |
• C. albicans
• Progressive azole resistance |
Attention deficit and hyperactivity disorder, frequent warts & respiratory tract infections in childhood only, asthma & migraines | Mild lymphopaenia | Polyclonal increase in IgA | ↓ | WT/R274W |
| III.6 | Infancy | Oral candidiasis in infancy, majority coinciding with courses of anti‐bacterials for respiratory tract infection | • Daily azole prophylaxis started in infancy, stopped at 22 y/o with no recurrence of candidiasis | – | Iron deficiency | n | n | n | WT/WT |
| III.7 | Neonatal | Oral candidiasis in neonatal period | • Daily azole prophylaxis started in infancy, stopped at 15 y/o with no recurrence of candidiasis | – | Iron deficiency | n | n | n | WT/WT |
| III.8 | Neonatal |
• Infancy:severe & frequent oral and cutaneous disease • Childhood:more persistent oral disease, nail involvement |
• Azoles used for treatment & prophylaxis started in infancy • Prophylaxis stopped at 12 y/o with no adverse effect • Pulsed i.v. ambisome for acute episodes from 12 y/o |
• C. albicans
• Progressive azole resistance • Pan‐azole resistance 12 y/o |
Iron deficiency | n | n | ↓ | WT/R274W |
y/o = years old; Ig = immunoglobulin; i.v. = intravenous; n = normal; WT = wild‐type.
Patient samples for Th17 assays
Fresh blood samples were acquired from healthy controls (n = 19; median age = 44 years; age range = 18–62 years; M : F = 7 : 12), patients with CMC (n = 7; median age 28 years; age range 1–47 years; M : F = 4 : 3; see Table 2 for characteristics); and one STAT1 wild‐type individual from the kindred with CMC due to STAT1 GOF (III.7 in Fig. 1a and Table 1; age = 17 years; female). All samples were processed within 24 h.
Table 2.
Demographic, phenotypical and genetic data of patients with chronic mucocutaneous candidiasis (CMC) used for subsequent T helper type 17 (Th17) cell assays.
| Age | Sex | Diagnosis | Clinical/immunological phenotype | Genetic mutation |
|---|---|---|---|---|
| 47 years | Female | Familial STAT‐1 GOF | Patient II.7 from family cohort (see Table 1) |
Heterozygous STAT1 c.C820T; p.R274W Found on whole exome sequencing, reported previously 16, 17 |
| 35 years | Male | Familial STAT‐1 GOF | Patient III.1 from family cohort (see Table 1) |
Heterozygous STAT1 c.C820T; p.R274W Found on whole exome sequencing, reported previously 16, 17 |
| 25 years | Male | Familial STAT‐1 GOF | Patient III.2 from family cohort (see Table 1) |
Heterozygous STAT1 c.C820T; p.R274W Found on whole exome sequencing, reported previously 16, 17 |
| 33 years | Male | Sporadic STAT1 mutation |
Presented in infancy with CMC Recurrent otitis media and chest infections with resultant bronchiectasis in childhood Bilateral cerebral abscesses due to Streptococcus pneumoniae at age 16 years Selective immunoglobulin A deficiency and a specific polysaccharide antibody deficiency Treated with anti‐fungals and immunoglobulin therapy |
Heterozygous STAT1 c.G629A; p.R210K Missense mutation in coiled‐coil domain found in several unrelated patients presenting with CMC (unpublished data) |
| 40 years | Female | Hyper‐immunoglobulin E syndrome | Delayed shedding of primary teeth, bone fractures, characteristic facies, Staphylococcal skin abscesses and respiratory tract infections with pneumatocoele formation and bronchiectasis and CMC |
Heterozygous STAT3 c.C1144T; p.R382W Described previously in several unrelated patients 6, 7 |
| 1 year | Female | Interleukin‐12 receptor B1 deficiency |
Presented in 1st year of life with disseminated Bacillus Calmette–Guérin and CMC Severely impaired production of interferon‐γ to all stimuli with no up‐regulation in response to interleukin‐12 co‐stimulation |
Homozygous IL12RB1 c.C790T; p.Q264X Predicted to result in truncated protein |
| 13 years | Male | Autoimmune polyglandular syndrome 1 |
Presented in early childhood with episodic mucocutaneous candidiasis, ectodermal dystrophy and enteropathy Strongly positive serum auto‐antibodies directed against IL‐17A, IL‐17F, IL‐22 and tryptophan hydroxylase Reduction in serotonin positive and chromogranin positive cells on gastrointestinal histology |
Compound heterozygous AIRE c.C769T/c.964del13 13 bp deletion in exon 8 found in over 70% of British APS1 patients 31 |
STAT‐1 = signal transducer and activator of transcription‐1; GOF = gain‐of‐function; IL = interleukin; bp = base pairs.
Figure 1.
