

Abstract
Generation of a CD8+ response to extracellular antigen requires processing of the antigen by antigen presenting cells (APC) and cross-presentation to CD8+ T cell receptors via MHC class I molecules. Cross-presentation is facilitated by efficient antigen uptake followed by immune-complex-mediated maturation of the APCs. We hypothesize that improved antigen uptake of a glycopeptide sequence containing a CD8+ T cell epitope could be achieved by delivering it on a liposome surface decorated with an immune complex-targeting ligand, an L-Rhamnose (Rha) epitope. We synthesized a 20 amino acid glycopeptide TSAPDT(GalNAc)RPAPGSTAPPAHGV from the variable number tandem repeat region of the tumor marker MUC1 containing an N-terminal azido moiety and a tumor-associated α-N-acetyl galactosamine (GalNAc) at the immunogenic DTR motif. The MUC1 antigen was attached to Pam3Cys, a Toll-like receptor-2 ligand via copper (I)-catalyzed azido-alkyne cycloaddtion (CuAAc) chemistry. The Rha-decorated liposomal Pam3Cys-MUC1-Tn 4 vaccine was evaluated in groups of C57BL/6 mice. Some groups were previously immunized to generate anti-Rha antibodies. Anti-Rha antibody expressing mice that received the Rha liposomal vaccine showed higher cellular immunogenicity compared to the control group while maintaining a strong humoral response.
Graphical Abstract
Introduction
Detection of tumors or viral infections is always challenging to the immune system because the virus or the disregulated oncogene hides inside the autologous cells, making the humoral immune mechanism inactive against them.1 This problem is solved by the major histocompatibility complex class I (MHC I) antigen processing mechanism.1 Generally, MHC I molecules display autologous peptides which are ignored by the immune system, but during viral infection or in the course of neoplast formation, the respective peptide displayed on MHC I is recognized by the receptors of CD8+ T cells and they are stimulated to produce an immune response.1, 2 So, in order to make an antitumor vaccine more effective, the antigen has to be optimally presented to CD8+ T cells by MHC I molecules.3 Unfortunately, such antigens are often too poorly presented by the MHC I molecules to induce strong CD8+ T cell responses. To overcome this problem, liposomes have been successfully used as carriers of a range of viral, bacterial and tumor antigens, and their efficient delivery to the antigen presenting cells (APCs) by this method is also well known.4–9
Mucins have become attractive targets for potential development of antitumor immunotherapies because in various adenocarcinomas and in premalignant lesions leading to these cancers, human mucin 1 (MUC1) reaches the cell surface aberrantly glycosylated. Cancer derived-MUC1 molecules contain a GalNAcα1-Ser/Thr modification (Tn antigen), and other truncated O-glycans due to a lack of core 1,3-galactosyltransferase.10 The changed glycosylation exposes immunogenic peptide epitopes to the immune system. These peptide and glycopeptide antigens can be processed and presented to the immune system to induce MUC1-specific cytotoxic T lymphocytes (CTLs).11 Some examples of cancers that contain the MUC1 modification are breast, lung, ovary, colon, rectum and pancreatic adenocarcinomas.12–15 Further, CTLs specific for epitopes on MUC1 have been found in patients with breast,16 ovarian,17 and pancreatic18 cancers and MUC1-specific antibodies are associated with a survival benefit.19–21 As a result, a 2009 report from the National Cancer Institute (NCI) ranked MUC1 second out of 75 tumor-associated antigens on the basis of their therapeutic function, immunogenicity, specificity, oncogenicity, expression levels and % positive cells, stem cell expression, number of patients with antigen positive cancers, number of epitopes and cellular location of expression.22 Based on these reports, numerous laboratories have explored innovative approaches to increasing the immunogenicity of MUC1 to develop an immunotherapeutic that could kill cancer cells or even be used in the context of immunoprevention.23–35 Goydos et al. demonstrated that a synthetic MUC1-based vaccine could safely be used in humans36 and a phase I clinical study was reported with a 100 amino acid long MUC1-based vaccine against advanced pancreas and bile duct cancer.37, 38 In addition, MUC1 peptide pulsed DCs have been used safely as a vaccine in humans with advanced metastatic breast and ovarian cancer.39
Dendritic cells (DCs), which are some of the most important APCs, have the ability to take up exogenously encountered antigens by phagocytosis or by receptor-mediated endocytosis process, and present the antigens via MHC I molecules to CD8+ T cells in a process called antigen cross-presentation.3, 40 The Fc receptors (FcRs) on the surface of the DCs play a major role in antigen uptake and processing. For example, Fcγ receptors (FcγRs) binding to immune complexes facilitate immune-complex-mediated maturation of the DCs, resulting in presentation of antigens to CD4+ helper T cells on MHC II, presentation of antigens to CD8+ cytotoxic T cells on MHC I followed by CD8+ T cell activation, and presentation of antigens to regulatory T cells.41 Herein, we attempt to develop a MUC1-based tumor immunotherapeutic with an effector CD8+ T cell epitope as well as B cell and CD4+ T cell epitopes. We incorporated these elements on a liposome surface that would trigger FcR-mediated uptake as well as produce anti-MUC1 antibodies. The latter antibodies can also bind tumor cells and kill them through antibody dependent cell-mediated cytotoxicity (ADCC), again, mediated through FcγRs on myeloid cells.42
It is widely recognized that abundant and specific natural antibodies for a number of foreign antigens are widely distributed in the human population. One of these is the Galα1-3Galβ1-4GlcNAc-R (α-Gal) epitope.43, 44 Galili and co-workers have shown that in vivo interaction of α-Gal epitopes (in model vaccines against HIV gp120 and flu virus) with naturally occurring anti-α-Gal antibodies in 1,3-galactosyltransferase knockout mice leads to anti-α-Gal antibody-dependent antigen uptake, mediated through FcγRs, that resulted in a greater than 100-fold increase in immunogenicity.45, 46 Studies by Bovin, Gildersleeve and others on human serum identified additional naturally-occurring anti-carbohydrate antibodies.47–49 From those studies, we became interested in antibodies specific for the xenoantigen Rha. Anti-Rha antibodies were among the most abundant in humans and can also be generated in readily available non-transgenic mice.9, 50, 51 In fact, Rha is now one of the most effective natural antibody eliciting molecules known52 and Rha has been conjugated to lipids to recruit anti-Rha antibodies to tumor cells.53, 54
Previously, we have shown that the anti-Rha antibodies generated in mice can recognize a Rha moiety carried by a MUC1 antigen, and on a MUC1-based liposomal vaccine leading to better MHC class II presentation to CD4+ T cells.9, 50 In these studies, we asked whether bringing the anti-Rha antibodies into action via an anti-Rha–FcγR mediated mechanism41 would increase the cross-presentation of MUC1 epitopes on MHC I by DCs. In addition to IgM antibodies, natural anti-Rha antibodies are mostly IgG3 in the mouse9 or mostly IgG2 in humans.52 An advantage is that these IgG isotypes generally bind poorly to inhibitory Fc receptors, thus enhancing the immune response.55 To explore this hypothesis we delivered the antigen incorporated on a liposome surface.
