

Summary
This open‐label multi‐centre study evaluated Gammaplex® 5%, a human intravenous immunoglobulin (IVIG) 5% liquid, in 25 children and adolescent patients (aged 3–16 years) with primary immunodeficiency diseases (PIDs). Subjects received Gammaplex 5% (at doses of 300–800 mg/kg/infusion) for 12 months, with a 3‐month follow‐up. The primary efficacy end‐point was the incidence of serious acute bacterial infections (SABIs) during the 12‐month treatment period. Secondary objectives assessed safety and tolerability. Nineteen males and six females were treated using the same infusion schedule as their prior IVIG treatment (14 and 11 subjects on 21‐ and 28‐day dosing schedules, respectively). Two SABIs of pneumonia were reported, resulting in an annual SABI event rate of 0·09 [upper one‐sided 99% confidence interval (CI) = 0·36]. Twenty‐one subjects (84%) experienced ≥ 1 infection during the study, with a median infective episode per subject/year of 3·08 (range = 0–10·4). Sixteen subjects (64%) missed ≥ 1 day of nursery or school because of infection or other illness. All trough immunoglobulin G levels exceeded 7·00 g/l after 15 weeks (mean = 9·69 g/l; range = 7·04–15·35 g/l). Product‐related adverse events occurred in 14 subjects (56%); none were serious. Of 368 total infusions, 97 (26%) were associated temporally with an adverse event (≤ 72 h after infusion), regardless of causality. Laboratory test results and adverse‐reaction data showed no evidence of product‐related haemolysis or thromboembolic events. These data demonstrate that Gammaplex 5% is effective in preventing SABIs and well tolerated in children and adolescents with PID.
Keywords: Gammaplex® 5%, immunodeficiency disease, intravenous immunoglobulin
Introduction
Primary immunodeficiency diseases (PIDs) are a heterogeneous group of disorders characterized by an intrinsic defect in the immune system. Many are characterized by hypogammaglobulinaemia and/or specific defective antibody production and increased susceptibility to infection 1. On average, untreated patients with PID experience approximately ≥ 4 serious acute bacterial infections (SABIs) per year 2, and the majority of primary immunodeficiencies are undiagnosed or diagnosed only after the individual has experienced recurrent infections 3. A US survey of > 1300 patients with PID found that only 27% of patients were diagnosed initially with PID by 6 years of age, and only 40% of patients were diagnosed by 18 years of age; furthermore, 49% of patients reported permanent functional impairment prior to diagnosis, and the number of permanent functional impairments was correlated directly with length of delayed diagnosis 4. Therefore, early diagnosis and effective immunoglobulin G (IgG) treatment in children and adolescents is particularly important to reduce the rate of infection and prevent sequelae 1.
The standard of care for PID characterized by hypogammaglobulinaemia is replacement therapy with intravenous or subcutaneous immunoglobulin (IVIG and SCIG, respectively) 5, which decreases the frequency of serious infections and hospitalizations and improves quality of life among patients with PID 6, 7. Thus, IgG treatment is given to patients with PID at regular intervals to restore serum IgG to normal levels and minimize infections 3, 5.
Gammaplex® 5% [intravenous immune globulin (human) 5% liquid; Bio Products Laboratory Ltd, Elstree, UK], a highly purified, unmodified human IgG product manufactured using only plasma from healthy US donors, is approved for the treatment of PID and idiopathic thrombocytopenic purpura (ITP) 8, 9. The manufacturing process includes three distinct virus inactivation/reduction steps (solvent/detergent, 20 nm virus filtration and terminal low pH incubation).
Although IgG replacement therapy has been administered to patients with PID for decades 5, published data examining the efficacy and tolerability of IVIG in paediatric PID subjects are limited. Most licensed IVIGs have included a small number of children in their pivotal regulatory studies, but their results are rarely analysed separately from the adult population 10. The efficacy and safety of Gammaplex 5% have been demonstrated previously in a 12‐month, open‐label, multi‐centre, Phase 3 study of 50 patients with PID; however, only seven children or adolescents (aged 9–17 years) were included in this study 11. Here, we report the results of a prospective, non‐comparative, open‐label clinical trial to evaluate the efficacy, safety and tolerability of Gammaplex 5% in 25 paediatric subjects (aged 2–16 years) with PID.
Materials and methods
Study design
This was a multi‐centre, open‐label, non‐randomized Phase 4 study of Gammaplex 5%. Subjects were treated for 12 months, with follow‐up visits conducted at 10–14 days and 3 months after the last infusion. This non‐comparative study was designed in line with applicable regulatory guidelines 2, 12, and was approved by an institutional review board/ethics committee at each of nine study centres in the United States (seven), Chile (one) and Israel (one). Informed consent was obtained from each subject's parent/guardian, and assent was obtained from each subject where appropriate.