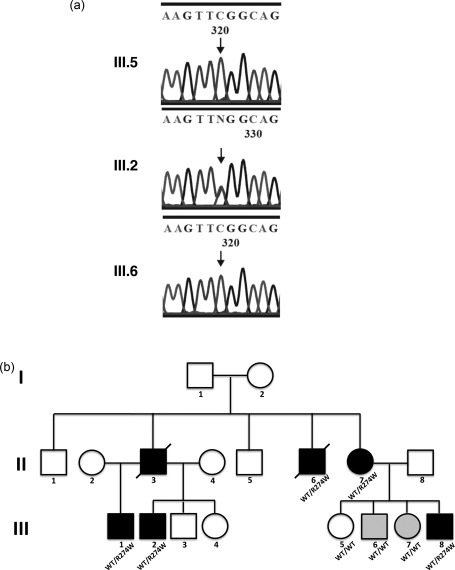
STAT1 mutation in chronic mucocutaneous candidiasis (CMC) family. Whole exome sequencing (WES) identified a CMC‐associated variant in STAT1 (c.C820T) in four of the affected individuals (a). Sanger sequencing confirmed the presence of this variant (and in one additional individual not exome sequenced) with plots shown for an unaffected (III.5), and an affected individual (III.2) and one family member originally misclassified as ‘affected’ (III.6) but found to have wild‐type STAT1, leading to a review of their diagnosis (b). Family pedigree with STAT1 genotype shown; light grey shading denotes individuals originally misclassified as ‘affected’.
Ethical approval for genetic testing and Th17 assays using patient samples was gained under the Genetic and Functional Characterisation of Patients with Primary Immune Deficiencies, Infectious and Inflammatory Conditions Study (REC 12/SC/0044); approval for healthy donor samples used in the Th17 assays was gained from the National Research Ethics Service (REC 14/SC/0025).
Whole exome sequencing
Genomic DNA was extracted from whole blood using QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions and quantified by fluorescence using the Quant‐iT PicoGreen kit (Invitrogen, Carlsbad, CA, USA). The whole exome was captured using TruSeq technology and sequencing was performed on 5 µg DNA on the Illumina HiSeq2000 (Core Genomics; Wellcome Trust Centre for Human Genetics, Oxford, UK). Briefly, the DNA was fragmented, end‐repaired, A‐tailed and adapter‐ligated before size selection and amplification. The libraries were multiplexed, captured and quality checked (QC'd) before paired‐end sequencing over one lane of a HiSeq flow cell. The data were aligned to the reference genome. Variants detected in the exon capture target region were filtered to remove synonymous variants and those present in the 1000 Genomes Project (PMID 23128226) at a frequency greater than 5%.
Sanger sequencing
Primers were designed using Primer3 software (http://bioinfo.ut.ee/primer3/) to amplify the region containing the identified STAT1 variant (forward: CCCTCCACAAACTCTCTTGC, reverse: AGCCTGGGTGATAGGTGAGA). Twenty‐five ng DNA was amplified using a 25‐µl polymerase chain reaction (PCR) reaction containing 2·5 µl 10× buffer, 1·5 mM MgCl2, 0·2 mM dNTP, 250 nM primer (each direction) and 0·5 U platinum Taq DNA polymerase (Invitrogen). Cycling conditions were an initial denaturation step at 94ºC for 2 min followed by 30 cycles of 94ºC for 30 s, 56ºC for 30 s and 72ºC for 40 s. Four µl of the PCR product was cleaned using 0·1 µl exonuclease I (ExoI) and 0·1 µl calf intestinal phosphatase (CIP) with 0·4 µl New England Biolabs (NEB) buffer 3 in an 8‐µl reaction. The reaction was incubated at 37ºC for 30 min before inactivation at 95ºC for 5 min; 3·5 µl cleaned‐up template DNA was sequenced using Big Dye Terminator 3·1 according to the manufacturer's instructions (Applied Biosystems, Foster City, CA, USA).
In‐vitro assay for Th17 cells using intracellular cytokine staining for IL‐17A
PBMCs were isolated from whole blood using lymphoprep (Stemcell Technologies, Cambridge, UK) density centrifugation, diluted in RPMI‐1640 (Sigma‐Aldrich, St Louis, MO, USA) + 10% fetal bovine serum (Sigma‐Aldrich) to achieve a final concentration of 1 × 106/ml, and plated in a 24‐well plate (1 ml/well). Forty ng/ml PMA and 10−5 M ionomycin were used for stimulation, with the addition of 1 µg/ml Golgiplug (BD Biosciences, Oxford, UK) after 1 h. Cells were incubated for 6 h at 37°C and washed in phosphate‐buffered saline (PBS) before staining for extracellular markers with anti‐CD4‐Qdot605 (Invitrogen) and anti‐CD8‐allophycocyanin (APC)‐H7 (BD Biosciences) for 20 min on ice. Fixation and permeabilization was carried out according to the manufacturer's instructions (eBioscience IC fixation buffer, 10xPerm buffer; eBioscience, San Diego, CA, USA), and cells stained with anti‐IL‐17A‐AF488 (clone: eBio64DEC17; eBioscience) for 1 h before being analysed using flow cytometry (LSRII). The percentage of IL‐17A‐positive cells was measured within the CD4+ population.
