

Abstract
AIM: To study the effect of anti-copper treatment for survival of hepatic cells expressing different ATP7B mutations in cell culture.
METHODS: The most common Wilson disease (WD) mutations p.H1069Q, p.R778L and p.C271*, found in the ATP7B gene encoding a liver copper transporter, were studied. The mutations represent major genotypes of the United States and Europe, China, and India, respectively. A human hepatoma cell line previously established to carry a knockout of ATP7B was used to stably express WD mutants. mRNA and protein expression of mutant ATP7B, survival of cells, apoptosis, and protein trafficking were determined.
RESULTS: Low temperature increased ATP7B protein expression in several mutants. Intracellular ATP7B localization was significantly impaired in the mutants. Mutants were classified as high, moderate, and no survival based on their viability on exposure to toxic copper. Survival of mutant p.H1069Q and to a lesser extent p.C271* improved by D-penicillamine (DPA) treatment, while mutant p.R778L showed a pronounced response to zinc (Zn) treatment. Overall, DPA treatment resulted in higher cell survival as compared to Zn treatment; however, only combined Zn + DPA treatment fully restored cell viability.
CONCLUSION: The data indicate that the basic impact of a genotype might be characterized by analysis of mutant hepatic cell lines.
Keywords: ATP7B, D-penicillamine, Zinc, Mutations, Wilson disease, Therapy
Core tip: Copper overload in Wilson disease (WD) is treated with anti-copper therapy. However, the effect of treatment has not been studied using human hepatic cells lacking the ATP7B copper transporter. Using a previously established ATP7B KO cell line, we generated stable mutant cell lines to study functional analysis and response to zinc (Zn) and D-penicillamine (DPA). All mutants showed individual response rates following copper and anti-copper treatment. Highest rescue from copper toxicity was observed after combined Zn and DPA treatment. The study provides novel insights into genotype-phenotype correlations and genotype-specific treatment of WD.
INTRODUCTION
Wilson disease (WD; MIM No. 277900) is an autosomal recessive disorder resulting from mutation of the ATP7B gene[1]. More than 600 mutations of ATP7B are known (www.hgmd.org). Due to improved genetic diagnosis, novel mutations are being found around the world. The WD gene consists of 21 exons that span a genomic region of about 80 kb and is located on the long arm of chromosome 13 (13q14.1)[2,3]. ATP7B encodes a large membrane protein of 1465 amino acids that was characterized to be a copper (Cu) transporting P-type adenosine triphosphatase (ATPase) which has high homology to the amino acid sequence of the gene responsible for Menkes disease (MIM No. 300011). Apart from involvement of ATP7B in causing rare inherited disease, its role in Cu homeostasis is central to the function of important biochemical pathways[4]. Of note, anti-copper therapy effective for treatment of WD patients was recently recognized to represent an alternative for the treatment of other diseases[5,6]. The effect of anti-copper treatment for hepatocytes, the major target cell of the disease, and the correlation to individual WD genotypes have yet to be determined.
ATP7B is mainly expressed in the liver and to a lesser extent in the brain and other organs. ATP7B has two functions in the liver which are central for Cu homeostasis[4,7]. ATP7B protein transports Cu into the trans Golgi network (TGN) where the metal is transferred to apoceruloplasmin that is finally released as ceruloplasmin into the blood. Excess Cu is sequestered by ATP7B into vesicles that are subsequently released from the body via bile canaliculi. ATP7B protein normally resides in the TGN but is believed to traffic to the endocytic vesicles under high Cu conditions for biliary Cu excretion. Impaired ATP7B function due to mutations in the ATP7B gene results in toxic Cu accumulation, ultimately leading to cell death.
Hallmarks of WD include Cu accumulation in the liver and the brain, a low ceruloplasmin activity, and the presence of Kayser-Fleischer (KF) corneal rings[1]. Diagnosis is difficult since individual abnormalities could be absent or at borderline. A wide spectrum of clinical presentations is observed, including liver damage and/or neurological symptoms, ranging from asymptomatic phenotypes which show only mild abnormalities of Cu homeostasis, to patients having liver cirrhosis, acute liver failure, or severe neurological disability. Onset of disease is also highly variable and is often observed at childhood but also in adolescence and even in late adults[8]. Unrelated proteins that mediate uptake, delivery, and efflux of Cu have been implicated to modify disease; however, an understanding of the molecular mechanisms that are involved in the complex, highly variable phenotype, including the toxic events observed in hepatocytes, is far from being achieved.
The type and location of ATP7B mutation has been suggested to be one determinant of the disease phenotype indicating that individual mutations of ATP7B may be linked to a phenotype. However, prognosis of the disease related to a specific WD mutation expressed in patients has not been established. Therefore, there is a pressing need for an optimal treatment regimen of unknown genotypes in various regions, including China where novel genotypes have increasingly been identified[9-12]. The majority of WD mutations are missense but deletions and insertions are also observed. Since most WD patients carry compound heterozygous ATP7B mutations that may modulate the phenotypic expression of an individual mutation, the analysis of homozygous mutations has helped to explore links of ATP7B genotype and phenotype, e.g., in studies of large families in specific regions[13,14]. Recently, functional characterization of individual ATP7B mutations expressed in various cell lines was suggested to improve the understanding of the clinical impact of a given mutation[15-17].
WD is a fatal disease when not appropriately treated by anti-coppering drugs and can lead to early death. The two most commonly used drugs for therapy of WD are D-penicillamine (DPA) and zinc (Zn) that involve de-coppering by chelation or metallothionein induction, respectively[18]. Both lines of drugs are associated with side effects and the correct choice of treatment, including the survival of hepatocytes, is still under debate. In a significant portion of patients with WD, treatment is not effective leading to discontinuation of therapy[18]. Chelation therapy seems to be most beneficial in European WD patients, whereas Chinese patient benefit most from Zn treatment[19-21]. Whether poor response to therapy may relate to the genotype is not known, since a standard therapy regimen for WD has not been established.
We addressed the functional characterization of ATP7B mutations following anti-copper treatment in a novel hepatic ATP7B knockout (KO) cell line that was previously established by us[22]. The effect of Cu on intracellular trafficking, viability, and apoptosis was studied.
MATERIALS AND METHODS
Cell culture
HepG2 (human hepatocellular carcinoma) cells purchased from American Type Culture Collection (ATCC) and derivatives of ATP7B knockout cells[22] were cultured in RPMI media (Lonza) containing 10% fetal bovine serum (FBS) and supplemented with 100 U/mL penicillin/streptomycin (PAA). Cell lines were maintained in 5% CO2 at 37 °C in a humidified chamber.