Subject selection
Included subjects had PID and hypogammaglobulinaemia and/or antibody deficiency, were aged 2–16 years inclusive, and weighed ≥ 10 kg. All subjects were required to be receiving an IVIG treatment prior to study entry, with doses required to be 300–800 mg/kg per infusion, unchanged by ± 50% of the mean dose for ≥ 3 months prior to study entry, and administered every 21 or 28 days. At study entry, trough serum IgG levels were required to be ≥ 6·00 g/l and to be ≥ 3·00 g/l higher than pre‐IVIG therapy levels.
Subjects were excluded for the following reasons: they were receiving long‐term, high‐dose steroids or immunomodulatory/immunosuppressive drugs; they had selective IgA deficiency or a history of antibodies to IgA; they had secondary immunodeficiency; or they had a history of severe anaphylaxis to blood products, liver or renal disease, or deep vein thrombosis or thrombotic complications of IVIG therapy.
Treatment
Trough IgG levels were recorded before starting Gammaplex 5% and prior to most infusions during the study. Gammaplex 5% was administered intravenously at 300–800 mg/kg per infusion every 21 or 28 days (± 4 days) following the subject's previous IVIG schedule. Subjects on the 21‐ and 28‐day infusion schedules received up to 17 and 13 infusions of Gammaplex 5%, respectively. Infusions began at 0·01 ml/kg/min (0·5 mg/kg/min) for the first 15 min. If tolerated, the infusion rate was increased every 15 min to 0·02 ml/kg/min (1·0 mg/kg/min), 0·04 ml/kg/min (2·0 mg/kg/min), 0·06 ml/kg/min (3·0 mg/kg/min) and 0·08 ml/kg/min (4·0 mg/kg/min) thereafter. If an adverse event (AE) of moderate or severe intensity occurred, the infusion rate was reduced until symptoms subsided, then resumed at a tolerable rate.
Efficacy, safety and pharmacokinetic (PK) outcomes
The primary outcome was the number of SABIs per subject per year. SABIs were defined as bacterial pneumonia, bacteraemia or sepsis, osteomyelitis/septic arthritis, visceral abscess and bacterial meningitis 2. Secondary outcomes included days off from school/nursery, acute care visits or hospitalizations, non‐serious infections and antibiotic use. Safety and tolerability were assessed using AEs, vital signs and clinical laboratory tests.
PK parameters were also assessed. Trough levels of total IgG and IgG subclasses (IgG1, IgG2, IgG3 and IgG4) were determined at every infusion and at the first follow‐up visit using turbidimetry. Trough levels of IgG against specific antigens (Cytomegalovirus, Haemophilus influenzae B and Streptococcus pneumoniae serotypes 4, 6B, 9V, 14, 18C, 19F and 23F) were determined at screening; before infusions 7, 8, 9, 10 and 12; and, for subjects on a 21‐day schedule, before infusions 14 and 16. H. influenzae B and S. pneumoniae antibodies were determined by enzyme immunoassays and microsphere photometry, respectively. IgG half‐life was determined using a non‐compartmental method (WinNonlin®; Pharsight Corporation, Mountain View, CA, USA). Blood samples for PK assessments were obtained between infusions seven and eight (28‐day schedule) or infusions nine and 10 (21‐day schedule); sampling times were immediately pre‐ and post‐infusion, 1 and 24 h post‐infusion and 2, 4, 7, 14, 21 and 28 days post‐infusion. If a subject was on a 21‐day schedule, the next infusion after the PK sampling was delayed until the 28‐day PK blood sample had been obtained.
Diary cards
The subject or parent/guardian completed diary cards to record the following outcomes: AEs, oral temperature, any presumed infection, physician/emergency room (ER) visits, school/nursery days missed because of infection or illness and concomitant medication (including antibiotics).
Data analysis
To detect an annual SABI incidence of 0·5 per subject 2, 25 eligible subjects were planned for enrolment (with ≥ 4 subjects aged 2–5 years, ≥ 4 subjects aged 6–11 years and ≥ 8 subjects aged 12–16 years) to obtain 20 evaluable children 12. All analyses were conducted on the intent‐to‐treat (ITT) population, defined as all enrolled subjects who received ≥ 1 infusion of Gammaplex 5%.
The SABI rate was calculated from the total number of observed infections and the total number of subject years on study. The SABI event rate and the upper bound of its one‐sided 99% confidence interval (CI) were estimated using the exact method for a one‐sample Poisson rate.
Secondary efficacy and safety end‐points were summarized descriptively and expressed per subject per year where applicable. All AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA), version 8·1. An adverse reaction (AR) was defined as a treatment‐emergent AE that began during an infusion or within 72 h after completion of an infusion, that was considered by investigators to be possibly, probably or definitely related to study drug, or for which the investigator's causality assessment was either missing or indeterminate. A one‐sided 95% exact confidence limit was computed for the proportion of subjects experiencing ≥ 1 temporally associated AR; if the upper bound was < 40%, then the incidence of infusion‐related AEs associated with the study drug was considered acceptable in accordance with regulatory guidelines 2.