Ex‐vivo assay for Th17 cells using surface staining for CCR6, CXCR3, CCR4 and CD161
One hundred µl of EDTA whole blood was stained with the following monoclonal antibodies: anti‐CD3‐peridinin chlorophyll (PerCP), anti‐CD4‐APC‐H7, anti‐CCR6‐Alexa Fluor 647, anti‐CXCR3‐phycoerythrin‐cyanin 7 (PE‐Cy7), anti‐CCR4‐BV421 and anti‐CD161‐fluorescein isothiocyanate (FITC) (BD Biosciences). Samples were incubated for 15 min at room temperature in the dark, after which 1 ml of FACSLyse solution (BD Biosciences) was added for 5 min. Samples were subsequently washed with PBS (Oxoid, Basingstoke, UK), fixed with 1% paraformaldehyde and analysed by flow cytometry (BD FACSCanto II) using FACSDiva software (BD Biosciences). Samples were gated initially to acquire 30 000 CD3+ T cell events, from which CD3+CD4+ helper T cells were selected, with subsequent analysis of CCR6, CXCR3, CCR4 and CD161 expression. All populations were expressed as a percentage of CD3+CD4+ T cells. T, B and natural killer (NK) cell enumeration was performed using TrucountTM tubes (BD Biosciences), from which absolute counts for CCR6+CXCR3–CCR4+CD161+ T helper cells were calculated.
Statistics
Statistical analysis was performed using Microsoft Excel and GraphPad prism version 5 software (GraphPad PRISM, San Diego, CA, USA). Normal ranges were established for percentage and absolute counts of CCR6+CXCR3–CCR4+CD161+CD4+ T cells by determining the 5th and 95th percentiles of the healthy control cohort. Receiver operating characteristic (ROC) curve analysis was used to assess analytical sensitivity and specificity of the ex‐vivo assay. In order to assess the correlation between the results obtained from the in‐vitro and ex‐vivo assays a Pearson's correlation coefficient was calculated using paired samples; an r 2 value of > 0·50 was considered to represent a positive correlation. For all analyses, a P‐value of < 0·05 was considered statistically significant.
Results
Whole exome sequencing reveals a gain‐of‐function mutation in STAT1
We identified a family in which CMC segregates as an autosomal‐dominant trait; their clinical and immunological phenotypes are summarized in Table 1. It is notable that two patients developed oropharyngeal carcinoma and that all the patients maintained on long‐term azole prophylaxis grew progressively resistant strains of C. albicans.
Whole exome sequencing (WES) data were generated for seven family members (II.7, III.1, III.2, III.5, III.6, III.7 and III.8). The QC'd data were screened for reported pathogenic CMC associated variants. This identified a missense variant in STAT1 (c.C820T), leading to an arginine to tryptophan substitution in the coiled‐coil domain (p.R274W) in four affected individuals (Fig. 1a). Sanger sequencing (Fig. 1b) was used to confirm the mutation in these individuals and in one additional affected family member (II.6) who had not been exome sequenced. This mutation has been described in the context of autosomal‐dominant CMC to confer a gain of STAT‐1 function and hinder the development and function of Th17 cells 16, 17.
Two family members (III.6 and III.7), wild‐type for STAT1, were originally misdiagnosed with CMC in early infancy on the basis of mild, discrete episodes of oral candidiasis in the context of a family history of CMC (Fig. 1a). However, in contrast to other affected family members, they did not have any further infections after starting azole prophylaxis in infancy, and have subsequently been found to have normal numbers of Th17 cells (Table 1). Importantly, the STAT1 variant that was identified ultimately to underlie CMC in this kindred would not have been identified if these two individuals were assigned as ‘affected’ in the analysis.
Characteristics of patients with CMC used in Th17 assays
Demographic, clinical, immunological and genetic data related to the patients with CMC whose samples were used for the Th17 assays detailed below are shown in Table 2.
Patients with CMC have significantly reduced IL‐17A producing CD4+ T cells compared to healthy controls
IL‐17A+CD4+ T cells were measured using flow cytometry following in‐vitro stimulation with PMA and ionomycin in 13 healthy controls, one STAT1 wild‐type individual from the kindred with CMC due to STAT‐1 GOF (III.7 in Fig. 1a and Table 1) and five patients with CMC. The latter consisted of three patients from the STAT‐1 GOF kindred, one patient with a sporadic STAT1 mutation (c.G629A; p.R210K), that has been found in several unrelated patients presenting with CMC (unpublished data), and one patient with HIGE due to a dominant negative mutation in STAT3 (Table 2). Consistent with previously published data, we found that the percentage of IL‐17A‐producing cells within the CD4+ population was reduced significantly (P = 0·0002) in the CMC patients [median = 0·21%; interquartile range (IQR) = 0·15–0·30%] compared to healthy controls (median = 0·83%; IQR = 0·73–1·16%) (Fig. 2). The STAT1 wild‐type individual (III.7) from the kindred with a GOF mutation in STAT1 was found to have normal numbers of IL‐17A‐producing T cells (2·04% of CD4+ T cells).