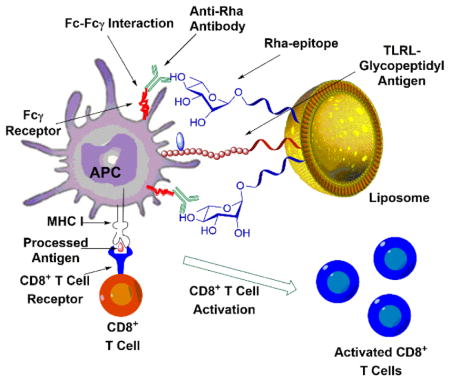
In these studies we show that a glycosylated MUC1-derived glycopeptide anchored in a liposome decorated with Rha epitopes elicits both an improved humoral and cellular immune response in mice previously vaccinated with a Rha antigen (Figure 1). Antigen uptake and presentation was evaluated in groups of C57BL/6 mice by evaluating anti-MUC1 antibody production, CD8+ T cell proliferation, interferon gamma (INFγ) production, and cytotoxic T cell-mediated apoptosis of cancer cells. We believe these studies are relevant to the development of many other types of vaccines for humans based on the high natural abundance of anti-Rha in the human population.
Figure 1.

Mechanism of Anti-Rha-mediated enhancement of cellular and humoral immune response.
Results and Discussion
Antigen Design
We sought an antigenic sequence that would contain a CD8+ T cell epitope which could be presented on MHC I and would be a sequence that still contained B cell and CD4+ helper T cell epitopes. Ninkovic et al. have previously proposed that the monoglycosylated 21-mer GP3: AHGVTSAPDT(GalNAc)RPAPGSTAPPA represents an ideal peptide for development as an anti-tumor vaccine candidate in humans.56 These claims are based on experiments showing that the GP3 is processed by immunoproteasomes or by cathepsin-L to produce a 10-mer glycopeptide SAPDT(GalNAc)RPAPG as a major product. SAPDT(GalNAc)RPAPG was consequently shown to strongly bind to empty MHC class I HLA-A*0201 molecules. The latter is the most frequent MHC class I allele found in Caucasians. The same study showed that the SAPDTRPAPG peptide and the respective threonine-modified glycopeptide stimulated human cytotoxic T-cells in vitro. Earlier studies defined the preferred processing sites for GP3: AHGVTSAPDT(GalNAc)RPAPGSTAPPA (preferred cleavage sites of immunoproteasomes are underlined), showed the sequence to be efficiently processed, and determined that substrates glycosylated at the DTR motif were the best source of glycopeptides that would potentially bind to MHC I.57 We then considered the implications of stimulation of cytotoxic T-cells in C57BL/6 mice which contain the MHC class I H-2KbDb alleles. Apostolopoulos et al. have shown that the MUC1 8-mer SAPDT(GalNAc)RPA binds more strongly to the MHC class I mouse allele H-2Kb compared with the corresponding unglycosylated peptide. Further, SAPDT(GalNAc)RPA binds to H-2Kb with high affinity and generates T cells both in vitro and in vivo, which recognize both SAPDTRPA and SAPDT(GalNAc)RPA peptides.58 Findings made by Finn and coworkers suggest that MUC1 variable number tandem repeats (VNTRs) containing TACAs were more potent at breaking self-tolerance in MUC1 transgenic mice than the unglycosylated VNTR, since they are more ‘foreign’-like epitopes in comparison to the unglycosylated MUC1.59, 60 Similar findings were made by Boons, who was able to demonstrate that a MUC1-based antigen having GalNAc, again at the DTR site, is more effective in MHC I stimulation of CD8+ T cell activation in MUC1-transgenic mice compared to the non-glycosylated MUC1 sequence.61 Based on these considerations we selected a 20-mer GalNAc modified glycopeptide related to GP3, TSAPDT(GalNAc)RPAPGSTAPPAHGV (Figure 2), a sequence we hypothesized would contained an MHC-I epitope for both humans and potentially the C57BL/6 mice used in this study. However, we were uncertain at the onset of the antigen design phase as to whether or not this specific glycopeptide would be processed to produce either CD8+ SAPDT(GalNAc)RPA or CD4+ helper T cell epitopes in C57BL/6 mice.
Figure 2.

Design of the antigen: Structure of the TLR-2 agonist, linker, and MUC1 glycopeptide sequence. Known human and mouse CD8+ T cell epitopes (boxes).
Small self-like glycopeptides are not highly immunogenic.62 One approach to enhance immunogenicity has been to conjugate the peptides to synthetic bacterial lipoproteins.63–66 Pam3CysSS is a synthetically prepared domain of the N-terminal region of Braun’s lipoprotein.67 It has been shown to be able to stimulate virus-specific CTL responses against influenza virus-infected cells when conjugated with influenza nucleoprotein peptides 147–158 from influenza nucleoprotein64 and to elicit protective antibodies against foot-and-mouth disease when coupled to the antigenic determinant of VP1.63 We used the Pam3Cys triazole-linker moiety because we demonstrated in Sarkar et. al that it was sufficient to serve both as an anchor to the liposome and to act as an adjuvant (Figure 2).9,68 The epitope is expected to interact with Toll Like Receptor-2 (TLR-2) similar to Pam3CysSK4.69
Synthesis of an Azido-Functionalized MUC1-Tn Sequence
An azido functionality was appended to the N-terminus of the peptide as a chemical handle for CuAAc conjugation with a propargylated Pam3Cys. We synthesized the 20 amino acid sequence, N3-linker-TSAPDT(GalNAc)RPAPGSTAPPAHGV (2) for our model vaccine, Scheme 1. The protected azide-containing peptide was prepared by the Fmoc-strategy on an Omega 396 synthesizer (Advanced ChemTech, Louisville, KY) using solid phase chemistry. The coupling of the amino acids, the acetyl protected Tn-Thr and the azide functionality was achieved by HOBt and DIC in the presence of NMP starting with a preloaded Fmoc-Ala-Wang resin. Fmoc deprotections were accomplished by treatment with 25% piperidine in dimethylformamide. A modified Reagent K cocktail consisting of 88% TFA, 3% thioanisole, 5% ethanedithiol, 2% water and 2% phenol was used for simultaneous resin cleavage and deprotection of the glycopeptide. The acetyl protections on the N-acetyl galactal survived the cleavage conditions. At the end of cleavage, the cocktail mixture was filtered through a Quick-Snap and collected in 20 mL ice-cold butane ether. The peptide was allowed to precipitate for an hour at −20 °C, centrifuged, and washed twice with ice-cold methyl-t-butyl ether. The precipitate was dissolved in 25% acetonitrile and lyophilized to complete dry powder affording peptide 2. Quality of peptides was analyzed by analytical reverse phase HPLC and MALDI-TOF. The acetyl deprotection was achieved by treating the azidopeptide 1 with 6 mM sodium methoxide in methanol. The deprotected peptide 2 was purified by size exclusion chromatography using Bio-Gel (P-2, 45–90 μm) and water as solvent followed by lyophilization.
Scheme 1.

Synthesis of Pam3Cys-MUC1-Tn.