PK parameters were calculated from both absolute and baseline‐adjusted serum total IgG values using a non‐compartmental method; baseline‐adjusted profiles were derived by subtracting the pre‐dose serum concentration for each individual from all post‐dose measurements. Certain PK parameters were only calculated if a terminal monoexponential decline could be identified unambiguously.
Results
Study subjects
Of the 25 subjects enrolled and included in the ITT population (Fig. 1), 14 and 11 were on 21‐ and 28‐day infusion schedules, respectively. The majority of subjects (n = 24; 96·0%) completed the study and returned for both follow‐up visits. One subject on a 21‐day infusion schedule withdrew consent and discontinued from the study after the fourth infusion (88 days on treatment). The PK analyses excluded two subjects due to study discontinuation (n = 1) and school commitments resulting in limited PK samples (n = 1).
Figure 1.
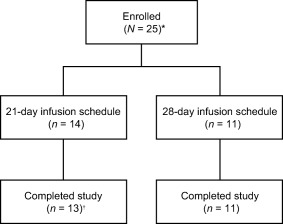
Subject disposition. *All enrolled patients were included in the intent‐to‐treat population. †One subject withdrew consent and discontinued the study after four infusions (88 days on treatment).
Subject demographic and baseline characteristics are summarized in Table 1. Three subjects were 2–5 years of age, 12 subjects were 6–11 years of age and 10 subjects were 12–16 years of age. All subjects had received prior IVIG therapy, 56·0% (n = 14) had previously received a 10% IVIG product, and 60·0% (n = 15) had a documented AR related to their prior IVIG therapy. Twenty‐two subjects (88·0%) had a diagnosis of common variable immunodeficiency and three (12·0%) were diagnosed with X‐linked agammaglobulinaemia. Prior to study entry, 15 subjects reported a medical history of chronic sinusitis and none had a history of bronchiectasis; none of these cases were reported as a SABI.
Table 1.
Subject demographic and baseline characteristics.
| Variable | Subjects (N = 25) |
|---|---|
| Age (years) | |
| Mean (s.d.) | 10·4 (3·84) |
| Median (range) | 11 (3–16) |
| Subjects in each age group, n (%) | |
| 2–5 years | 3 (12·0) |
| 6–11 years | 12 (48·0) |
| 12–16 years | 10 (40·0) |
| Gender, n (%) | |
| Male | 19 (76·0) |
| Female | 6 (24·0) |
| Diagnosis, n (%) | |
| Common variable immunodeficiency | 22 (88·0) |
| X‐linked and autosomal forms of agammaglobulinaemia | 3 (12·0) |
| Subjects with infection(s) related to prior IVIG therapy in past 6 months, n (%) | |
| 0 infections | 7 (28·0) |
| 1 infection | 7 (28·0) |
| 2 infections | 5 (20·0) |
| 3 infections | 4 (16·0) |
| ≥ 4 infections | 2 (8·0) |
| Ongoing infections, n (%) | 3 (12·0) |
IVIG = intravenous immunoglobulin; s.d. = standard deviation.
A total of 368 infusions of Gammaplex 5%, all within the dose range of 300–800 mg/kg, were given to the 25 subjects. Deviations from the specified infusion rates, including time between rate increments, occurred in 10 infusions in seven subjects. The mean dose per infusion for those on the 21‐ and 28‐day schedules was 545 mg/kg (range = 429–689 mg/kg) and 521 mg/kg (range =316–800 mg/kg), respectively. The mean infusion duration across all subjects was 182 min (range = 112–433 min), with a median of 175 min.
Infections
Two SABIs of pneumonia were reported (Table 2), with onset between the first infusion of Gammaplex 5% and the first follow‐up, leading to an annual SABI event rate of 0·09 (upper one‐sided 99% CI = 0·36). This SABI event rate meets the criteria for efficacy established by regulatory guidelines 2. Two additional events of mild presumed pneumonia were observed but were not considered to be SABIs by the investigators. Each event was diagnosed by the subject's local physician upon physical examination but without confirmation via X‐ray or blood test. Neither event resulted in hospitalization, nor did either meet any other criteria for seriousness. Of the 21 subjects who experienced ≥ 1 presumed infection during the study (Table 2), the infections recorded by 19 subjects (90·5%) did not meet SABI criteria. These presumed infections involved primarily the upper respiratory tract (e.g. sinusitis); no subjects withdrew from the study due to an infection.
Table 2.