Site-Directed Mutagenesis and generation of stable ATP7B mutant cell lines
Wild type ATP7B cDNA was cloned into pGCsamENATP7B retroviral vector encoding blasticidin resistance[23]. Site-directed mutagenesis was performed using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer’s instructions. Mutagenized cDNA was sequenced to confirm the presence of the selected variant and exclude secondary mutations. KO cells were transduced with the plasmid containing the WD mutation. KO cells harbouring wild type ATP7B plasmid were used as a control. Cells were selected in media containing 6 μg/mL blasticidin (Invitrogen).
Confocal staining
Cell lines were grown on cover slips coated with 0.1% gelatin to about 80% confluency. For studying Cu induced trafficking under low and high Cu conditions, cells were either maintained in RPMI basal cell culture media or treated by addition of 100 μmol/L Cu for 3 h. Cells were then washed with PBS, fixed using 4% paraformaldehyde (Electron Microscopy Sciences) and permeabilized with 0.25% triton X-100 (Sigma). Cells were blocked for 30 min with 3% bovine serum albumin (Sigma), followed by staining with primary anti-ATP7B (kind gift of Dr. I. Sandoval, Madrid, Spain) and anti-lamp2 (Santa Cruz Biotechnology, sc-18822) antibodies. Secondary antibody incubation was done using goat anti-rabbit 594 and goat anti-mouse 488 (Life technologies, A-11012 and A-11001, respectively) for 45 min. Confocal images were recorded with a Nikon A1 confocal laser scanning microscope and an ECLIPSE Ti with a CFI Plan Fluor 40 × (NA 1.3 oil) lens (Nikon, Japan). The images were analyzed using NIS-elements software.
Western blot analysis
Western blot was performed as described previously[22]. Briefly, cells were lysed in RIPA buffer containing 60 mmol/L Tris-HCl, 150 mmol/L NaCl, 2% Na-deoxycholate, 2% triton X-100, 0.2% SDS and 15 mmol/L EDTA supplemented with protease inhibitors (Roche, Complete Mini, EDTA-free). Protein concentration was determined by Bradford assay (Bio-Rad, Protein Assay). 10 μg of protein was loaded onto a 9% SDS gel and blotting was performed using a 0.45 μmol/L nitrocellulose membrane (GE Healthcare). Polyclonal anti-rabbit ATP7B antibody was used for protein detection. HSC70 (Santa Cruz Biotechnology, sc-1059) staining was used as a protein loading control. Densitometric analysis was performed using ImageJ 1.46 software (http://imagej.nih.gov/ij/docs/guide/). Relative expression was normalized to KO cells expressing wild type ATP7B.
MTT assay
104 cells were seeded in triplicates in a 96 well plate and cultivated overnight in 100 μL RPMI media lacking phenol red (PAA). Cells were then exposed to different Cu concentrations (CuCl2; Sigma Aldrich). After 48 h, viability was determined by MTT assay as previously described[22]. For testing drug efficacy, cells were preincubated with 200 μmol/L Zn (ZnCl2; AppliChem) for 2 h[24] and washed twice with phosphate buffered saline (PBS, PAA) prior to Cu addition. D-penicillamine (Dako) was added at 6.25 mmol/L simultaneously with Cu. Cells were incubated for 48 h. Cell rescue was determined as percentage of treated, viable compared to untreated cells.
Determination of apoptosis
106 cells were incubated with 100 μmol/L Cu at 37 °C for 24 h. Supernatants and cells were collected and subjected to Annexin-V and propidium iodide staining (Roche Annexin-V-FLUOS kit) and analyzed using flow cytometry (Epics XL.MCL, Beckman Coulter).
Real-time PCR analysis
PCR analysis was performed as described previously[25]. Briefly, isolation of total RNA was performed by RNeasy kit (Qiagen). 1 μg of RNA was transcribed using SuperScript II (Invitrogen) according to the instructions of the manufacturer. For quantitative real time PCR (qPCR) the RT product was incubated with SYBR Green PCR Core Plus (Eurogentec, Belgium) and 150 nmol/L of primers. PCR was analyzed on the ABI Prism 7900 HT Sequence Detection System (PE Applied Biosystems). Each sample was tested in three independent experiments. Ct values were normalized to the expression of the house-keeping GAPDH gene (ΔΔct method).
Statistical analysis
Statistical analysis was performed by Kruskal-Wallis one-way ANOVA and Student’s t-test using SPSS 22.0 software. Data are given as mean ± SE.
RESULTS
Establishment of hepatic cells expressing ATP7B mutations
In order to analyze the impact of various WD mutants for survival of hepatic cells, we generated stable cell lines in an ATP7B knockout (KO) human hepatoma cell line[22]. The three major ATP7B mutations reported from WD patients around the world (p.H1069Q, p.R778L and p.C271*) were chosen for this study (Supplementary Table 1)[26-30]. In addition, mutations from a recently identified cohort of Western India (p.L795F, p.T977M, p.M573fs, and p.E122fs) were also included[26]. The selected four missense and three mutations encoding incomplete reading frames are located throughout different functional domains of ATP7B (Figure 1). The presence of the desired mutations and the absence of any other aberration within ATP7B were confirmed in the cell lines by sequencing of chromosomal DNA (data not shown). In order to characterize ATP7B mRNA expression of the cell lines, real time PCR analysis was performed indicating that expression was in the same range as compared to KO cells expressing wild type ATP7B (KO.wt) (Figure 2A). To explore the stability of ATP7B protein expression in different mutants, Western blot analysis of the cell lines was performed. Cells were either maintained at 37 °C or at 30 °C prior to Western blot analysis to determine unstable ATP7B expression that is known to be augmented by lower temperature[31]. At 37 °C, only mutant cell line KO.L795F showed ATP7B expression levels similar to KO.wt cells (Figure 2B). Cell lines KO.H1069Q and KO.T977M showed a lower ATP7B protein expression. Lowest ATP7B expression was observed for cell line KO.R778L. Cell lines KO.M573fs, KO.C271* and KO.E122fs that have abrogated reading frames showed no detectableATP7B protein. Incubation of cells at 30 °C prominently increased protein expression in cell lines KO.L795F, KO.H1069Q and KO.T977M (Figure 2C). Relative increase of protein expression was highest for cell line KO.R778L, although overall expression was still low (Supplementary Table 2).
Figure 1.