Figure 2.
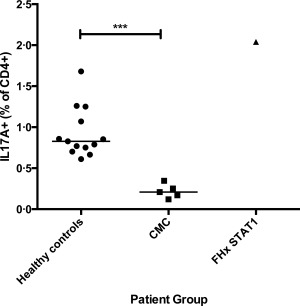
Graph showing significant reduction in the percentage of interleukin (IL)−17A‐producing CD4+ T cells in patients with chronic mucocutaneous candidiasis (CMC) compared to healthy controls after in‐vitro stimulation of peripheral blood mononuclear cells (PBMCs) for 6 h with phorbol myristate acetate (PMA) and ionomycin (P = 0·0002). Patients with CMC included four individuals with STAT1 mutations and one with hyper‐immunoglobulin (Ig)E syndrome (HIGE) syndrome due to signal transducer and activator of transcription‐3 (STAT‐3) deficiency. Also included was a STAT1 wild‐type individual (III.7) with a family history of CMC due to STAT‐1 gain‐of‐function (GOF) (FHx STAT1).
Patients with CMC have significantly reduced numbers of CCR6+CXCR3–CCR4+CD161+ helper T cells compared to healthy controls
IL‐17‐producing T cells have been shown to express CCR6, CCR4 and CD161 and not CXCR3 21, 22, 23, 24. We sought to develop an ex‐vivo assay using these cell surface markers (Fig. 3) and to assess whether this could be used to discriminate between patients with known defects in Th17 immunity underlying CMC from healthy controls.
Figure 3.
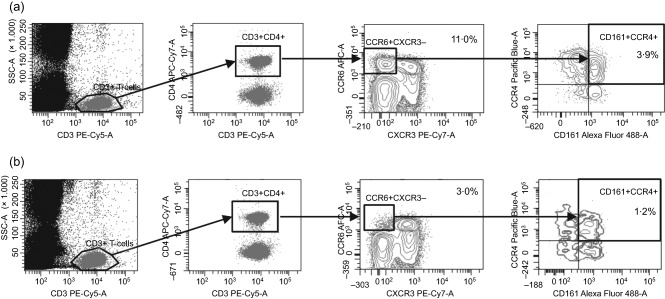
Fluorescence activated cell sorter (FACs) plots depicting gating strategy employed for the detection of CCR6+CXCR3–CCR4+CD161+ helper T cells. Cells were gated initially to acquire 30 000 CD3+ events. CD4+ T cells were then selected based on co‐expression of CD3 and CD4. Cells that were positive for CCR6 but negative for CXCR3 were then gated, and finally cells that were double‐positive for CCR4 and CD161 were selected from the CD3+CD4+CCR6–CXCR3– T cell population. Sample plots are shown for (a) one healthy control and (b) one patient with CMC due to signal transducer and activator of transcription‐1 (STAT‐1) gain‐of‐function (GOF). The relative counts depicted denote the percentage of CCR6+CXCR3–CCR4+CD161+ cells within the CD4+ T cell population.
We tested whole blood samples from 19 healthy controls and seven patients with CMC, consisting of three patients from the STAT‐1 GOF kindred, one with a sporadic STAT1 variant, one with IL‐12RB1 deficiency, one with HIGE syndrome due to STAT‐3 deficiency and one patient with APS1 and autoantibodies against IL‐17A (Table 2). We also tested one STAT1 wild‐type individual (III.7) from the kindred with CMC due to STAT‐1 GOF (Fig. 4).
Figure 4.
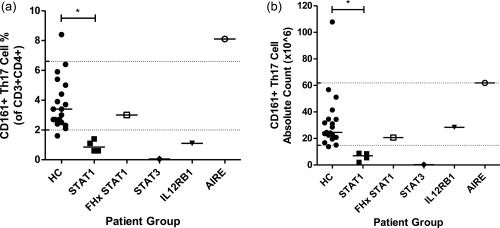
Graphs showing reduction in CCR6+CXCR3–CCR4+CD161+ T helper cells in patients with chronic mucocutaneous candidiasis (CMC) compared to healthy controls. (a) Graph showing % of CCR6+CXCR3–CCR4+CD161+ cells within the CD4+ T cell population in healthy controls versus individual patient groups. (b) Graph showing absolute counts (×106/l) of CCR6+CXCR3–CCR4+CD161+ CD4+ T cells in healthy controls versus individual patient groups. HC = healthy controls; STAT1 = CMC due to STAT1 mutation; FHx STAT1 = STAT1 wild‐type individual (III.7) from the kindred CMC due to signal transducer and activator of transcription‐1 (STAT‐1) gain‐of‐function (GOF); STAT3 = hyper‐immunoglobulin (Ig)E syndrome due to STAT‐3 deficiency; IL 12RB1 = IL‐12RB1 deficiency; AIRE = APS1 due to AIRE deficiency; *P < 0·05.