Infections during Gammaplex 5% therapy.
| Variable | Subjects (N = 25) |
|---|---|
| Subjects with SABIs, n (%) | 2 (8·0) |
| Subjects recording infection during study, n (%) | 21 (84·0) |
| Overall upper respiratory tract infection | 16 (64·0) |
| Sinus infection | 10 (40·0) |
| Lower respiratory tract infection | 7 (28·0) |
| Gastrointestinal infection | 4 (16·0) |
| Urinary tract infection | 1 (4·0) |
| Other infection* | 8 (32·0) |
| Infectious episodes per subject per year† | |
| Mean (s.d.) | 3·20 (2·7) |
| Median (range) | 3·08 (0–10·4) |
| Number of infectious episodes per subject per year, n (%)† | |
| None | 4 (16·0) |
| > 0 to < 3 | 7 (28·0) |
| 3 to < 5 | 9 (36·0) |
| 5 to < 10 | 4 (16·0) |
| ≥10 | 1 (4·0) |
*Other infections (recorded for ≤ 4 subjects each) included influenza, otitis media, herpes simplex, tonsillar disorder, staphylococcal infection and oral fungal infection.
†If the patient recorded a possible infection on his/her diary card for any day, an infection was presumed whether or not it was confirmed by a health‐care professional. Infections per subject per year were calculated as number of infections × 365/number of days in study. SABI = serious acute bacterial infection; s.d. = standard deviation.
Antibiotics
Twenty‐one subjects (84·0%) received a total of 79 courses of therapeutic systemic antibiotics, and six subjects (24·0%) received 11 courses for prophylaxis. The mean [standard deviation (s.d.); range] number of days for which subjects received systemic and therapeutic systemic antibiotic medications was 110·6 (s.d. = 137·10; range = 5–355) days and 32·0 (s.d. = 28·28; range = 5–101) days, respectively. Prior to entering the study, three subjects were on prophylactic antibiotics, which continued for the duration of the study.
Fever
Nine subjects (36·0%) recorded an AE of fever (> 38°C); of these, fevers in three subjects (12·0%) were judged by the investigators to be related possibly to the study drug.
Visits to the physician and/or hospital ER and hospitalization
Unscheduled physician and ER visits occurred at a mean rate of 3·5 (median = 3·1) and 0·5 (median = 0) per subject per year, respectively (Table 3). The majority of patients (88·0%) did not require hospitalization during the study. For the three subjects admitted to the hospital (once each), each hospital stay was ≤ 4 days in length. One subject had five ER visits for the following reasons: lobar pneumonia (a SABI); left side pain; pain in the knees/feet and dizziness; general malaise, increased cough and acute sinusitis; and shortness of breath and wheezing. Each of these five events was moderate in intensity and was not considered related to the study drug.
Table 3.
Physician and hospital ER visits.
| Variable | Physician visit | Hospital ER visit |
|---|---|---|
| Number of visits per year due to infection or other medical problem* | ||
| Mean (s.d.) | 3·5 (4·00) | 0·5 (1·13) |
| Median (range) | 3·1 (0–16) | 0 (0–5) |
| Number of visits per year due to infection or other medical problem, n (%)* | ||
| 0 | 7 (28·0) | 17 (68·0) |
| > 0 to < 7 | 14 (56·0) | 8 (32·0) |
| 7 to < 14 | 3 (12·0) | 0 |
| 14 to < 21 | 1 (4·0) | 0 |
| ≥ 21 | 0 | 0 |
*Calculated by the number of visits listed each week in diary cards × 365/number of days of diary data. ER = emergency room; s.d. = standard deviation.
Days off school or nursery
Sixteen subjects (64·0%) missed ≥ 1 day from school or nursery due to infection or other illness. Subjects missed a mean of 4·2 (s.d. = 8·28) days per year, with a median of 1·1 (range = 0–32) days; most subjects (88·0%) missed < 7 days of school or nursery (Fig. 2).
Figure 2.
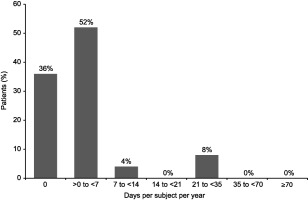
Percentage of patients by number of days off from school/nursery per subject per year due to infection or other illness (N = 25).
IgG concentrations
The geometric mean half‐life for total IgG (based on absolute IgG concentrations) was 35·2 days and was comparable for the 21‐day (geometric mean half‐life, 33·2 days) and 28‐day (geometric mean half‐life, 38·8 days) infusion schedules.
Mean trough IgG ranges were comparable for the 21‐day (9·87–10·83 g/l) and 28‐day (8·22–8·82 g/l) dosing schedules (Fig. 3). After the second infusion, individual trough IgG levels for all subjects remained consistently above 7·00 g/l. While many subjects (n = 18; 72·0%) had an IgG trough level at some point during the study that was below the mean pre‐study value, no evidence of a systematic decline in IgG values over time was observed during Gammaplex 5% treatment. In addition, as the primary end‐point for SABI incidence was met, no evidence of treatment failing to provide infection prophylaxis was observed. Because pre‐study IgG values were assessed by different (local) laboratories, while IgG values during the study were assessed by a central laboratory, any differences in values could be artefactual.
Figure 3.