Synthesis of Pam3Cys-MUC1-Tn 4
Propargylated Pam3Cys 3 was prepared as previously reported (Scheme S2, supporting information).9 However, the copper (I)-catalyzed azido-alkyne cycloaddtion (CuAAc) between the azidopeptide 2 with alkyne 3 was more challenging than previously reported. Previously, CuSO4 in H2O:THF:MeOH was used as catalyst and solvent, respectively.9, 70 However, these conditions worked poorly with azido peptide 2. Moreover, the conditions also promoted methyl ester formation in the MUC1 peptide,9 observed by mass spectrometry. We explored various cycloaddition conditions (Table S1, supporting information) and found CuI, TBTA, Na-ascorbate and DIEA in H2O:THF:DMF (1:1:2) to be far more effective with substrate 2, (Scheme 1), to afford Pam3Cys-MUC1-Tn 4
Synthesis of CD8+ T cell epitope 5
To overcome possible inefficient in vitro processing of the whole 20 amino acid Pam3Cys-MUC1-Tn 4 by the tumor in a CTL assay, we prepared the putative 8 amino acid residue CD8+ T cell epitope SAPDTnRPA (5) using solid phase peptide synthesis on Wang resin and purified through Bio-Gel P-2 (45–90 μm) with water as eluent followed by lyophilization (Scheme S1, supporting information). When added to EL-4 cells, this peptide should bind to MHC I and stimulate the same cytolytic T cells that are induced when the 20 residue peptide is taken up, processed, and cross-presented on MHC I by antigen presenting cells.
Synthesis of Rha-TEG-Cholesterol
Rha-TEG-Cholesterol was prepared as previously described and used in formulation of the liposomes (Sarkar et al., 2013).
Synthesis of a Rha-Ficoll Conjugate
To evaluate the effect of Rha in enhancing antigen uptake, we needed to generate anti-Rha antibodies in mice, since naïve mice do not have anti-Rhamnose antibodies (Sarkar et al., 2010). We achieved this by vaccinating mice using a Rha-Ficoll conjugate with equivolume Alum emulsion. We choose Ficoll as the carrier for Rha because it is well known for producing a T-independent antibody response. The Rha-Ficoll was prepared as previously described (Sarkar et al., 2013).
Liposome Formulation
We prepared two different batches of liposomes for the four groups of mice. For the preparation of the normal liposomes 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) (80%) and cholesterol (20%) were used (Batch 2). For the Rha-displaying liposomes, 10% Rha-TEG-Cholesterol was mixed with 10% cholesterol, keeping the other components the same (Batch 1). The liposomes were formulated by the extrusion method in a total lipid concentration of 30 mM. The concentration of Pam3Cys-MUC1-Tn 4 was 10 nanomolar in both kinds of liposomes.
Immunological Results
Preliminary Study
To check the antigenicity of our vaccine, we first started with two C57BL/6 mice (H-2KbDb) and primed with Pam3Cys-MUC1-Tn incorporated on the Batch 2 liposomes. The mice were boosted twice with the vaccine at 14 day intervals. On the seventh day after the second boost, the mice and control non-immunized mice were bled and the sera were pooled to check the anti-MUC1 antibody titer. An ELISA was performed to demonstrate that the mice were producing anti-MUC1 antibody (Figure S1, supporting information). The mice were then sacrificed on the same day and the CD8+ T cells were isolated. A T-cell proliferation assay was performed to ensure that the vaccine could stimulate the generation of CD8+ T cells. Isolated spleen CD8+ T cells were cultured with different concentrations of the liposomal Pam3Cys-MUC1-Tn 4 in the presence of C57BL/6 DCs. A considerable amount of T-cell proliferation (Figure S2, supporting information) demonstrated the presence of at least one CD8+ T-cell epitope in the vaccine.
Evaluation of Effect of Rha-TEG-Cholesterol-formulated Liposomal Vaccines
Four groups of six female C57BL/6 mice were used for this study (Table 1).
Table 1.
Vaccination plan for the groups of mice.
| Group A | Group B | Group C | Group D | |
|---|---|---|---|---|
| Immunization | - | Rha-Ficoll | - | Rha-Ficoll |
| Vaccination | Liposomal Pam3Cys-MUC1-Tn | Liposomal Pam3Cys-MUC1-Tn with Rha-TEG-Cholesterol | ||
Anti-Rha Antibody Generation in Mice
To test our hypothesis, we again generated anti-Rha antibodies in groups of mice. Mice of Groups B and D were injected subcutaneously (day 0) with 100 μL equivolume emulsion of Rha-Ficoll and Alum (100 μg of Rha-Ficoll per mouse). The mice were boosted with the same composition at 14 day intervals up to the fourth boost. On the seventh day after the fourth boost, blood was pooled from the mice and sera were isolated. An ELISA was performed to determine the anti-Rha antibody titer. The Rha-Ficoll immunized mice showed about 25-fold higher titer than non-immunized mice against microtiter plates coated with Rha-BSA (Figure S3, supporting information). Control wells were coated with BSA.
Tumor Antigen Vaccination
Once mice were producing anti-Rha, the second step was to vaccinate all the groups of mice with the corresponding liposome formulations containing the Pam3Cys-MUC1-Tn 4 tumor antigen (Table 1). The one goal of our study was to evaluate the production of antibodies against the antigen and the stimulation of CD4+ helper T-cells. The other major goal was to evaluate whether or not CD8+ T cells were being activated, resulting in cytokine production and anti-tumor cytotoxicity. Here we have evaluated the effect of the Rha ligand on antigen uptake and presentation using CD8+ T cell proliferation, cytotoxicity and interferon gamma (IFNγ) production.
Anti-MUC1 Antibody Production
The anti-MUC1 antibody titer of all four groups of mice was evaluated by ELISA. The study was performed on pooled sera isolated from two mice of each group after the 3rd boost of the vaccine. The ELISA shows a considerable amount of anti-MUC1 antibody production by each group of mice, with over 10-fold higher titer in Goup D mice (Figure 3). This demonstrated that CD4+ T cell help was stimulated more effectively by the targeting through Rha.
Figure 3.

Anti-MUC1 antibody titer for all groups of mice.
CD8+ T Cell Proliferation Study
A CD8+ T cell proliferation study was performed to evaluate whether enhanced uptake of the Rha-TEG-Cholesterol-formulated liposomal vaccine resulted in more effective CD8+ T cell priming in vivo. We used the eight amino acid CD8+ T cell epitope 5 (Scheme S1, supporting information), which could be generated in vivo from the 20 amino acid MUC1-Tn peptide, to restimulate primed CD8+ T cells in vitro. To determine the concentration of CD8+ T cell epitope 5 required to activate CD8+ T cells, we first performed a T cell proliferation assay with the CD8+ T cells from Group D in the presence of DCs pulsed with peptide concentrations of 2.5, 5, 10, and 20 μg/mL. The assay indicates a continuous increase in proliferation with the increase of peptide concentration (Figure 4). In the proliferation assay with all four groups, peptide 5 was used at 25 μg/mL concentration. The comparative study indicates that Group D is the only group that shows enhanced peptide specific proliferation relative to the other groups (Figure 5).
Figure 4.

CD8+ T cell proliferation of Group D at different peptide concentrations.
Figure 5.

CD8+ T cell proliferation of all four groups at 25 μg/mL peptide.
Interferon Gamma (IFNγ) Production
Following antigen stimulation, most CD8+ T cells release interferon gamma (IFNγ). The production of IFNγ is higher for primed CD8+ T cells. We performed an IFNγ-ELISA assay to evaluate if enhanced uptake of the Rha-TEG-Cholesterol-formulated liposomal vaccine resulted in more primed CD8+ T cells. Isolated CD8+ T cells from each group of mice were cultured with DCs pulsed with CD8+ T cell epitope 5. After 24 hours incubation at 37°C, the supernatant was collected for ELISA analysis.. The assay indicated 4.6- to 10-fold higher CD8+ T cell epitope 5 specific IFNγ production for Group D, compared to the other three groups (Figure 6).
Figure 6.