There were no statistically significant differences between healthy control and patient CD4+ T cell counts (data not shown). However, it must be noted that the absolute CD4+ T cell count for the patient with IL‐12RB1 deficiency was higher than all other individuals tested. This patient was 15 months old at the time of venepuncture and her CD4+ T cell count was in keeping with the age‐specific normal range. As such, in order to interpret fully the absolute counts for CCR6+CXCR3–CCR4+CD161+ T helper cells obtained for this patient, the establishment of a paediatric normal range would perhaps be more appropriate, but was beyond the scope of this study, and this patient's results have been excluded from further analysis of absolute counts.
Within the healthy controls, CCR6+CXCR3–CCR4+CD161+ T cells measured 24·6 × 106/l (IQR = 20·9–34·5 × 106/l) and made up 3·4% (IQR = 2·5–5·0%) of CD4+ T cells; normal ranges were calculated using the 5th and 95th percentiles for both percentage (normal range = 2·0–6·6%) and absolute counts (normal range = 14·9–61·9 ×106/l) (Fig. 4a). The patient with APS1 and CMC secondary to autoantibodies against IL‐17 demonstrated increased relative amounts and, to a lesser extent, absolute counts of CCR6+CXCR3–CCR4+CD161+ T helper cells, compared to both healthy controls and the CMC cohort. Excluding this patient, within the rest of the CMC cohort CCR6+CXCR3–CCR4+CD161+ T cells measured 7·0 × 106/l (IQR = 1·6–13·7 × 106/l) and made up 0·8% (IQR = 0·4–1·2%) of CD4+ T cells (Fig. 4b).
A significant decrease was observed between the healthy controls and patients with CMC, excluding the patient with APS1, for both percentage and absolute counts of CCR6+CXCR3–CCR4+CD161+ T helper cells (P = 0·0003 and 0·0008, respectively; data not shown). When analysed further by patient subgroup, a significant reduction in both percentage and absolute counts was observed in the patients with CMC due to STAT‐1 GOF (P < 0·05) but not in the STAT1 wild‐type family member (III.7), and an appreciable reduction was also observed in the patient with HIGE syndrome due to STAT‐3 deficiency (Fig. 4).
Numbers of CCR6+CXCR3–CCR4+CD161+ helper T cells correlate with numbers of IL‐17A‐producing Th17 cells
We next sought to assess the relationship between the number of CCR6+CXCR3–CCR4+CD161+ helper T cells measured ex‐vivo and the numbers of IL‐17A‐producing Th17 cells measured using the in‐vitro assay. Paired samples from 15 healthy controls and five patients with CMC were run on each assay (Fig. 5). The latter group consisted of four patients with STAT‐1 GOF and one patient with HIGE due to STAT‐3 deficiency. The results obtained using the ex‐vivo assay correlated positively with those obtained using the in‐vitro technique (r 2 = 0·5706, P = 0·0001), suggesting that these cell surface markers could be used as a surrogate marker for IL‐17‐producing T cells.
Figure 5.
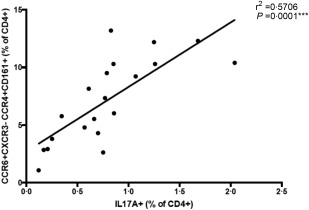
Correlation plot showing positive correlation (r 2 = 0·5706, P = 0·0001) between the percentage of CCR6+CXCR3–CCR4+CD161+ and interleukin (IL)‐17A producing CD4+ T cells measured using paired samples in healthy controls and patients with chronic mucocutaneous candidiasis (CMC).
Examination of CCR6+CXCR3– helper T cells is sufficient to identify patients with CMC
In order to optimize the assay for use in the diagnostic setting we investigated whether the removal of CD161, CCR4 and CXCR3 from the antibody staining panel would affect the performance of the assay. Based on the rationale that the absolute counts for the patient with IL‐12RB1 deficiency should be analysed using a paediatric normal range, this patient was excluded from our calculations for absolute count analytical sensitivity and specificity. The patient with APS1 was also excluded as, in contrast to the other patients with CMC analysed, they demonstrated increased relative and absolute counts of CCR6+CXCR3–CCR4+CD161+ T helper cells. Optimal cut‐offs were selected in order to allow maximum discrimination between healthy controls and patients with CMC. Using ROC curve analysis we were able to demonstrate 100% sensitivity and specificity in the cohort tested, using CCR6, CXCR3, CCR4 and CD161 expression to define Th17 percentage and absolute counts (Table 3). Assay performance was not affected by the removal of CD161 or CCR4 from the antibody panel, but was impaired by the further removal of CXCR3 (Table 3). Based on these data we conclude that the enumeration of CCR6+CXCR3– T helper cells is sufficient to identify patients with CMC due to Th17 deficiency.