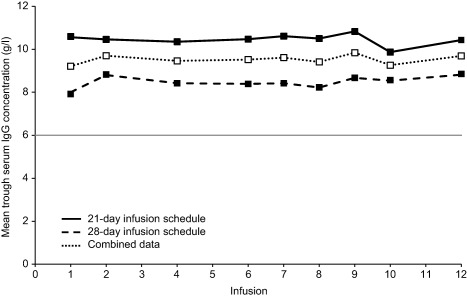
Mean immunoglobulin (Ig)G trough (pre‐dose) levels following Gammaplex 5% infusion to infusion 12. Because the last infusion for subjects on the 28‐day dosing schedule was infusion 12, data for infusions 13–17 (available for the 21‐day dosing schedule only) are not shown. Threshold at 6·00 g/l (minimum trough serum IgG level for inclusion in study) is indicated by the grey line. Screening (visit 1) blood samples were not included, as not all sampling was at trough (some were taken mid‐cycle). Patients on the 21‐day schedule had their 10th infusion delayed to accommodate the 28‐day blood sampling, as illustrated by the consistent dip in concentrations at infusion 10.
Combining 21‐ and 28‐day schedules, mean levels of IgG1 (range = 4·96–5·34 g/l), IgG2 (range = 3·01–3·32 g/l), IgG3 (range = 0·42–0·50 g/l) and IgG4 (range = 0·10–0·11 g/l) remained stable during Gammaplex 5% therapy. Throughout the study, trough IgG levels to specific antigens remained stable, with no increasing or decreasing trends (Table 4).
Table 4.
Trough levels of IgG antibodies to specific antigens.
| Visit (infusion) | |||
|---|---|---|---|
| Specific antigens | 1 (screen) | 14 (infusion 12) | Follow‐up 1 |
| CMV IgG (AU/ml) | |||
| n | 24 | 21 | 20 |
| Mean (s.d.) | 38·50 (9·757) | 39·15 (14·093) | 45·62 (22·761) |
| Median (range) | 37·00 (19·0–58·0) | 36·00 (20·0–83·9) | 40·00 (30·0–126·6) |
| S. pneumoniae serotype 14 (µg/ml) | |||
| n | 25 | 24 | 23 |
| Mean (s.d.) | 9·12 (5·284) | 7·09 (4·865) | 7·27 (3·805) |
| Median (range) | 8·60 (0·5–21·4) | 5·80 (2·0–24·4) | 6·80 (2·4–21·1) |
| S. pneumoniae serotype 18C (µg/ml) | |||
| n | 25 | 24 | 23 |
| Mean (s.d.) | 2·16 (1·668) | 1·91 (1·691) | 2·92 (4·327) |
| Median (range) | 1·60 (0·2–7·4) | 1·25 (0·4–6·7) | 1·50 (0·4–21·4) |
| S. pneumoniae serotype 19F (µg/ml) | |||
| n | 25 | 24 | 23 |
| Mean (s.d.) | 12·41 (14·228) | 11·77 (13·940) | 13·36 (12·797) |
| Median (range) | 7·50 (0·4–53·1) | 6·55 (1·3–60·5) | 8·10 (1·7–49·4) |
| S. pneumoniae serotype 23F (µg/ml) | |||
| n | 25 | 24 | 23 |
| Mean (s.d.) | 21·59 (13·198) | 19·48 (11·951) | 20·67 (9·370) |
| Median (range) | 17·00 (5·3–46·5) | 17·35 (4·0–52·3) | 20·20 (5·3–41·9) |
| S. pneumoniae serotype 4 (µg/ml) | |||
| n | 25 | 24 | 23 |
| Mean (s.d.) | 2·21 (1·987) | 2·12 (2·455) | 2·61 (2·723) |
| Median (range) | 1·50 (0·4–10·0) | 1·75 (0·4–12·9) | 1·90 (0·5–11·2) |
| S. pneumoniae serotype 6B (µg/ml) | |||
| n | 25 | 24 | 23 |
| Mean (s.d.) | 15·73 (20·814) | 6·71 (4·537) | 15·87 (42·143) |
| Median (range) | 9·80 (1·8–105·5) | 5·65 (2·1–23·2) | 6·80 (2·8–208·4) |
| S. pneumoniae serotype 9V (µg/ml) | |||
| n | 25 | 24 | 23 |
| Mean (s.d.) | 7·14 (5·739) | 6·27 (4·084) | 7·14 (3·722) |
| Median (range) | 5·20 (1·3–28·0) | 5·35 (1·2–20·1) | 5·90 (1·6–18·3) |
| H. influenzae type b (mg/l) | |||
| n | 25 | 23 | 23 |
| Mean (s.d.) | 3·59 (2·189) | 3·41 (2·048) | 3·77 (2·156) |
| Median (range) | 3·02 (1·3–9·0) | 2·81 (1·3–9·0) | 2·96 (1·2–9·0) |
H. influenzae = Haemophilus influenzae; IgG = immunoglobulin G; s.d. = standard deviation; S. pneumoniae = Streptococcus pneumoniae.