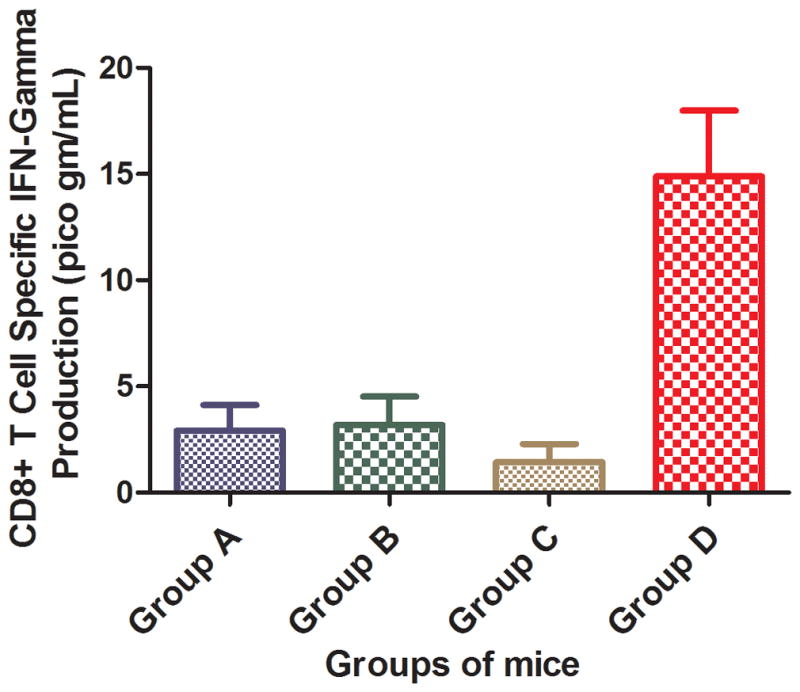
CD8+ T cell specific IFNγ production of all four groups at 25 μg/mL peptide.
JAM Assay for CTLs
One approach to generating a more effective antitumor immunotherapeutic vaccine may be to stimulate CD8+ T cells to specifically induce apoptosis of tumor cells. We assayed for cytotoxic CD8+ T cells from Group B and D mice, both of which contained anti-Rha antibodies. Radiolabeled EL4 tumor cells were pulsed with CD8+ T cell epitope 5, followed by incubation with the CD8+ T cells using EL4 to CD8+ T cell ratios of 1:100 and 1:50. Both groups showed killing of the EL4 cells by apoptosis induction; however, Group D showed about twice the cytotoxicity (Figure 7).
Figure 7.

Apoptosis of EL4 cells induced by CD8+ T cells from Groups B and D. The ratio of EL4 cells to CD8+ T cells is 1:100 or 1:50.
Conclusion
A new, fully synthetic, glycopeptide-based tumor antigen has been synthesized, Pam3Cys-MUC1-Tn 4. The antigen was successfully formulated into liposomes along with a Rha-TEG-Cholesterol designed to bind anti-Rha antibodies which are found abundantly in human serum and can be generated in mice. In these studies anti-Rha antibodies were generated in C57BL/6 mice by Rha-Ficoll immunization to mimic human natural anti-Rha antibodies. The liposomal Pam3Cys-MUC1-Tn vaccine formulated with Rha-TEG-Cholesterol resulted in improved antibody and CD8+ T cell response against the Pam3Cys-MUC1-Tn antigen in anti-Rha expressing mice compared to mice lacking anti-Rha antibodies. This result suggests that the Rha epitope in the vaccine can form immune complexes with the anti-Rha antibodies in vivo followed by efficient antigen uptake in DCs, leading to cross-presentation on MHC class I molecules. It is important to note that our CD8+ assays only measured T cells recognizing the CD8+ epitope contained in the added 8 residue peptide. Other T cells to additional cross-presented epitopes may have been generated. The use of anti-Rha antibodies in immune-complex mediated maturation of antigen presenting cells to induce a CD8+ T cell response is a novel discovery. The Rha targeting allowed use of an antigen that was otherwise only weakly cross-presented and enhanced its ability to elicit CD8+ T cells while still generating a humoral response. The vaccine model contains separate Rha and antigen epitopes that gives the vaccine platform a wide range of applicability.
Experimental Procedures
General Methods
Most fine chemicals including copper salts, cholesterol, L-Rhamnose, DIEA, DMF and other dry solvents were purchased from Acros Organics. Boron trifluoride-etherate was obtained from Aldrich and all the other solvents were purchased from Fischer Scientific. 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and Ficoll 400 were acquired from Avanti Polar Lipids Inc. (Alabaster, AL). Silica gel (230–400 mesh) for flash column chromatography was purchased from Sorbent Technologies; and thin-layer chromatography (TLC) precoated plates were obtained from EMD. TLC plates (silica gel 60, f254) were visualized under UV light or by charring (5% H2SO4–MeOH) or by staining with ninhydrin. Flash column chromatography was performed on silica gel (230–400 mesh) using solvents as received. Tris [(1-benzyl-1H-1,2,3-triazol-4-yl)methyl] amine (TBTA) was obtained from Anaspec (San Jose, CA). Preloaded Fmoc-L-Ala-Wang resin, and all other Fmoc-L-amino acids were procured from Anaspec (San Jose, CA) or from Chem-Impex Inc. Imject Alum was purchased from Thermo Scientific. Goat anti-mouse IgG/IgM antibodies were obtained from Sigma. Dynabead kit for CD8+ T cell isolation was purchased from Invitrogen (a division of Thermo Fisher Scientific). All other secondary antibodies were obtained from Jackson Immunoresearch Laboratories (West Grove, PA). Female C57BL/6 mice (6–8 weeks old) were obtained from the Jackson Laboratory. All mice were maintained in the AAALAC-accredited animal facility at the University of Toledo Health Science Campus under a specific pathogen-free environment. All mouse experiments were performed according to NIH guidelines with approval of the UT Institutional Animal Care and Use Committee.
Synthesis of Glycopeptide Azide 1
Glycopeptide azide 1 was synthesized on an Omega 396 synthesizer (Advanced ChemTech, Louisville, KY) by Fmoc strategy using solid-phase chemistry. The peptide synthesis was performed on the 100 μmol scale by coupling amino acid esters of HOBt using DIC as the coupling agent. A 6-fold excess of Nα-Fmoc amino acid esters of HOBt in N-methylpyrrolidinone (NMP) were used in the synthesis. A 1:1 ratio of amino acid to DIC was used in all the coupling reactions. Deprotection of the Nα-Fmoc group was accomplished by treatment with 25% piperidine in dimethylformamide twice: first for 5 min and then a second time for 25 min. The final coupling employed 6-azido-hexanoic acid was treated the same as the other Fmoc amino acids in the peptide synthesizer. Approximately 0.5 g of Fmoc-Thr(Ac3-alpha-GalNAc)-OH was used in the synthesis. After coupling all the required amino acids, the peptide was cleaved from the solid support and globally deprotected (except the acetyl protections on the carbohydrate moiety) by gentle magnetic stirring in a 10 mL round bottomed flask in the presence of 4 mL of a modified reagent K cocktail (88% TFA, 3% thioanisole, 5% ethanedithiol, 2% water, and 2% phenol) for 2.5 h under N2 atmosphere. On completion, the cocktail mixture was filtered through a Quick-Snap column and the filtrate was collected in 20 mL ice-cold butane ether. The peptide was precipitated for an hour at −20°C, centrifuged, and washed twice with ice cold methyl-t-butyl ether. The precipitate was dissolved in 25% acetonitrile and lyophilized to obtain the glycopeptide 1 as a white powder. The identity of the peptide was confirmed by MALDI-TOF (matrix assisted laser desorption ionization time-of-flight) mass spectrometer, model 4800 from Applied Biosystems. MALDI-TOF: [M+H] m/z calcd for C100H156N29O37, 2355.12; found, 2355.217 (Figure S4A, supporting information). The peptide was purified using a Vydac reverse-phase preparative column (C18) using a gradient of 5% to 90% acetonitrile (Figure S4B, supporting information).