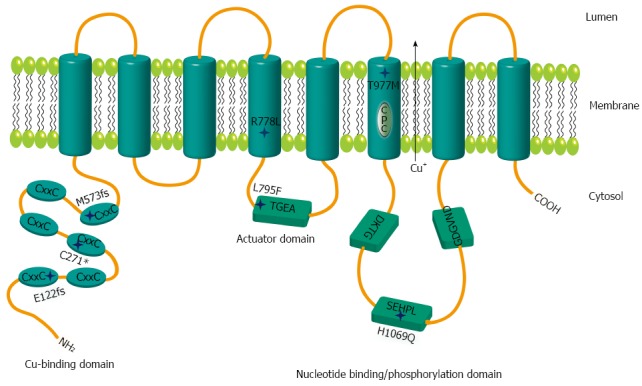
Schematic representation of mutations within ATP7B. Relative positions of the mutations are shown by stars. The six metal binding domains at the N terminal are indicated by CxxC. Sequence motifs TGEA, DKTG, SEHPL, GDGVND, and CPC depict conserved elements of copper (Cu) ATPases.
Figure 2.
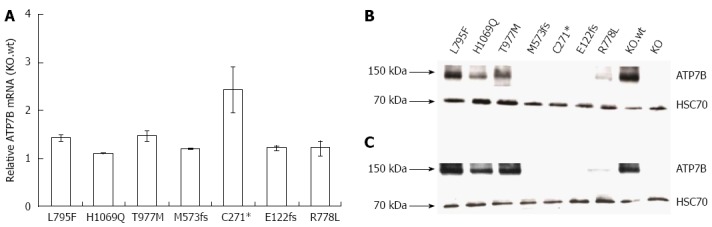
ATP7B mRNA and protein expression in mutant cell lines. A: Relative ATP7B mRNA expression in mutant cell lines was determined. The expression was normalized to GAPDH housekeeping gene. Gene expression as factor fold change relative to KO cells expressing wild type was calculated; B and C: Representative Western blots showing ATP7B protein at 37 °C and 30 °C, respectively.
ATP7B trafficking in hepatic mutant cell lines
Trafficking of ATP7B from TGN to vesicular compartments upon exposure of cells to high Cu levels has been reported[31]. We addressed whether trafficking of ATP7B in response to elevated Cu is impaired in the mutant hepatic cells[32]. Subcellular trafficking of ATP7B was determined under two different Cu conditions (Figure 3) using lamp2, a late endosome-lysosome marker[33,34]. First we established that trafficking of exogenous ATP7B in KO.wt was similar to HepG2 cells (Supplementary Figure 1). Using low Cu conditions, KO.wt cells showed a perinuclear localization of ATP7B. Upon exposure of cells to 0.1 mmol/L Cu, KO.wt cells displayed a dispersed punctate staining pattern of ATP7B distant from the perinuclear region towards the cytosolic vesicular compartment. Co-localization of ATP7B with lamp2 was predominantly observed in KO.wt cells under high Cu conditions. A similar staining pattern was observed with HepG2 cells (data not shown). In cell line KO.L795F ATP7B staining was found to be dispersed in the cytoplasm under both low and high Cu conditions, although a higher co-localization with lamp2 was observed at high Cu. As compared to KO.wt cells, no co-localization with vesicle-like structures was seen in cell line KO.L795F. ATP7B staining pattern of cell line KO.H1069Q was partially dispersed. A high overlap with lamp2 was observed. Only minor differences as compared to low Cu were observed upon exposure to high Cu. Cell line KO.T977M showed a perinuclear staining pattern of ATP7B at low Cu. Under high Cu conditions, ATP7B was somewhat dispersed with only a slight overlap with lamp2. Cell line KO.R778L showed a diffuse staining pattern suggesting an extensively mislocalized ATP7B dispersed throughout the cell. No co-localization of ATP7B was observed with lamp2 in cell line KO.R778L. The cell lines KO.M573fs, KO.C271*, KO.E122fs and KO did not show any ATP7B-specific staining (data not shown).
Figure 3.
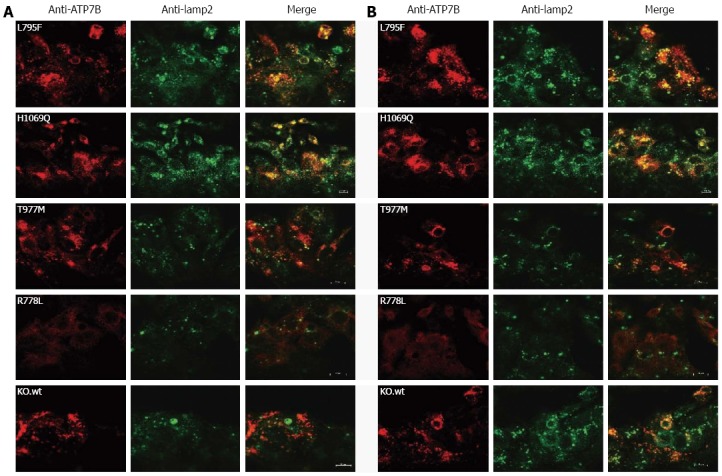
Localization of ATP7B mutant protein in stably transfected KO cells. Co-localization studies were performed with lamp2 at low copper (Cu) (A) and in the presence of 100 μmol/L Cu (B). Bars represent 10 μm. A representative photograph is shown for each mutant cell line.
Viability of hepatic mutant cell lines following copper exposure
Since most ATP7B mutant cell lines showed defects in trafficking, we further assessed whether expression of ATP7B mutations can lead to escape of cells from toxic Cu. The viability of the cell lines at various Cu concentrations was determined (Figure 4). Cell line KO.L795F showed high viability at 0.25 mmol/L Cu (89.0% ± 1%) which slowly decreased to 16.9% ± 5% at 1.5 mmol/L Cu concentration. Cell lines KO.H1069Q and KO.T977M displayed lower levels of Cu resistance with a viability of 78.2% ± 3% and 47.3% ± 2%, respectively at 0.25 mmol/L Cu. Viability significantly dropped in these mutants below 2.0% ± 1% at highest Cu concentration. In contrast, the three mutant cell lines encoding incomplete reading frames and KO.R778L showed Cu resistance similar to KO cells[22] with values below 16.2% ± 5% at 0.25 mmol/L Cu that further dropped to almost 0.0% at higher Cu concentrations. Values calculated for IC50 (Supplementary Table 2) suggested that only cell lines KO.L795F, KO.H1069Q and KO.T977M displayed significant survival after toxic Cu exposure.
Figure 4.