Safety and tolerability
The total number of temporally associated AEs was 143 (a rate of 0·39 AEs per infusion). Of 368 total infusions, 97 (26·4%) were associated with an AE up to 72 h after the infusion (regardless of causality). The upper 95% confidence limit for this proportion was 0·304 (30·4%), which meets the criteria for this safety end‐point established by regulatory guidelines [i.e. is lower than the target upper 95% confidence limit of 0·4 (40·0%)] 2. Approximately half (51·7%; 74 of 143) of the infusion‐associated AEs were not considered product‐related by clinicians. The proportion of infusions associated temporally with product‐related AEs was highest at infusions 1 (n = 7; 28·0%), 2 (n = 5; 20·0%) and 3 (n = 6; 24·0%); thereafter, the proportions decreased to 0% by infusion 17 (Fig. 4). AEs were considered product‐related in 14 of 25 subjects (56·0%); of these subjects, two experienced AEs (headache, fatigue and myalgia) that were considered definitely related to study drug. No serious product‐related AEs were observed, and no subjects withdrew from treatment due to an AE. Laboratory test results and AE data showed no evidence of product‐related haemolysis or thromboembolic events. Table 5 summarizes ARs occurring in ≥ 3 subjects.
Figure 4.
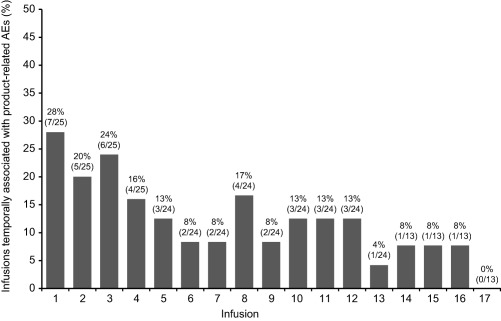
Percentage of infusions associated temporally with product‐related adverse events (AEs) to infusion 17. Bars are labelled as % (n/N), where N = the number of infusions administered and n = the number of administered infusions that were associated temporally with product‐related AEs.
Table 5.
ARs occurring in ≥ 3 subjects (N = 25).
| Preferred term | Subjects (N = 25) n (%) | Infusions (n = 368) n (%) |
|---|---|---|
| Headache | 11 (44·0) | 21 (5·7) |
| Sinusitis | 6 (24·0) | 8 (2·2) |
| Hypotension | 4 (16·0) | 10 (2·7) |
| Tachycardia | 3 (12·0) | 5 (1·4) |
| Pyrexia | 3 (12·0) | 4 (1·1) |
| Hypertension | 3 (12·0) | 4 (1·1) |
| Fatigue | 3 (12·0) | 4 (1·1) |
| Infusion‐site reaction | 3 (12·0) | 4 (1·1) |
| Dry skin | 3 (12·0) | 3 (0·8) |
| Nasal congestion | 3 (12·0) | 3 (0·8) |
| Upper respiratory tract infection | 3 (12·0) | 3 (0·8) |
| Abdominal pain | 3 (12·0) | 2 (0·5) |
An adverse reaction (AR) was defined as a treatment‐emergent adverse event that began during an infusion or within 72 h after completion of an infusion, that was considered by investigators to be possibly, probably or definitely related to study drug, or for which the investigator's causality assessment was either missing or indeterminate.
Discussion
Consistent with previous results in a mixed adult and paediatric population 11, this open‐label, non‐comparative study showed that Gammaplex 5% is efficacious and well tolerated in children and adolescent patients with PID. The primary efficacy variable was the annual incidence of SABIs. Of the 25 paediatric subjects, two developed a SABI, resulting in an annual SABI event rate of 0·09 per subject (upper one‐sided 99% CI = 0·36). In patients receiving regular administration of IVIG, a SABI frequency of < 0·5 per patient per year has been observed 2. The SABI rate reported in this study is consistent with that observed rate. In addition, the SABI event rate observed here meets the efficacy criteria established by regulatory guidelines (i.e. a rate of < 1·0 per patient per year is ‘adequate to provide substantial evidence of efficacy’ 2).
While published data on the incidence of SABIs in young children treated with IVIG are limited, these results are broadly in agreement with those published for another IVIG product in which a SABI event rate of 0·11 per subject per year (upper one‐sided 99% CI = 0·37) was reported 10. Additional analyses of SABI rates were performed by combining data from the present study with data from a previous study of Gammaplex 5% in 50 predominantly adult subjects with PID 11. As no SABIs were reported in the previous study, the mean SABI event rate per subject per year for the combined patient population (n = 75) is 0·03 (upper one‐sided 99% CI = 0·12). If the additional two events of mild pneumonia in the present study were also included as SABIs in the combined patient population, the mean event rate would be 0·06 per subject per year (upper one‐sided 99% CI = 0·17). This SABI event rate remains below regulatory efficacy guidelines 2 and is similar to SABI event rates reported for other IVIG products 13, 14, 15.