Synthesis of Glycopeptide Azide 2
Glycopeptide 1 (5 mg, 2.24 μmol) was taken in 2 mL dry methanol and 12 μL of freshly prepared 1 M sodium methoxide was added to the solution. The reaction was monitored by MALDI-TOF analysis. On completion, the reaction was neutralized with solid carbon dioxide. The solution was concentrated and purified by Bio-Gel (P-2, fine 45–90 μm, 12 g) size exclusion chromatography (column bed length: 30 cm, diameter: 2.5 cm) using deionized water as eluent. Lyophilization of the elutant afforded 2 as a white powder (4.7 mg, 100%). MALDI-TOF: [M+H] m/z calcd for C94H150N29O34, 2229.0895; found, 2229.336 (Figure S5, supporting information).
Synthesis of Pam3Cys-MUC1-Tn 4
CuI (134 μg, 0.54 μmol) and TBTA (0.857 mg, 1.62 μmol) were dissolved in H2O-THF (1:1, 0.40 mL). Na-ascorbate (0.80 mg, 4.04 μmol) was added to the solution followed by stirring for 5 minutes. Compound 3 (1.27 mg, 1.35 μmol) in THF (0.40 mL) was added to the reaction mixture and stirred for 15 minutes followed by the addition of a solution of compound 2 (1 mg, 0.45 μmol) in H2O-DMF (1:3, 0.4 mL). The reaction mixture was stirred at 20 °C under N2 atmosphere for 16 h. The reaction mixture was concentrated, dissolved in CHCl3, washed with 7.5% aqueous citric acid solution, dried over sodium sulfate and the solvent was evaporated to afford compound 4 as a light yellow solid (1.9 mg, 100%). MALDI-TOF: [M+H] m/z calcd for C151H256N31O40S, 3175.86; found 3175.809 (Figure S6, supporting information).
Synthesis of CD8+ T-Cell Epitope 5
The CD8+ T-Cell epitope 5 was synthesized manually by assembling the amino acids on Fmoc-Ala-preloaded Wang resin by Fmoc strategy using solid-phase chemistry. The reactions were performed in a 20 mL syringe reactor cartridge with agitation provided by a stream of N2. The peptide synthesis was performed by coupling HOBt esters of Fmoc-protected amino acids in situ using PyBOP as the coupling agent in presence of diisopropylethyl amine (DIPEA). Deprotection of the N-α-Fmoc group was achieved by treatment with 25% piperidine in dimethylformamide thrice: first for 5 min then a second and third time for 10 mins each. After the synthesis, the resin cleavage as well as global deprotection (except the acetyl protections on the carbohydrate moiety) was accomplished by treatment with TFA-H2O-TIPS (95:2.5:2.5), for 4 h under N2 atmosphere. The mixture was filtered, washed with the cleavage cocktail (1–2 mL), followed by CH2Cl2. The filtrate was concentrated to dryness under vacuum. The dry peptide (5 mg, 2.24 μmol) was dissolved in 2 mL of dry methanol, and 12 μL of freshly prepared 1 M sodium methoxide was added and the reaction mixture was stirred at ambient temperature under N2 atmosphere for 4 h. The reaction was neutralized with solid carbon dioxide, concentrated and purified by Bio-Gel (P-2, fine 45–90μm, 12 g) size exclusion chromatography (column bed length: 30 cm, diameter: 2.5 cm) using deionized water as solvent. Lyophilization of the eluants afforded 5 as a white powder (4.7 mg, 100%). MALDI-MS: [M+H] m/z calcd for C94H150N29O34, 1017.48; found, 1017.940 (Scheme S1, supporting information).
Liposome Formulation
Different lipid stock solutions were prepared in chloroform in separate glass vials and aliquots of the stock solutions were mixed in proportions to obtain a solution with a total lipid concentration of 30 mM in a total volume of 2 mL (Batch 1: DPPC 80%, cholesterol 10%, Rha-TEG-Cholesterol 10%, and Pam3Cys-MUC1-Tn 0.69μM; Batch 2: DPPC 80%, cholesterol 20%, Pam3Cys-MUC1-Tn 0.69 μM). A constant stream of nitrogen was used to evaporate the chloroform and the resulting lipid films were dried under vacuum for 12 h. 2 mL of HEPES buffer (pH = 7.4) was then added to hydrate the dry lipid films and the suspensions were incubated at 43 °C for 40 min. The suspensions were subjected to 10 freeze–thaw cycles (dry ice/acetone and water at 40 °C). Final liposomes were prepared by extrusion (21 times) using a LipoFast Basic fitted with a 100 nm polycarbonate membrane to control the liposome size.
Preliminary Study
Immunization
Two female C57BL/6 mice (6–8 weeks old, The Jackson Laboratory) were primed (day 0) and boosted three times (days 14, 28 and 42) with 100 μL intraperitoneal injections of Pam3Cys-MUC1-Tn conjugate 10 (10 nm per injection) incorporated on liposome (Batch 2) in PBS.
Anti-MUC1 Antibody ELISA
96-well plates (Immulon 4 HBX) were coated with MUC1-Tn conjugate 2 (15 μg/mL) in 0.01 M phosphate buffered saline (PBS) and incubated over night at 4 °C. The plates were washed 5 times with PBS containing 0.1% Tween-20. Blocking was achieved by incubating the plates for 1 h at room temperature with BSA in PBS (1 mg/mL). The plates were then washed 5 times and incubated for 1 h with serum dilutions of BSA/PBS. Unbound antibody in the serum was removed by washing and the plates were incubated for 1 h at room temperature with Horseradish Peroxidase (HRP) goat antimouse IgG + IgM (Sigma) diluted 5000 times in PBS/BSA. The plates were washed and TMB (3,3′,5,5′-tetramethylbenzidine) one component HRP microwell substrate (Bio FX, Owings Mills, MD) was added and allowed to react for 10–20 min. Absorbance was recorded at 620 nm and was plotted against log10[1/serum dilution].
CD8+ T-Cell Proliferation Assay
On day 49, selected mice were sacrificed and the spleens were removed and placed in 5 mL of freshly prepared spleen cell culture medium (DMEM with 10% fetal calf serum) as described previously.9 Single cell suspensions were prepared using modified sterile glass homogenizers. The cells were washed three times with culture medium and from the cell suspension; the CD8+ T cells were isolated using a Dynabead FlowComp™ Mouse CD8 kit (Invitrogen). The cell concentration was brought to 5×106 cells/mL. 100μL aliquots of the spleen cell suspensions were added to 96-well plates (5×105 cells per well). The DC suspension cultured from the bone marrow of a non-immunized C57BL/6 mouse was pulsed with the antigen by incubating with the Pam3Cys-MUC1-Tn liposomes at antigen concentrations from 0.02 to 2 μg/mL at 37°C for 4 h. 100 μL aliquots of the pulsed DCs were added to the wells containing the CD8+ T cells (5×104 DCs per well). The plates were incubated at 37 °C for 4 days. On day 4, the cells were pulsed with radioactive [3H]-thymidine (40 μCi/mL, 25 μL per well) and incubated overnight at 37°C in presence of 5% CO2. The cells were harvested on glass-fiber filters and incorporation was determined by measurements on a Top Count scintillation counter (Packard, Downers Grove, IL).