Table 3.
Performance characteristics of ex‐vivo assay for T helper type 17 (Th17) cells based on receiver operating characteristic (ROC) curve analysis for (a) percentage (%) and (b) absolute counts (×106/l) using full or condensed antibody panels.
| (a) | Optimal cut‐off | Sensitivity% | Specificity% | AUC |
|---|---|---|---|---|
| CCR6+CXCR3–CCR4+CD161+ | <1·5 | 100·0 | 100·0 | 1·000 |
| CCR6+CXCR3–CCR4+ | <3·6 | 100·0 | 100·0 | 1·000 |
| **CCR6+CXCR3–** | <5·0 | 100·0 | 100·0 | 1·000 |
| CCR6+ | <15·0 | 83·3 | 100·0 | 0·900 |
| (b) | Optimal cut‐off | Sensitivity% | Specificity% | AUC |
|---|---|---|---|---|
| CCR6+CXCR3–CCR4+CD161+ | <11·3 | 100·0 | 100·0 | 1·000 |
| CCR6+CXCR3–CCR4+ | <28·7 | 100·0 | 100·0 | 1·000 |
| **CCR6+CXCR3–** | <32·9 | 100·0 | 100·0 | 1·000 |
| CCR6+ | <122·0 | 100·0 | 85·0 | 0·970 |
AUC = area under the curve.
Discussion
WES has become an important tool in the identification of molecular defects underlying PIDs. However, analysis of sequencing data relies on robust clinical and immunological phenotyping in order to assign cases as ‘affected’ or ‘unaffected’. The kindred presented highlights this potential pitfall as the mutation in STAT1 that was ultimately identified was not present in all the individuals originally assigned as ‘affected’. The absence of this known variant in individuals III.6 and III.7, alongside functional data demonstrating normal amounts of IL‐17A‐producing and CCR6+CXCR3–CCR4+CD161+ T helper cells, has led to a revision of their disease status and withdrawal of long‐term anti‐fungal prophylaxis to no adverse effect.
The clinical data presented highlight the risks of oropharyngeal carcinoma and the development of pan‐azole resistance in those receiving long‐term prophylaxis. Oral and oesophageal squamous cell carcinomas have been described in CMC due to STAT‐1 GOF and APS1 16, 17, 25. The relative contributions of the underlying molecular defect plus other genetic modifiers, chronic inflammation, the strain of Candida and co‐factors such as smoking and alcohol remain unclear. However, a case can be made for regular surveillance for these types of cancer. Whilst the propensity for the development of carcinoma at the site of CMC could be seen as an indication for long‐term anti‐fungal prophylaxis, the development of persistent resistance to fluconazole has been noted previously in the context of CMC due to APS1 26, 27. Risk factors include treatment with more than six courses of fluconazole per year and long‐term low‐dose treatment 26. It has therefore been suggested that patients should be treated with short pulses of therapeutic dose anti‐fungals for acute flares in order to limit the development of anti‐fungal resistance 27 and sensitivities of colonizing strains should be monitored 26.
We have presented laboratory data showing evidence of Th17 cell dysregulation in patients with CMC using intracellular cytokine staining for IL‐17A and an ex‐vivo technique measuring cell surface expression of CCR6, CXCR3, CCR4 and CD161. Indeed, intracellular cytokine staining for IL‐17A, following in‐vitro stimulation with PMA and ionomycin, has been used previously to identify Th17 cells in patients with CMC 28. Whilst the data that we have presented show that this is an effective means of detecting Th17 deficiency in patients with CMC due to STAT‐1 GOF and HIGE syndrome, this method of Th17 cell detection is long and labour‐intensive, and therefore not highly conducive to use in a routine diagnostic immunology laboratory. As such, an ex‐vivo staining method would provide a more readily accessible and rapid means of identifying and measuring Th17 cells. Previous studies have demonstrated the use of CCR6, CXCR3, CCR4 and CD161 in the identification of IL‐17‐producing T helper cells 21, 23, 24. However, to our knowledge, no studies have investigated the combined use of all these cell surface markers in one ex‐vivo assay.
We have shown a positive correlation between the percentage of IL‐17A‐producing CD4+ T cells detected after in‐vitro stimulation of PBMCs followed by intracellular cytokine staining, and the percentage of CCR6+CXCR3–CCR4+CD161+ T helper cells detected after ex‐vivo cell surface staining. From these results we conclude that the enumeration of CCR6+CXCR3–CCR4+CD161+ T helper cells in unstimulated whole blood could be used as a surrogate measure of IL‐17‐producing helper T cells. The use of percentage rather than absolute counts may obviate the need to produce age‐specific normal ranges.