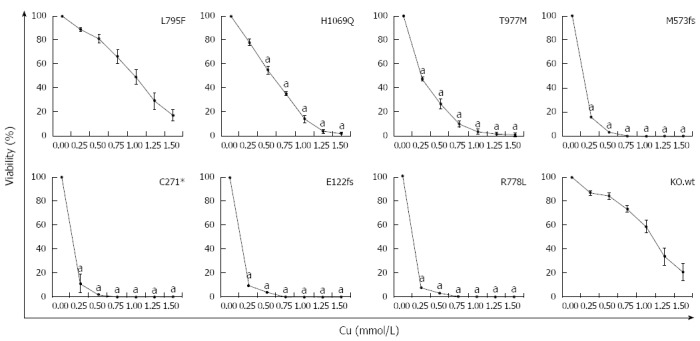
Survival of ATP7B mutant cell lines after copper exposure. Cell survival of ATP7B mutant cell lines relative to control (no copper) was determined by MTT assay after 48 h copper (Cu) exposure. Data is represented as mean ± SE of at least three independent experiments. Significance (aP < 0.05) as compared to KO.wt.
Induction of apoptosis
Cu is also known as an inducer of apoptosis[35]. Having shown that WD mutations affect cell survival, we assessed induction of apoptosis following Cu exposure (Figure 5). Experiments were carried out at 0.1 mmol/L Cu since significant necrosis was observed at higher Cu concentrations (data not shown). KO.wt and HepG2 cells showed similar rates of apoptosis (Supplementary Figure 2). Induction of apoptosis was low (11.1% ± 1%) for cell line KO.L795F underscoring that cell line KO.L795F can resist high concentrations of Cu as shown above. All other cell lines displayed higher rates of apoptosis (range 17.1% ± 1% to 24.4% ± 3%) suggesting that although cell lines KO.H1069Q and KO.T977M can escape high Cu, significant levels of apoptosis are induced by Cu.
Figure 5.
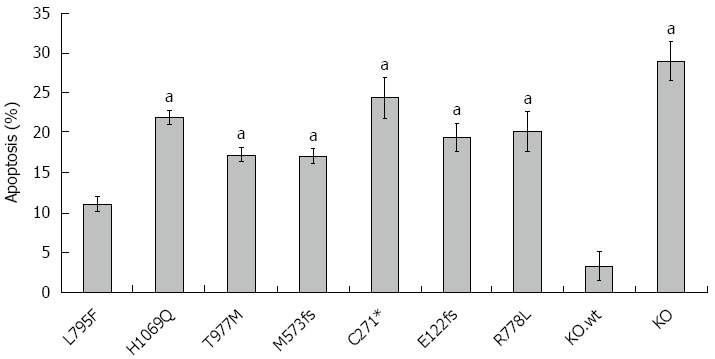
Rate of apoptosis in ATP7B mutant cell lines after copper exposure. Cells were exposed to 100 μmol/L Cu for 24 h. Induction of apoptosis was determined by Annexin-V staining followed by flow cytometry analysis. Data is represented as mean ± SE of at least three independent experiments. aP < 0.05 vs KO cells expressing wild type ATP7B.
Zinc and DPA treatment of mutant cell lines
We addressed whether the individual rates of survival and apoptosis observed in the ATP7B mutant cell lines may also have an impact on the response to Zn and DPA treatment. In order to determine the treatment response for different ATP7B mutants, KO mutant cell lines were exposed to Zn, DPA and Zn + DPA at different Cu concentrations (Figure 6). Upon treatment, all mutant cell lines showed a significant higher survival at Cu concentrations ≥ 0.25 mmol/L (mean 55.1% ± 3%; median 63.1%) as compared to untreated control (mean 18.1% ± 4%; median 3.6 %) indicating that all three treatments are highly efficient. Overall, Zn treatment alone was less effective (mean 35.6% ± 5%) and significantly outperformed by DPA treatment (mean 57.8% ± 5 %). Combined treatment Zn + DPA resulted in highest cell survival (mean 69.7% ± 2 %) suggesting a synergistic effect of both modalities as compared to single treatments as shown before for KO cells[22].
Figure 6.
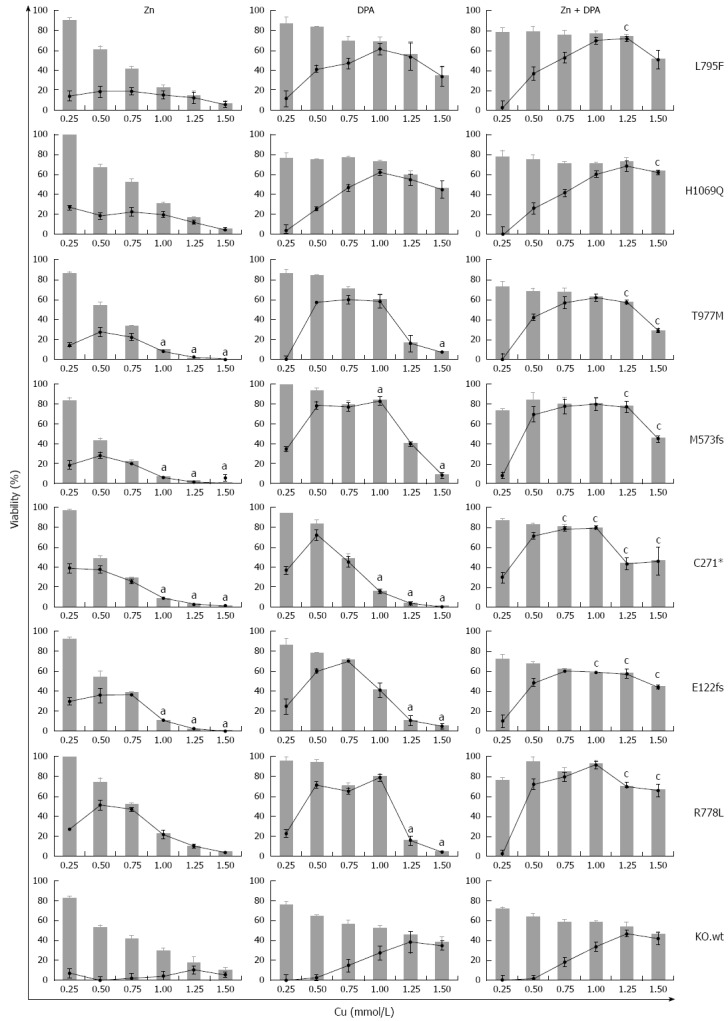
Survival of ATP7B mutant cell lines after zinc and D-penicillamine treatment. Cells were treated with zinc (Zn), D-penicillamine (DPA), and Zn + DPA followed by determination of viability at 48 h. Bar graphs indicate cell viability after treatment relative to untreated cells [0 mmol/L copper (Cu)]. Relative increase of viability by individual treatment (gain) is indicated by line graph. Data is represented as mean ± SE of at least three independent experiments. Viability of KO cells expressing ATP7B wild type is shown as control. aP < 0.05 vs KO.wt cells, cP < 0.05 vs DPA treatment.