Evaluation of Effect of Rha-TEG-Cholesterol-formulated Liposomal Vaccines
Immunization
Twenty-four female C57BL/6 mice were used in this study. They were divided into four groups as A, B, C and D, each group containing six mice. Groups B and D were injected subcutaneously (day 0) with 100 μL equivolume emulsion of Rhamnose-Ficoll (Rha-Ficoll) and Imject Alum (100 μg of Rha-Ficoll per mouse). The mice were boosted with the same composition on day 14, 28, 56 and 70. The mice were bled on day 77 and the sera were pooled to check for the presence of anti-Rha antibodies.
ELISA for Anti-Rha Antibody Titer
96-well plates (Immulon 4 HBX) were coated with Rha-BSA conjugate (2 μg/mL) in PBS and incubated overnight at 4 °C. The ELISA was continued as described above.
Vaccination
Vaccination was started on day 82 after Rha-Ficoll immunization. Two different batches of liposomes were prepared, Batch 1 and Batch 2. Group A and C were primed and boosted twice in 14 days intervals with 100 μL of Batch 2 liposomes per mouse (100 μL subcutaneous injection, 10 nm Pam3Cys-MUC1-Tn per mouse). Groups B and D were primed and boosted thrice in 14 days intervals with 100 μL of Batch 1 liposomes/mouse (100 μL subcutaneous injection, 10 nm Pam3Cys-MUC1-Tn per mouse). After such preparation, the mice were kept in rest and two mice from each group were boosted with Rha-Ficoll (Groups B and D only, composition and amount as mentioned earlier) followed by liposomal vaccines (all groups, 12 days after the last Rha-Ficoll boost, composition and amount are same as stated above) prior to different assays.
Anti-MUC1 Antibody Titers ELISA
Mice from each group were bled after the third boost. The sera from individual mice in a group were pooled. The ELISAs were performed as described above.
CD8+ T Cell Proliferation Assay with All Groups of Mice
Two mice from each group were sacrificed on the seventh day after the fourth boost of vaccine and cells were isolated from spleen and lymph nodes as described earlier. From the cell suspension, the CD8+ T cells were isolated using a Dynabead FlowComp™ Mouse CD8 kit (Invitrogen). The CD8+ T cell concentration was brought to 4×105 cells/mL and 100 μL aliquots were added to a 96 well plate (2×104 cells/well). 100 μL of the DC suspension (2×103 cells/mL, 1×104 cells/well) cultured from the bone marrow of a non-immunized C57BL/6 mouse was added to the wells containing CD8+ T cells, and also to the empty wells as control. Thus the DC to CD8+ T cell ratio was 1:10 and total volume was 200 μL per well. Combinations of CD8+ T cells with DCs were pulsed with CD8+ T cell epitope 5 with effective concentration of 25 μg/mL and incubated for 6 days at 37 °C. On day 5, the cells were pulsed with radioactive[3H]thymidine (40 μCi/mL, 25 μL per well) and at the end of day 6, the cells were harvested on glass-fiber filters and incorporation was determined by measurements on a Top Count scintillation counter (Packard, Downers Grove, IL). A preliminary assay to determine the optimal peptide concentration used cells from one group D mouse and a range of peptide concentrations from 2.5 to 20 μg/mL.
Interferon Gamma (IFNγ) ELISA
CD8+ T cells (5×105 cells/mL) isolated from two mice from each group were distributed on a 6-well plate (500 μL per well). The DC suspension (5×104 cells/mL, 2.5×104 cells/well) cultured from the bone marrow of a non-immunized C57BL/6 mouse was added to the wells containing CD8+ T cells. Thus the DC to CD8+ T cell ratio was 1:10 and total volume was 1 mL per well. The combination of CD8+ T-cells with DCs were pulsed with CD8+ T cell epitope 5 with effective concentration of 25 μL/mL and incubated for 24 hours at 37 °C. After 24 hour, the supernatant was collected and IFNγ production was measured by using Murine IFNγ Mini ELISA Development Kit (Peprotech).
JAM Assay for Apoptosis
Two mice from each group were sacrificed on the seventh day after the fourth boost of vaccine and cells were isolated from spleen and lymph nodes as described earlier.71 The CD8+ T cells were isolated as before. C57BL/6 EL4 lymphoma cells (ATCC TIB 39) were freshly grown in DMEM medium and the concentration of the cell suspension was brought to 1×105 cells/mL. The cell suspension was pulsed with [3H]-thymidine (40 μCi/mL) for 3 hours. The cell suspension was then divided into two equal halves after washing and one half of the EL4 cells were pulsed with CD8+ T cell epitope 5 and incubated 12 hours at 37 °C in the presence of 5% CO2. Both preparations were then washed thrice and the EL4 cell concentrations were again brought to 1×105 cells/mL. 100 μL aliquots of the EL4 cell suspensions were then added to 96-well plates (1×104 cells per well). Each group of CD8+ T cell suspensions was prepared with concentrations of 1×107 and 0.5×107 cells/mL. 100 μL aliquots from each concentration were added to different wells containing EL4 cells with or without peptide pulsing. 100 μL of 2 μM staurosporin was added to some of the wells instead of CD8+ T cells as a positive control. The negative controls were EL4 cells alone and EL4 cells pulsed with CD8+ T cell epitope 5, without any CD8+ T cells. The plate was incubated for 6 hours at 37 °C in the presence of 5%. CO2. The cells were then harvested on glass-fiber filters and incorporation was determined by measurements on a Top Count scintillation counter (Packard, Downers Grove, IL). Percent specific killing was calculated with-respect-to negative control EL4 cells pulsed with CD8+ T cell epitope 5 (without CD8+ T cells).
Supplementary Material
Acknowledgments
We acknowledge the National Institutes of Health (Grant number GM094734-01 and GM094734-02 to S.J.S. and K.A.W.) for financial assistance.