Further analysis has shown that the enumeration of CCR6+CXCR3– T helper cells alone, irrespective of CCR4 and CD161 expression, is in fact sufficient to identify patients with CMC due to molecular defects known to impair IL‐17 immunity. Interestingly, it is only the subsequent removal of CXCR3 which adversely affects assay performance. In line with our findings, a previous study has shown that whilst patients with CMC have significantly fewer CCR6+IL‐17A+ T cells than healthy controls after in‐vitro stimulation with PMA and ionomycin, there were no significant differences in the numbers of CCR6 or CCR4 single‐positive or CCR6 CCR4 double‐positive T cells 28. Alongside the reduction in CCR6+CXCR3– T helper cells, one of the STAT‐1 GOF patients in our study showed a simultaneous increase in CCR6+CXCR3+ T helper cells (data not shown); this could be responsible for the observed reduction in assay performance when using CCR6 alone, and may represent a compensatory mechanism for their inability to produce CCR6+CXCR3– Th17 cells.
We have also been able to demonstrate that the ex‐vivo assay is not only capable of identifying patients with CMC due to STAT‐1 GOF, but also patients with distinct molecular aetiologies of CMC leading to impaired IL‐17 immunity, such as STAT‐3 and IL‐12RB1 deficiency. We therefore believe that this assay could be used as a screening assay for the detection of Th17 deficiency in patients presenting with CMC. Furthermore, the assay may also be used to identify patients with CMC due to the presence of neutralizing autoantibodies against IL‐17, as evidenced by the detection of increased amounts of CCR6+CXCR3–CCR4+CD161+ T helper cells in the patient with APS1. This finding could be explained by a peripheral expansion of Th17 cells in order to overcome the effects of neutralizing autoantibodies and is consistent with a previous study showing that PBMCs from patients with APS1 produce more IL‐17, and that patients have increased numbers of IL‐17A‐producing T helper cells compared with healthy controls after stimulation with Candida 29. Given that this study is limited by low patient numbers and a predominance of one particular molecular defect from a single kindred, the scope of the assay's utility would, of course, need to be verified using a larger cohort of patients with distinct molecular aetiologies of primary CMC, and its predictive ability would be best assessed in a prospective study.
In conclusion, we believe that ex‐vivo staining of whole blood for CCR6+CXCR3– T helper cells provides a means of rapidly identifying patients with CMC due to Th17 deficiency. This could be used to guide targeted genetic investigation, thereby facilitating a more tailored and timely approach to patient diagnosis, which is increasingly important with the potential advent of new disease‐specific treatments such as ruxolitinib for STAT1 GOF mutations 30.
Disclosure
The authors have no disclosures to declare.
Acknowledgements
This report is independent research arising from a Healthcare Scientist Research Fellowship supported by the National Institute for Health Research. The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute of Health Research or the Department of Health. We would also like to acknowledge the NIHR Oxford Biomedical Research Centre, the Wellcome Trust Centre for Human Genetics (Oxford, UK) including High‐Throughput Genomics Group, and the European Research Council under the European Union's Seventh Framework Programme; and Mr Adam Burns and Dr Anna Schuh at the Oxford BRC Haemato‐Molecular Diagnostic Laboratory for help with sequencing for II.6.
References
- 1. McDonald DR. TH17 deficiency in human disease. J Allergy Clin Immunol 2012; 129:1429–35; quiz 36–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lilic D. Unravelling fungal immunity through primary immune deficiencies. Curr Opin Microbiol 2012; 15:420–6. [DOI] [PubMed] [Google Scholar]
- 3. Engelhardt KR, Grimbacher B. Mendelian traits causing susceptibility to mucocutaneous fungal infections in human subjects. J Allergy Clin Immunol 2012; 129:294–305; quiz 6–7. [DOI] [PubMed] [Google Scholar]
- 4. Ma CS, Chew GY, Simpson N et al Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med 2008; 205:1551–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Milner JD, Brenchley JM, Laurence A et al Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper‐IgE syndrome. Nature 2008; 452:773–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Holland SM, DeLeo FR, Elloumi HZ et al STAT3 mutations in the hyper‐IgE syndrome. N Engl J Med 2007; 357:1608–19. [DOI] [PubMed] [Google Scholar]
- 7. Minegishi Y, Saito M, Tsuchiya S et al Dominant‐negative mutations in the DNA‐binding domain of STAT3 cause hyper‐IgE syndrome. Nature 2007; 448:1058–62. [DOI] [PubMed] [Google Scholar]