The present study also included several secondary efficacy variables to support the conclusion that Gammaplex 5% is efficacious in a paediatric population. For example, 64% of subjects missed days at school or nursery, with a mean of 4·2 days missed per subject per year, which is lower than that reported for Gammaplex 5% in adults with PID (8·73 days per subject per year) 11. The annual rate for physician visits is slightly lower in children and adolescents compared with adults (3·5 and 5·6, respectively), as is the annual rate of ER visits (0·3 and 0·4, respectively) 11. The annual incidence of non‐serious infections in children is very similar to that reported in adults (3·20 versus 3·28, respectively) 11 and is also comparable to the incidence of non‐serious infections described for other IVIG products 13, 16, 17.
Trough IgG levels exceeded 7·00 g/l for all subjects after 15 weeks of Gammaplex 5% treatment and remained fairly stable over time. The median trough total IgG levels before infusion (range = 9·26–10·44 g/l; excluding infusion 1) were within the range for subjects without PID and that have been shown previously to confer good protection against infection in adults with PID 17. The mean absolute IgG half‐life following Gammaplex 5% infusion was 35·2 days, which is consistent with the half‐life reported for other IVIG preparations in adults with PID 3, 18. Also, the IgG concentration versus time profiles in children and adolescents were similar to those reported for Gammaplex 5% in adults 11, suggesting that 21‐ and 28‐day Gammaplex 5% dosing schedules are appropriate for children, adolescents and adults with PID.
The safety of Gammaplex 5% in children and adolescents was also comparable to that observed with Gammaplex 5% in predominantly adult subjects with PID 11, with the incidence of product‐related AEs in each study being 56 and 48%, respectively. Regulatory guidelines specify a safety limit for the proportion of infusions with ≥ 1 temporally associated AEs of an upper one‐sided 95% confidence limit of 0·40 (i.e. 40% of infusions) 2. In this study, the upper one‐sided 95% confidence limit for infusions with temporally associated AEs was 0·26 (26%). Although the infection rate was an efficacy end‐point in this study, any infections reported at the time of infusions were also reported as AEs. This conservative approach of not excluding infections from the calculated incidence for temporally associated AEs resulted in a rate that not only remained below the specified regulatory limit of 40% 2 but also compares well with that reported for Gammaplex 5% (24%) in a predominantly adult population 11.
Overall, compliance with the planned regimen of Gammaplex 5% dose and incremental infusion rates was favourable; all doses were within the planned range of 300–800 mg/kg, with a mean dose per infusion of 536 mg/kg. The infusions were generally well tolerated, as none of the 25 subjects discontinued the study prematurely because of an AE, no subjects were hospitalized because of AEs possibly related to Gammaplex 5%, and no evidence of any thromboembolic events was observed during the study.
In conclusion, the results of this study demonstrate that Gammaplex 5% is effective in preventing SABIs in children and adolescents with PID associated with significant hypogammaglobulinaemia and/or antibody deficiency, both 21‐ and 28‐day dosing schedules are appropriate in this population and that Gammaplex 5% is suitable for the management of both paediatric and adult patients with PID.
Disclosure
This study was funded by Bio Products Laboratory Ltd. I.R.M. has served as a clinical trial investigator for Bio Products Laboratory. S.G. has received research support from Baxter and CSL Behring, and has served as an adviser and speaker for Baxter and a clinical trial investigator for Baxter, Bio Products Laboratory, and Octapharma. M.S.B. and N.H. are contractors for Bio Products Laboratory Ltd. J.N.M. has served as a clinical trial investigator for Bio Products Laboratory, a consultant for Baxter and Prometic, and an iDMC member for MacroCure and Octapharma.
Acknowledgements
The study was supported by Bio Products Laboratory Ltd (Elstree, UK) and co‐ordinated by INC Research Inc. (Raleigh, NC, USA). The investigators for this study were as follows: Todd Green MD (Children's Hospital of Pittsburgh, Pittsburgh, PA, USA); Sudhir Gupta MD, PhD (University of California, Irvine, Irvine, CA, USA); Anne‐Marie Irani MD (Virginia Commonwealth University Health System, Richmond, VA, USA); Robyn J. Levy MD (Family Allergy and Asthma Center, PC, Atlanta, GA, USA); Isaac R. Melamed MD (IMMUNOe, Centennial, CO, USA); James N. Moy MD (Rush University Medical Center, Chicago, IL, USA); Carmen Luz Navarrete Suárez MD (Roberto del Rio Children's Hospital, Santiago, Chile); Raz Somech MD, PhD (Edmond and Lily Safra Children's Hospital, Tel Hashomer, Israel); and Daniel Suez MD (Allergy, Asthma and Immunology Clinic, PA, Irving, TX, USA). The authors thank the patients and their caregivers for their participation in the study and all staff at the study centres for their contributions. Bio Products Laboratory Ltd provided funding for medical writing and editorial support in the development of this manuscript. Morgan C. Hill PhD and Roger J. Hill PhD, of QXV Communications (Haddam, CT, USA) revised the manuscript based on input from authors, and Dena McWain of QXV Communications copyedited and styled the manuscript per journal requirements.