Some of the mutations showed unique response rates to the treatments. Relative increase of viability by Zn treatment (gain) was highest for mutant cell line KO.R778L (mean 26.9% ± 8%). Following DPA treatment, only cell lines KO.L795F and KO.H1069Q showed full restoration of viability as compared to KO.wt cells. Relative increase of viability by DPA treatment was mostly gradual (≤ 1.0 mmol/L Cu) for cell lines KO.L795F and KO.H1069Q whereas most of the other mutant cell lines, in particular KO.M573fs, showed a plateau of high values (> 65%) at Cu concentrations of 0.5-1.0 mmol/L Cu. Relative increase of viability by combination as compared to DPA treatment was highest in cell line KO.C271* (mean 57.8% ± 6% and 29.2% ± 11%, respectively).
DISCUSSION
Our results indicate that the analysis of hepatic cell lines resulted in a functional classification of WD mutations. Mutant cell lines showed different degrees of cell survival and characteristic responses upon treatment with Zn and DPA. The findings provide first in vitro evidence that survival of hepatic cells following anti-copper treatment may depend on the ATP7B mutation, an observation that may further lead to a better prognosis of unknown WD mutants and improved knowledge on WD genotype to phenotype associations.
HepG2 cells are widely used for analysis of Cu toxicity and apoptosis[36,37]. By using ATP7B KO cells derived from HepG2[22], we assessed viability of hepatic cells without interference by intrinsic ATP7B or impairment by hybrid protein sequences[31,37] reflecting a situation close to human WD hepatocytes. Mutant p.H1069Q was observed in this study to have a moderately reduced impact on cell survival underlining previous results obtained in other systems[17,37]. Localisation of protein p.H1069Q was reported to be close to the ER[17,31,37]. The observed stabilization of p.H1069Q protein expression in hepatic cells by low temperature confirmed previous results obtained in human kidney cells[31] suggesting that improper folding of the mutant protein is not cell type specific. Mutant p.R778L did not show detectable levels of cell survival in contrast to the other missense mutants underscoring that it may be difficult to predict activity from sequence analysis alone. ATP7B expression and improvement of survival were not detected for the three mutations encoding incomplete reading frames underlining previous observations[16,17,38].
Based on cell survival, WD mutants could be classified into groups having high (p.L795F, p.H1069Q), moderate (p.T977M), and no survival (p.M573fs, p.C271*, p.E122fs and p.R778L). Homozygous mutation p.L795F was associated in WD patients from India with late onset and neurological disease[26]. For mutation p.H1069Q an association with late and neurological presentation has been observed[8,30,39,40]. A high cell survival may thus be associated to a relatively late, predominantly neurologic phenotype, possibly due to the almost wild type activity of ATP7B that can prevent severe disease, at least in the early years. Of note, in two patients having compound heterozygous mutation p.L795F together with frameshift mutation p.M769fs, late onset of disease was significantly reduced[26] suggesting that one allele of mutation p.L795F is sufficient to prevent early onset of disease when combined with a putative non-functional mutation. Homozygous mutation p.T977M was observed in Indian WD patients who had an early onset of disease (around year 4) predominantly affecting liver and resulting in moderate neurological impairment[26]. In European WD patients, mutation p.T977M was however relatively mild[41]. On the other hand, ATP7B frameshift and nonsense mutations have been implicated to result in early onset and a more severe manifestation of WD[42-44]. The phenotypes that were associated with homozygous mutations p.M573fs, p.C271* and p.E122fs are mostly severe including high disease burden and relatively early onset of diseases, at least for p.M573fs and p.C271* (around years 7-10). Overall disease burden according to the Tier 1 score[26] was highest for mutation p.E122fs within this group, except that osseomuscular disease was completely missing in patients having this mutation. Mutation p.R778L has a high frequency in WD patients from Asia, especially in China, Japan, South Korea and was associated with severe hepatic disease and with a relatively early onset (about 10 years)[9,45-49], however, a later onset of disease mostly associated with cerebral symptoms was noted in a different cohort[50]. In addition to the common genotypes, there are an increasing number of novel genotypes identified, especially in China, which may benefit from genotype-specific therapy approaches[10,12].
Zn and DPA are effective treatments for WD[51,52] and are implemented in current guidelines for treatment. While DPA is reducing overall Cu load in the body due to chelation, the first line defence of Zn is thought to take place in the intestine where Cu is expelled via metallothionein. The mechanism of both drugs at the level of the hepatocyte has been explored[24,53], however, to the best of our knowledge this is the first study on Zn and DPA in human hepatic cells expressing mutant ATP7B. The results indicate that the survival of hepatic cells following treatment might depend on the genotype of ATP7B. With the exception of mutant p.R778L, efficacy of Zn treatment for hepatic cells is mostly confined to low Cu concentrations. Previous data indicate that metallothionein is induced in hepatic cells even when ATP7B is absent[22] suggesting that high metallothionein expression is insufficient for rescue of cells.
Our study indicates as shown before for KO cells[22] that combined treatment by Zn and DPA resulted in the highest survival of hepatic cells suggesting that a synergism by both drugs is needed to resist maximal liver Cu concentrations that can be up to 3 mg/g dry weight[1]. While our own experience with combined treatment is limited to small WD patient cohorts and standardized treatment regimen has not been established, a possible translation of our results awaits further in vivo studies and regimens that minimize interference of both drugs[18,51,54]. Notably, in support of our in vitro findings WD patients having mutation p.R778L were observed to benefit largely from Zn monotherapy corroborating the importance of Zn treatment in different Asian countries, including China[20,21,55]. In addition, our finding of a general reduced hepatic cell survival after Zn treatment as compared to DPA is corroborated by clinical findings from large WD cohorts[19,52] and also from Indian patients (AA, MB, unpublished data). Apart from the WD genotype, many variables are likely to contribute to the phenotype and drug response, including the genetic background in different populations, diet, type of disease manifestation, and local preferences of anti-Cu treatment regimens. In addition, combination of the two drugs in vitro and in vivo may act by different mechanism. Given the limitations of our in vitro study, functional characterization of cells following Cu exposure and Zn/DPA treatment may however indicate the basic impact of a given WD genotype.
ACKNOWLEDGMENTS
We thank Oksana Nadzemova for technical support and Alexander Zibert for mathematical analysis.