- 8. Al Khatib S, Keles S, Garcia‐Lloret M et al Defects along the T(H)17 differentiation pathway underlie genetically distinct forms of the hyper IgE syndrome. J Allergy Clin Immunol 2009; 124:342–8, 8.e1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang Q, Davis JC, Dove CG, Su HC. Genetic, clinical, and laboratory markers for DOCK8 immunodeficiency syndrome. Dis Markers 2010; 29:131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Q, Davis JC, Lamborn IT et al Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med 2009; 361:2046–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Engelhardt KR, McGhee S, Winkler S et al Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal‐recessive form of hyper‐IgE syndrome. J Allergy Clin Immunol 2009; 124:1289–302.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Puel A, Cypowyj S, Bustamante J et al Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin‐17 immunity. Science 2011; 332:65–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Beaucoudrey L, Puel A, Filipe‐Santos O et al Mutations in STAT3 and IL12RB1 impair the development of human IL‐17‐producing T cells. J Exp Med 2008; 205:1543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Puel A, Döffinger R, Natividad A et al Autoantibodies against IL‐17A, IL‐17F, and IL‐22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med 2010; 207:291–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kisand K, Bøe Wolff AS, Podkrajsek KT et al Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17‐associated cytokines. J Exp Med 2010; 207:299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu L, Okada S, Kong XF et al Gain‐of‐function human STAT1 mutations impair IL‐17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 2011; 208:1635–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van de Veerdonk FL, Plantinga TS, Hoischen A et al STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med 2011; 365:54–61. [DOI] [PubMed] [Google Scholar]
- 18. Yamazaki Y, Yamada M, Kawai T et al Two novel gain‐of‐function mutations of STAT1 responsible for chronic mucocutaneous candidiasis disease: impaired production of IL‐17A and IL‐22, and the presence of anti‐IL‐17F autoantibody. J Immunol 2014; 193:4880–7. [DOI] [PubMed] [Google Scholar]
- 19. Smeekens SP, Plantinga TS, van de Veerdonk FL et al STAT1 hyperphosphorylation and defective IL12R/IL23R signaling underlie defective immunity in autosomal dominant chronic mucocutaneous candidiasis. PLOS ONE 2011; 6:e29248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mizoguchi Y, Tsumura M, Okada S et al Simple diagnosis of STAT1 gain‐of‐function alleles in patients with chronic mucocutaneous candidiasis. J Leukoc Biol 2014; 95:667–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Acosta‐Rodriguez EV, Rivino L, Geginat J et al Surface phenotype and antigenic specificity of human interleukin 17‐producing T helper memory cells. Nat Immunol 2007; 8:639–46. [DOI] [PubMed] [Google Scholar]
- 22. Annunziato F, Cosmi L, Santarlasci V et al Phenotypic and functional features of human Th17 cells. J Exp Med 2007; 204:1849–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cosmi L, De Palma R, Santarlasci V et al Human interleukin 17‐producing cells originate from a CD161+CD4+ T cell precursor. J Exp Med 2008; 205:1903–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Crome SQ, Clive B, Wang AY et al Inflammatory effects of ex vivo human Th17 cells are suppressed by regulatory T cells. J Immunol 2010; 185:3199–208. [DOI] [PubMed] [Google Scholar]
- 25. Rautemaa R, Hietanen J, Niissalo S, Pirinen S, Perheentupa J. Oral and oesophageal squamous cell carcinoma–a complication or component of autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APECED, APS‐I). Oral Oncol 2007; 43:607–13. [DOI] [PubMed] [Google Scholar]
- 26. Rautemaa R, Richardson M, Pfaller M, Koukila‐Kähkölä P, Perheentupa J, Saxén H. Decreased susceptibility of Candida albicans to azole antifungals: a complication of long‐term treatment in autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APECED) patients. J Antimicrob Chemother 2007; 60:889–92. [DOI] [PubMed] [Google Scholar]
- 27. Rautemaa R, Richardson M, Pfaller MA, Perheentupa J, Saxén H. Activity of amphotericin B, anidulafungin, caspofungin, micafungin, posaconazole, and voriconazole against Candida albicans with decreased susceptibility to fluconazole from APECED patients on long‐term azole treatment of chronic mucocutaneous candidiasis. Diagn Microbiol Infect Dis 2008; 62:182–5. [DOI] [PubMed] [Google Scholar]
- 28. Eyerich K, Foerster S, Rombold S et al Patients with chronic mucocutaneous candidiasis exhibit reduced production of Th17‐associated cytokines IL‐17 and IL‐22. J Invest Dermatol 2008; 128:2640–5. [DOI] [PubMed] [Google Scholar]
- 29. Ng WF, von Delwig A, Carmichael AJ et al Impaired T(H)17 responses in patients with chronic mucocutaneous candidiasis with and without autoimmune polyendocrinopathy‐candidiasis‐ectodermal dystrophy. J Allergy Clin Immunol 2010; 126:1006–15, 15.e1‐4. [DOI] [PubMed] [Google Scholar]
- 30. Higgins E, Al Shehri T, McAleer MA et al Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain‐of‐function signal transducer and activator of transcription 1 (STAT1) mutation. J Allergy Clin Immunol 2015; 135:551–3. [DOI] [PubMed] [Google Scholar]
- 31. Pearce SH, Cheetham T, Imrie H et al A common and recurrent 13‐bp deletion in the autoimmune regulator gene in British kindreds with autoimmune polyendocrinopathy type 1. Am J Hum Genet 1998; 63:1675–84. [DOI] [PMC free article] [PubMed] [Google Scholar]