References
- 1. Wood P, Stanworth S, Burton J et al Recognition, clinical diagnosis and management of patients with primary antibody deficiencies: a systematic review. Clin Exp Immunol 2007; 149:410–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.US Department of Health and Human Services. Guidance for industry: safety, efficacy, and pharmacokinetic studies to support marketing of immune globulin intravenous (human) as replacement therapy for primary humoral immunodeficiency. Rockville, MD: US Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research, 2008.
- 3. Ballow M, Notarangelo L, Grimbacher B et al Immunodeficiencies. Clin Exp Immunol 2009; 158:14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Immune Deficiency Foundation . Primary immunodeficiency diseases in America: 2007, the third national survey of patients. Towson, MD: Immune Deficiency Foundation, 2009. [Google Scholar]
- 5. Orange JS. Clinical update in immunoglobulin therapy for primary immunodeficiency diseases In: Clinical focus on primary immunodeficiencies. Towson, MD: Immune Deficiency Foundation, 2011. (Issue 14):1–9. [Google Scholar]
- 6. Bonagura VR. Using intravenous immunoglobulin (IVIG) to treat patients with primary immune deficiency disease. J Clin Immunol 2013; 33:90–4. [DOI] [PubMed] [Google Scholar]
- 7. Torgerson TR. Overview of routes of IgG administration. J Clin Immunol 2013; 33:87–9. [DOI] [PubMed] [Google Scholar]
- 8. Bio Products Laboratory Ltd. Gammaplex [immune globulin intravenous (human), 5% liquid] [US prescribing information]. Elstree: Bio Products Laboratory Ltd, 2014. [Google Scholar]
- 9. Bio Products Laboratory Ltd. Gammaplex, a sterile liquid of 5% w/v normal immunoglobulin [UK summary of product characteristics]. Elstree: Bio Products Laboratory Ltd, 2015. [Google Scholar]
- 10. Church JA, Borte M, Taki H et al Efficacy and safety of Privigen in children and adolescents with primary immunodeficiency. Pediatr Asthma Allergy Immunol 2009; 22:53–62. [Google Scholar]
- 11. Moy JN, Scharenberg AM, Stein MR et al Efficacy and safety of a new immunoglobulin G product, Gammaplex®, in primary immunodeficiency diseases. Clin Exp Immunol 2010; 162:510–15. 21070209 [Google Scholar]
- 12. European Medicines Agency . Guideline on the clinical investigation of human normal immunoglobulin for intravenous administration (IVIG). London: European Medicines Agency Committee for Medicinal Products for Human Use, 2010. [Google Scholar]
- 13. Stein MR, Nelson RP, Church JA et al. Safety and efficacy of Privigen, a novel 10% liquid immunoglobulin preparation for intravenous use, in patients with primary immunodeficiencies. J Clin Immunol 2009; 29:137–44. [DOI] [PubMed] [Google Scholar]
- 14. Berger M, Pinciaro PJ. Safety, efficacy, and pharmacokinetics of Flebogamma 5% [immune globulin intravenous (human)] for replacement therapy in primary immunodeficiency diseases. J Clin Immunol 2004; 24:389–96. [DOI] [PubMed] [Google Scholar]
- 15. Ochs HD, Pinciaro PJ. Octagam 5%, an intravenous IgG product, is efficacious and well tolerated in subjects with primary immunodeficiency diseases. J Clin Immunol 2004; 24:309–14. [DOI] [PubMed] [Google Scholar]
- 16. Church JA, Leibl H, Stein MR et al Efficacy, safety and tolerability of a new 10% liquid intravenous immune globulin [IGIV 10%] in patients with primary immunodeficiency. J Clin Immunol 2006; 26:388–95. [DOI] [PubMed] [Google Scholar]
- 17. Eijkhout HW, van Der Meer JW, Kallenberg CG et al The effect of two different dosages of intravenous immunoglobulin on the incidence of recurrent infections in patients with primary hypogammaglobulinemia. A randomized, double‐blind, multicenter crossover trial. Ann Intern Med 2001; 135:165–74. [DOI] [PubMed] [Google Scholar]
- 18. Alyanakian MA, Bernatowska E, Scherrmann JM, Aucouturier P, Poplavsky JL. Pharmacokinetics of total immunoglobulin G and immunoglobulin G subclasses in patients undergoing replacement therapy for primary immunodeficiency disorders. Vox Sang 2003; 84:188–92. [DOI] [PubMed] [Google Scholar]