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information. Supplementary data associated with this article is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.York IA, Rock KL. Antigen processing and presentation by the class I major histocompatibility complex. Annu Rev Immunol. 1996;14:369–96. doi: 10.1146/annurev.immunol.14.1.369. [DOI] [PubMed] [Google Scholar]
- 2.Sigal LJ, Crotty S, Andino R, Rock KL. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature. 1999;398:77–80. doi: 10.1038/18038. [DOI] [PubMed] [Google Scholar]
- 3.Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol. 2010;10:403–14. doi: 10.1038/nri2780. [DOI] [PubMed] [Google Scholar]
- 4.Alving CR, Wassef NM. Cytotoxic T lymphocytes induced by liposomal antigens: mechanisms of immunological presentation. AIDS Res Hum Retroviruses. 1994;10(Suppl 2):S91–4. [PubMed] [Google Scholar]
- 5.Foged C, Arigita C, Sundblad A, Jiskoot W, Storm G, Frokjaer S. Interaction of dendritic cells with antigen-containing liposomes: effect of bilayer composition. Vaccine. 2004;22:1903–13. doi: 10.1016/j.vaccine.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 6.Jerome V, Graser A, Muller R, Kontermann RE, Konur A. Cytotoxic T lymphocytes responding to low dose TRP2 antigen are induced against B16 melanoma by liposome-encapsulated TRP2 peptide and CpG DNA adjuvant. J Immunother. 2006;29:294–305. doi: 10.1097/01.cji.0000199195.97845.18. [DOI] [PubMed] [Google Scholar]
- 7.Lasic DD. Novel applications of liposomes. Trends Biotechnol. 1998;16:307–21. doi: 10.1016/s0167-7799(98)01220-7. [DOI] [PubMed] [Google Scholar]
- 8.Mallick AI, Singha H, Chaudhuri P, Nadeem A, Khan SA, Dar KA, Owais M. Liposomised recombinant ribosomal L7/L12 protein protects BALB/c mice against Brucella abortus 544 infection. Vaccine. 2007;25:3692–704. doi: 10.1016/j.vaccine.2007.01.066. [DOI] [PubMed] [Google Scholar]
- 9.Sarkar S, Salyer AC, Wall KA, Sucheck SJ. Synthesis and immunological evaluation of a MUC1 glycopeptide incorporated into l-rhamnose displaying liposomes. Bioconjug Chem. 2013;24:363–75. doi: 10.1021/bc300422a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ju TZ, Cummings RD. A unique molecular chaperone Cosmc required for activity of the mammalian core 1 beta 3-galactosyltransferase. Proc Natl Acad Sci U S A. 2002;99:16613–16618. doi: 10.1073/pnas.262438199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vlad AM, Muller S, Cudic M, Paulsen H, Otvos L, Hanisch FG, Finn OJ. Complex carbohydrates are not removed during processing of glycoproteins by dendritic cells: Processing of tumor antigen MUC1 glycopeptides for presentation to major histocompatibility complex class II-restricted T cells. J Exp Med. 2002;196:1435–1446. doi: 10.1084/jem.20020493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gendler SJ, Lancaster CA, Taylor-Papadimitriou J, Duhig T, Peat N, Burchell J, Pemberton L, Lalani EN, Wilson D. Molecular cloning and expression of human tumor-associated polymorphic epithelial mucin. J Biol Chem. 1990;265:15286–93. [PubMed] [Google Scholar]
- 13.Finn OJ, Jerome KR, Henderson RA, Pecher G, Domenech N, Magarian-Blander J, Barratt-Boyes SM. MUC-1 epithelial tumor mucin-based immunity and cancer vaccines. Immunol Rev. 1995;145:61–89. doi: 10.1111/j.1600-065x.1995.tb00077.x. [DOI] [PubMed] [Google Scholar]
- 14.Brossart P, Schneider A, Dill P, Schammann T, Grunebach F, Wirths S, Kanz L, Buhring HJ, Brugger W. The epithelial tumor antigen MUC1 is expressed in hematological malignancies and is recognized by MUC1-specific cytotoxic T-lymphocytes. Cancer Res. 2001;61:6846–50. [PubMed] [Google Scholar]
- 15.Ho SB, Niehans GA, Lyftogt C, Yan PS, Cherwitz DL, Gum ET, Dahiya R, Kim YS. Heterogeneity of mucin gene expression in normal and neoplastic tissues. Cancer Res. 1993;53:641–651. [PubMed] [Google Scholar]
- 16.Jerome KR, Barnd DL, Bendt KM, Boyer CM, Taylorpapadimitriou J, Mckenzie IFC, Bast RC, Finn OJ. Cytotoxic lymphocytes-T derived from patients with breast adenocarcinoma recognize an epitope present on the protein core of a mucin molecule preferentially expressed by malignant-cells. Cancer Res. 1991;51:2908–2916. [PubMed] [Google Scholar]
- 17.Ioannides CG, Fisk B, Jerome KR, Irimura T, Wharton JT, Finn OJ. Cytotoxic T-cells from ovarian malignant-tumors can recognize polymorphic epithelial mucin core peptides. J Immunol. 1993;151:3693–3703. [PubMed] [Google Scholar]
- 18.Barnd DL, Lan MS, Metzgar RS, Finn OJ. Specific, major histocompatibility complex-unrestricted recognition of tumor-associated mucins by human cytotoxic T cells. Proc Natl Acad Sci U S A. 1989;86:7159–63. doi: 10.1073/pnas.86.18.7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Mensdorff-Pouilly S, Verstraeten AA, Kenemans P, Snijdewint FGM, Kok A, Van Kamp GJ, Paul MA, Van Diest PJ, Meijer S, Hilgers J. Survival in early breast cancer patients is favorably influenced by a natural humoral immune response to polymorphic epithelial mucin. J Clin Oncol. 2000;18:574–583. doi: 10.1200/JCO.2000.18.3.574. [DOI] [PubMed] [Google Scholar]
- 20.Pinheiro SP, Hankinson SE, Tworoger SS, Rosner BA, McKolanis JR, Finn OJ, Cramer DW. Anti-MUC1 antibodies and ovarian cancer risk: Prospective data from the Nurses’ Health Studies. Cancer Epidem Biomar. 2010;19:1595–1601. doi: 10.1158/1055-9965.EPI-10-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamanaka Y, Suehiro Y, Fukui M, Shikichi K, Imai K, Hinoda Y. Circulating anti-MUC1 IgG antibodies as a favorable prognostic factor for pancreatic cancer. Int J Cancer. 2003;103:97–100. doi: 10.1002/ijc.10801. [DOI] [PubMed] [Google Scholar]
- 22.Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM, Matrisian LM. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–37. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palitzsch B, Hartmann S, Stergiou N, Glaffig M, Schmitt E, Kunz H. A fully synthetic four-component antitumor vaccine consisting of a mucin glycopeptide antigen combined with three different T-helper-cell epitopes. Angew Chem Int Ed Engl. 2014;53:14245–9. doi: 10.1002/anie.201406843. [DOI] [PubMed] [Google Scholar]
- 24.Keil S, Claus C, Dippold W, Kunz H. Towards the development of antitumor vaccines: A synthetic conjugate of a tumor-associated MUC1 glycopeptide antigen and a tetanus toxin epitope. Angew Chem Int Ed Engl. 2001;40:366–369. doi: 10.1002/1521-3773(20010119)40:2<366::AID-ANIE366>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 25.Vassilaros S, Tsibanis A, Tsikkinis A, Pietersz GA, McKenzie IFC, Apostolopoulos V. Up to 15-year clinical follow-up of a pilot Phase III immunotherapy study in stage II breast cancer patients using oxidized mannan-MUC1. Immunotherapy-Uk. 2013;5:1177–1182. doi: 10.2217/imt.13.126. [DOI] [PubMed] [Google Scholar]