COMMENTS
Background
Wilson disease (WD) is a rare autosomal recessive disorder of Cu accumulation, especially in the liver and brain, where Cu toxicity results in a varying presentation of liver disease, neurological, and psychiatric conditions. Lifelong treatment currently involves Cu chelating compounds and zinc salts. A standard anti-Cu treatment has not been established for therapy of WD. The rarity of disease and the high number of compound heterozygote mutations aggravate genotype to phenotype correlations.
Research frontiers
Specific ATP7B mutations are often found to be concentrated in certain geographical populations. Over 600 different mutations of ATP7B have so far been identified, and it is thought - although not proven - that the range of symptoms presented by individual WD patients and the response to therapy has partly a genetic basis.
Innovations and breakthroughs
Previous work allowed the functional characterization of WD genotypes in different in vitro cellular systems. In this report, using human hepatic cells that have no intrinsic ATP7B expression, the impact of the WD genotype with regard to treatment by standard anti-Cu drugs was addressed. Unique insights into the genotype-phenotype correlations of the most prominent WD mutations that refer to more than 50% of patients in some regions of the world are presented.
Applications
Findings on a WD genotype-specific response of hepatic cells to anti-Cu treatment are important to stimulate further clinical and molecular studies to assess genotype to phenotype correlations, genotype-specific treatment regimens, and the prognosis of novel genotypes.
Terminology
ATP7B mutations have been studied in various mammalian tissue culture cells, however, intrinsic ATP7B expression or non-hepatic cells have blurred a direct correlation of the results. The study of primary hepatocytes from WD patients is difficult, since such cells cannot be propagated in tissue culture. Hepatic cells expressing homozygote WD mutations may represent a novel platform to study the impact of individual mutants.
Peer-review
Interesting paper with the novel idea how to learn more about genotype/phenotype correlations and potential genotype influence on reaction to therapy in WD. Presented results provide for a first time that the response of hepatic cells depends on the WD mutation, most interesting after treatment with penicillamine and zinc.
Footnotes
Conflict-of-interest statement: The authors declare no conflict of interest associated with this manuscript.
Data sharing statement: No additional data are available.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: December 7, 2015
First decision: January 13, 2016
Article in press: February 22, 2016
P- Reviewer: Czlonkowska A, Pan JJ S- Editor: Yu J L- Editor: A E- Editor: Wang CH
References
- 1.Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson’s disease. Lancet. 2007;369:397–408. doi: 10.1016/S0140-6736(07)60196-2. [DOI] [PubMed] [Google Scholar]
- 2.Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327–337. doi: 10.1038/ng1293-327. [DOI] [PubMed] [Google Scholar]
- 3.Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B, Romano DM, Parano E, Pavone L, Brzustowicz LM. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993;5:344–350. doi: 10.1038/ng1293-344. [DOI] [PubMed] [Google Scholar]
- 4.Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007;87:1011–1046. doi: 10.1152/physrev.00004.2006. [DOI] [PubMed] [Google Scholar]
- 5.Brady DC, Crowe MS, Turski ML, Hobbs GA, Yao X, Chaikuad A, Knapp S, Xiao K, Campbell SL, Thiele DJ, et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature. 2014;509:492–496. doi: 10.1038/nature13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinelli D, Travaglini L, Drouin CA, Ceballos-Picot I, Rizza T, Bertini E, Carrozzo R, Petrini S, de Lonlay P, El Hachem M, et al. MEDNIK syndrome: a novel defect of copper metabolism treatable by zinc acetate therapy. Brain. 2013;136:872–881. doi: 10.1093/brain/awt012. [DOI] [PubMed] [Google Scholar]
- 7.Wu F, Wang J, Pu C, Qiao L, Jiang C. Wilson’s disease: a comprehensive review of the molecular mechanisms. Int J Mol Sci. 2015;16:6419–6431. doi: 10.3390/ijms16036419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferenci P, Członkowska A, Merle U, Ferenc S, Gromadzka G, Yurdaydin C, Vogel W, Bruha R, Schmidt HT, Stremmel W. Late-onset Wilson’s disease. Gastroenterology. 2007;132:1294–1298. doi: 10.1053/j.gastro.2007.02.057. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Zhou H, Guo H, Bai Y. Genetic and Clinical Analysis in a Cohort of Patients with Wilson’s Disease in Southwestern China. Arch Med Res. 2015;46:164–169. doi: 10.1016/j.arcmed.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Li XH, Lu Y, Ling Y, Fu QC, Xu J, Zang GQ, Zhou F, De-Min Y, Han Y, Zhang DH, et al. Clinical and molecular characterization of Wilson’s disease in China: identification of 14 novel mutations. BMC Med Genet. 2011;12:6. doi: 10.1186/1471-2350-12-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li K, Zhang WM, Lin S, Wen L, Wang ZF, Xie D, Wei M, Qiu ZQ, Dai Y, Lin MC, et al. Mutational analysis of ATP7B in north Chinese patients with Wilson disease. J Hum Genet. 2013;58:67–72. doi: 10.1038/jhg.2012.134. [DOI] [PubMed] [Google Scholar]
- 12.Wang LH, Huang YQ, Shang X, Su QX, Xiong F, Yu QY, Lin HP, Wei ZS, Hong MF, Xu XM. Mutation analysis of 73 southern Chinese Wilson’s disease patients: identification of 10 novel mutations and its clinical correlation. J Hum Genet. 2011;56:660–665. doi: 10.1038/jhg.2011.76. [DOI] [PubMed] [Google Scholar]
- 13.Barada K, El-Atrache M, El-Hajj II, Rida K, El-Hajjar J, Mahfoud Z, Usta J. Homozygous mutations in the conserved ATP hinge region of the Wilson disease gene: association with liver disease. J Clin Gastroenterol. 2010;44:432–439. doi: 10.1097/MCG.0b013e3181ce5138. [DOI] [PubMed] [Google Scholar]
- 14.Wu ZY, Wang N, Lin MT, Fang L, Murong SX, Yu L. Mutation analysis and the correlation between genotype and phenotype of Arg778Leu mutation in chinese patients with Wilson disease. Arch Neurol. 2001;58:971–976. doi: 10.1001/archneur.58.6.971. [DOI] [PubMed] [Google Scholar]
- 15.Braiterman LT, Murthy A, Jayakanthan S, Nyasae L, Tzeng E, Gromadzka G, Woolf TB, Lutsenko S, Hubbard AL. Distinct phenotype of a Wilson disease mutation reveals a novel trafficking determinant in the copper transporter ATP7B. Proc Natl Acad Sci USA. 2014;111:E1364–E1373. doi: 10.1073/pnas.1314161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huster D, Kühne A, Bhattacharjee A, Raines L, Jantsch V, Noe J, Schirrmeister W, Sommerer I, Sabri O, Berr F, et al. Diverse functional properties of Wilson disease ATP7B variants. Gastroenterology. 2012;142:947–956.e5. doi: 10.1053/j.gastro.2011.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Payne AS, Kelly EJ, Gitlin JD. Functional expression of the Wilson disease protein reveals mislocalization and impaired copper-dependent trafficking of the common H1069Q mutation. Proc Natl Acad Sci USA. 1998;95:10854–10859. doi: 10.1073/pnas.95.18.10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schilsky ML. Treatment of Wilson’s disease: what are the relative roles of penicillamine, trientine, and zinc supplementation? Curr Gastroenterol Rep. 2001;3:54–59. doi: 10.1007/s11894-001-0041-4. [DOI] [PubMed] [Google Scholar]
- 19.Weiss KH, Gotthardt DN, Klemm D, Merle U, Ferenci-Foerster D, Schaefer M, Ferenci P, Stremmel W. Zinc monotherapy is not as effective as chelating agents in treatment of Wilson disease. Gastroenterology. 2011;140:1189–1198.e1. doi: 10.1053/j.gastro.2010.12.034. [DOI] [PubMed] [Google Scholar]
- 20.Abuduxikuer K, Wang JS. Zinc mono-therapy in pre-symptomatic Chinese children with Wilson disease: a single center, retrospective study. PLoS One. 2014;9:e86168. doi: 10.1371/journal.pone.0086168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ni W, Dong QY, Zhang Y, Wu ZY. Zinc monotherapy and a low-copper diet are beneficial in patients with Wilson disease after liver transplantation. CNS Neurosci Ther. 2013;19:905–907. doi: 10.1111/cns.12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandhok G, Schmitt N, Sauer V, Aggarwal A, Bhatt M, Schmidt HH. The effect of zinc and D-penicillamine in a stable human hepatoma ATP7B knockout cell line. PLoS One. 2014;9:e98809. doi: 10.1371/journal.pone.0098809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sauer V, Siaj R, Todorov T, Zibert A, Schmidt HH. Overexpressed ATP7B protects mesenchymal stem cells from toxic copper. Biochem Biophys Res Commun. 2010;395:307–311. doi: 10.1016/j.bbrc.2010.03.158. [DOI] [PubMed] [Google Scholar]
- 24.Schilsky ML, Blank RR, Czaja MJ, Zern MA, Scheinberg IH, Stockert RJ, Sternlieb I. Hepatocellular copper toxicity and its attenuation by zinc. J Clin Invest. 1989;84:1562–1568. doi: 10.1172/JCI114333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siaj R, Sauer V, Stöppeler S, Spiegel HU, Köhler G, Zibert A, Schmidt HH. Dietary copper triggers onset of fulminant hepatitis in the Long-Evans cinnamon rat model. World J Gastroenterol. 2012;18:5542–5550. doi: 10.3748/wjg.v18.i39.5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aggarwal A, Chandhok G, Todorov T, Parekh S, Tilve S, Zibert A, Bhatt M, Schmidt HH. Wilson disease mutation pattern with genotype-phenotype correlations from Western India: confirmation of p.C271* as a common Indian mutation and identification of 14 novel mutations. Ann Hum Genet. 2013;77:299–307. doi: 10.1111/ahg.12024. [DOI] [PubMed] [Google Scholar]
- 27.Chuang LM, Wu HP, Jang MH, Wang TR, Sue WC, Lin BJ, Cox DW, Tai TY. High frequency of two mutations in codon 778 in exon 8 of the ATP7B gene in Taiwanese families with Wilson disease. J Med Genet. 1996;33:521–523. doi: 10.1136/jmg.33.6.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Curtis D, Durkie M, Balac (Morris) P, Sheard D, Goodeve A, Peake I, Quarrell O, Tanner S. A study of Wilson disease mutations in Britain. Hum Mutat. 1999;14:304–311. doi: 10.1002/(SICI)1098-1004(199910)14:4<304::AID-HUMU5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 29.Shah AB, Chernov I, Zhang HT, Ross BM, Das K, Lutsenko S, Parano E, Pavone L, Evgrafov O, Ivanova-Smolenskaya IA, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet. 1997;61:317–328. doi: 10.1086/514864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stapelbroek JM, Bollen CW, van Amstel JK, van Erpecum KJ, van Hattum J, van den Berg LH, Klomp LW, Houwen RH. The H1069Q mutation in ATP7B is associated with late and neurologic presentation in Wilson disease: results of a meta-analysis. J Hepatol. 2004;41:758–763. doi: 10.1016/j.jhep.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 31.van den Berghe PV, Stapelbroek JM, Krieger E, de Bie P, van de Graaf SF, de Groot RE, van Beurden E, Spijker E, Houwen RH, Berger R, et al. Reduced expression of ATP7B affected by Wilson disease-causing mutations is rescued by pharmacological folding chaperones 4-phenylbutyrate and curcumin. Hepatology. 2009;50:1783–1795. doi: 10.1002/hep.23209. [DOI] [PubMed] [Google Scholar]
- 32.Forbes JR, Cox DW. Copper-dependent trafficking of Wilson disease mutant ATP7B proteins. Hum Mol Genet. 2000;9:1927–1935. doi: 10.1093/hmg/9.13.1927. [DOI] [PubMed] [Google Scholar]
- 33.Polishchuk EV, Concilli M, Iacobacci S, Chesi G, Pastore N, Piccolo P, Paladino S, Baldantoni D, van IJzendoorn SC, Chan J, et al. Wilson disease protein ATP7B utilizes lysosomal exocytosis to maintain copper homeostasis. Dev Cell. 2014;29:686–700. doi: 10.1016/j.devcel.2014.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jain S, Farías GG, Bonifacino JS. Polarized sorting of the copper transporter ATP7B in neurons mediated by recognition of a dileucine signal by AP-1. Mol Biol Cell. 2015;26:218–228. doi: 10.1091/mbc.E14-07-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lang PA, Schenck M, Nicolay JP, Becker JU, Kempe DS, Lupescu A, Koka S, Eisele K, Klarl BA, Rübben H, et al. Liver cell death and anemia in Wilson disease involve acid sphingomyelinase and ceramide. Nat Med. 2007;13:164–170. doi: 10.1038/nm1539. [DOI] [PubMed] [Google Scholar]
- 36.Strand S, Hofmann WJ, Grambihler A, Hug H, Volkmann M, Otto G, Wesch H, Mariani SM, Hack V, Stremmel W, et al. Hepatic failure and liver cell damage in acute Wilson’s disease involve CD95 (APO-1/Fas) mediated apoptosis. Nat Med. 1998;4:588–593. doi: 10.1038/nm0598-588. [DOI] [PubMed] [Google Scholar]
- 37.Huster D, Hoppert M, Lutsenko S, Zinke J, Lehmann C, Mössner J, Berr F, Caca K. Defective cellular localization of mutant ATP7B in Wilson’s disease patients and hepatoma cell lines. Gastroenterology. 2003;124:335–345. doi: 10.1053/gast.2003.50066. [DOI] [PubMed] [Google Scholar]
- 38.Wan L, Tsai CH, Hsu CM, Huang CC, Yang CC, Liao CC, Wu CC, Hsu YA, Lee CC, Liu SC, et al. Mutation analysis and characterization of alternative splice variants of the Wilson disease gene ATP7B. Hepatology. 2010;52:1662–1670. doi: 10.1002/hep.23865. [DOI] [PubMed] [Google Scholar]
- 39.Figus A, Angius A, Loudianos G, Bertini C, Dessi V, Loi A, Deiana M, Lovicu M, Olla N, Sole G. Molecular pathology and haplotype analysis of Wilson disease in Mediterranean populations. Am J Hum Genet. 1995;57:1318–1324. [PMC free article] [PubMed] [Google Scholar]
- 40.Houwen RH, Juyn J, Hoogenraad TU, Ploos van Amstel JK, Berger R. H714Q mutation in Wilson disease is associated with late, neurological presentation. J Med Genet. 1995;32:480–482. doi: 10.1136/jmg.32.6.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waldenström E, Lagerkvist A, Dahlman T, Westermark K, Landegren U. Efficient detection of mutations in Wilson disease by manifold sequencing. Genomics. 1996;37:303–309. doi: 10.1006/geno.1996.0564. [DOI] [PubMed] [Google Scholar]
- 42.Gromadzka G, Schmidt HH, Genschel J, Bochow B, Rodo M, Tarnacka B, Litwin T, Chabik G, Członkowska A. Frameshift and nonsense mutations in the gene for ATPase7B are associated with severe impairment of copper metabolism and with an early clinical manifestation of Wilson’s disease. Clin Genet. 2005;68:524–532. doi: 10.1111/j.1399-0004.2005.00528.x. [DOI] [PubMed] [Google Scholar]
- 43.Merle U, Eisenbach C, Weiss KH, Tuma S, Stremmel W. Serum ceruloplasmin oxidase activity is a sensitive and highly specific diagnostic marker for Wilson’s disease. J Hepatol. 2009;51:925–930. doi: 10.1016/j.jhep.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 44.Panagiotakaki E, Tzetis M, Manolaki N, Loudianos G, Papatheodorou A, Manesis E, Nousia-Arvanitakis S, Syriopoulou V, Kanavakis E. Genotype-phenotype correlations for a wide spectrum of mutations in the Wilson disease gene (ATP7B) Am J Med Genet A. 2004;131:168–173. doi: 10.1002/ajmg.a.30345. [DOI] [PubMed] [Google Scholar]
- 45.Liu XQ, Zhang YF, Liu TT, Hsiao KJ, Zhang JM, Gu XF, Bao KR, Yu LH, Wang MX. Correlation of ATP7B genotype with phenotype in Chinese patients with Wilson disease. World J Gastroenterol. 2004;10:590–593. doi: 10.3748/wjg.v10.i4.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim EK, Yoo OJ, Song KY, Yoo HW, Choi SY, Cho SW, Hahn SH. Identification of three novel mutations and a high frequency of the Arg778Leu mutation in Korean patients with Wilson disease. Hum Mutat. 1998;11:275–278. doi: 10.1002/(SICI)1098-1004(1998)11:4<275::AID-HUMU4>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 47.Okada T, Shiono Y, Hayashi H, Satoh H, Sawada T, Suzuki A, Takeda Y, Yano M, Michitaka K, Onji M, et al. Mutational analysis of ATP7B and genotype-phenotype correlation in Japanese with Wilson’s disease. Hum Mutat. 2000;15:454–462. doi: 10.1002/(SICI)1098-1004(200005)15:5<454::AID-HUMU7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 48.Gu YH, Kodama H, Du SL, Gu QJ, Sun HJ, Ushijima H. Mutation spectrum and polymorphisms in ATP7B identified on direct sequencing of all exons in Chinese Han and Hui ethnic patients with Wilson’s disease. Clin Genet. 2003;64:479–484. doi: 10.1046/j.1399-0004.2003.00179.x. [DOI] [PubMed] [Google Scholar]
- 49.Wu Z, Wang N, Murong S, Lin M. Identification and analysis of mutations of the Wilson disease gene in Chinese population. Chin Med J (Engl) 2000;113:40–43. [PubMed] [Google Scholar]
- 50.Fan Y, Yu L, Jiang Y, Xu Y, Yang R, Han Y, Cui Y, Ren M, Zhao S. Identification of a mutation hotspot in exon 8 of Wilson disease gene by cycle sequencing. Chin Med J (Engl) 2000;113:172–174. [PubMed] [Google Scholar]
- 51.Chang H, Xu A, Chen Z, Zhang Y, Tian F, Li T. Long-term effects of a combination of D-penicillamine and zinc salts in the treatment of Wilson’s disease in children. Exp Ther Med. 2013;5:1129–1132. doi: 10.3892/etm.2013.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weiss KH, Thurik F, Gotthardt DN, Schäfer M, Teufel U, Wiegand F, Merle U, Ferenci-Foerster D, Maieron A, Stauber R, et al. Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin Gastroenterol Hepatol. 2013;11:1028–35.e1-2. doi: 10.1016/j.cgh.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 53.McQuaid A, Lamand M, Mason J. The interactions of penicillamine with copper in vivo and the effect on hepatic metallothionein levels and copper/zinc distribution: the implications for Wilson’s disease and arthritis therapy. J Lab Clin Med. 1992;119:744–750. [PubMed] [Google Scholar]
- 54.Brewer GJ. Zinc and tetrathiomolybdate for the treatment of Wilson’s disease and the potential efficacy of anticopper therapy in a wide variety of diseases. Metallomics. 2009;1:199–206. doi: 10.1039/b901614g. [DOI] [PubMed] [Google Scholar]
- 55.Wu ZY, Lin MT, Murong SX, Wang N. Molecular diagnosis and prophylactic therapy for presymptomatic Chinese patients with Wilson disease. Arch Neurol. 2003;60:737–741. doi: 10.1001/archneur.60.5.737. [DOI] [PubMed] [Google Scholar]