- 26.Beatty PL, Finn OJ. Preventing cancer by targeting abnormally expressed self-antigens: MUC1 vaccines for prevention of epithelial adenocarcinomas. Ann N Y Acad Sci. 2013;1284:52–6. doi: 10.1111/nyas.12108. [DOI] [PubMed] [Google Scholar]
- 27.Rong Y, Jin D, Wu W, Lou W, Wang D, Kuang T, Ni X, Qin X. Induction of protective and therapeutic anti-pancreatic cancer immunity using a reconstructed MUC1 DNA vaccine. BMC Cancer. 2009;9:191. doi: 10.1186/1471-2407-9-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang S, Graeber LA, Helling F, Ragupathi G, Adluri S, Lloyd KO, Livingston PO. Augmenting the immunogenicity of synthetic MUC1 peptide vaccines in mice. Cancer Res. 1996;56:3315–3319. [PubMed] [Google Scholar]
- 29.Abdel-Aal AB, Lakshminarayanan V, Thompson P, Supekar N, Bradley JM, Wolfert MA, Cohen PA, Gendler SJ, Boons GJ. Immune and anticancer responses elicited by fully synthetic aberrantly glycosylated MUC1 tripartite vaccines modified by a TLR2 or TLR9 agonist. Chembiochem. 2014;15:1508–13. doi: 10.1002/cbic.201402077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Westerlind U, Hobel A, Gaidzik N, Schmitt E, Kunz H. Synthetic vaccines consisting of tumor-associated MUC1 glycopeptide antigens and a T-cell epitope for the induction of a highly specific humoral immune response. Angew Chem Int Ed Engl. 2008;47:7551–6. doi: 10.1002/anie.200802102. [DOI] [PubMed] [Google Scholar]
- 31.Wilkinson BL, Day S, Malins LR, Apostolopoulos V, Payne RJ. Self-adjuvanting multicomponent cancer vaccine candidates combining per-glycosylated MUC1 glycopeptides and the Toll-like receptor 2 agonist Pam3CysSer. Angew Chem Int Ed Engl. 2011;50:1635–9. doi: 10.1002/anie.201006115. [DOI] [PubMed] [Google Scholar]
- 32.Gathuru JK, Koide F, Ragupathi G, Adams JL, Kerns RT, Coleman TP, Livingston PO. Identification of DHBcAg as a potent carrier protein comparable to KLH for augmenting MUC1 antigenicity. Vaccine. 2005;23:4727–33. doi: 10.1016/j.vaccine.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 33.Hanisch F. Design of a MUC1-based cancer vaccine. Biochem Soc Trans. 2005;33:705–708. doi: 10.1042/BST0330705. [DOI] [PubMed] [Google Scholar]
- 34.Acres B, Apostolopoulos V, Balloul JM, Wreschner D, Xing PX, Ali-Hadji D, Bizouarne N, Kieny MP, McKenzie IF. MUC1-specific immune responses in human MUC1 transgenic mice immunized with various human MUC1 vaccines. Cancer Immunol Immun. 2000;48:588–594. doi: 10.1007/PL00006677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaidzik N, Westerlind U, Kunz H. The development of synthetic antitumour vaccines from mucin glycopeptide antigens. Chem Soc Rev. 2013;42:4421–42. doi: 10.1039/c3cs35470a. [DOI] [PubMed] [Google Scholar]
- 36.Goydos JS, Elder E, Whiteside TL, Finn OJ, Lotze MT. A phase I trial of a synthetic mucin peptide vaccine: induction of specific immune reactivity in patients with adenocarcinoma. J Surg Res. 1996;63:298–304. doi: 10.1006/jsre.1996.0264. [DOI] [PubMed] [Google Scholar]
- 37.Yamamoto K, Ueno T, Kawaoka T, Hazama S, Fukui M, Suehiro Y, Hamanaka Y, Ikematsu Y, Imai K, Oka M. MUC1 peptide vaccination in patients with advanced pancreas or biliary tract cancer. Anticancer Res. 2005;25:3575–3579. [PubMed] [Google Scholar]
- 38.Ramanathan RK, Lee KM, McKolanis J, Hitbold E, Schraut W, Moser AJ, Warnick E, Whiteside T, Osborne J, Kim H, Day R, Troetschel M, Finn OJ. Phase I study of a MUC1 vaccine composed of different doses of MUC1 peptide with SB-AS2 adjuvant in resected and locally advanced pancreatic cancer. Cancer Immunol Immun. 2005;54:254–64. doi: 10.1007/s00262-004-0581-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brossart P, Wirths S, Stuhler G, Reichardt VL, Kanz L, Brugger W. Induction of cytotoxic T-lymphocyte responses in vivo after vaccinations with peptide-pulsed dendritic cells. Blood. 2000;96:3102–3108. [PubMed] [Google Scholar]
- 40.Tel J, Schreibelt G, Sittig SP, Mathan TS, Buschow SI, Cruz LJ, Lambeck AJ, Figdor CG, de Vries IJ. Human plasmacytoid dendritic cells efficiently cross-present exogenous Ags to CD8+ T cells despite lower Ag uptake than myeloid dendritic cell subsets. Blood. 2013;121:459–67. doi: 10.1182/blood-2012-06-435644. [DOI] [PubMed] [Google Scholar]
- 41.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 42.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 43.Galili U. Evolution and pathophysiology of the human natural anti-alpha-galactosyl IgG (anti-Gal) antibody. Springer Semin Immunopathol. 1993;15:155–71. doi: 10.1007/BF00201098. [DOI] [PubMed] [Google Scholar]
- 44.Galili U, Macher BA, Buehler J, Shohet SB. Human natural anti-alpha-galactosyl IgG. II The specific recognition of alpha (1–3)-linked galactose residues. J Exp Med. 1985;162:573–82. doi: 10.1084/jem.162.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abdel-Motal U, Wang S, Lu S, Wigglesworth K, Galili U. Increased immunogenicity of human immunodeficiency virus gp120 engineered to express Galalpha1-3Galbeta1-4GlcNAc-R epitopes. J Virol. 2006;80:6943–51. doi: 10.1128/JVI.00310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abdel-Motal UM, Guay HM, Wigglesworth K, Welsh RM, Galili U. Immunogenicity of influenza virus vaccine is increased by anti-gal-mediated targeting to antigen-presenting cells. J Virol. 2007;81:9131–41. doi: 10.1128/JVI.00647-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huflejt ME, Vuskovic M, Vasiliu D, Xu H, Obukhova P, Shilova N, Tuzikov A, Galanina O, Arun B, Lu K, Bovin N. Anti-carbohydrate antibodies of normal sera: findings, surprises and challenges. Mol Immunol. 2009;46:3037–49. doi: 10.1016/j.molimm.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 48.Oyelaran O, McShane LM, Dodd L, Gildersleeve JC. Profiling human serum antibodies with a carbohydrate antigen microarray. J Proteome Res. 2009;8:4301–10. doi: 10.1021/pr900515y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pazur JH, Erikson MS, Tay ME, Allen PZ. Isomeric, anti-rhamnose antibodies having specificity for rhamnose-containing, streptococcal heteroglycans. Carbohydr Res. 1983;124:253–63. doi: 10.1016/0008-6215(83)88461-4. [DOI] [PubMed] [Google Scholar]
- 50.Sarkar S, Lombardo SA, Herner DN, Talan RS, Wall KA, Sucheck SJ. Synthesis of a single-molecule L-rhamnose-containing three-component vaccine and evaluation of antigenicity in the presence of anti-L-rhamnose antibodies. J Am Chem Soc. 2010;132:17236–46. doi: 10.1021/ja107029z. [DOI] [PubMed] [Google Scholar]
- 51.Chen W, Gu L, Zhang W, Motari E, Cai L, Styslinger TJ, Wang PG. L-rhamnose antigen: a promising alternative to alpha-gal for cancer immunotherapies. ACS Chem Biol. 2011;6:185–91. doi: 10.1021/cb100318z. [DOI] [PubMed] [Google Scholar]
- 52.Jakobsche CE, Parker CG, Tao RN, Kolesnikova MD, Douglass EF, Spiegel DA. Exploring binding and effector functions of natural human antibodies using synthetic immunomodulators. ACS Chem Biol. 2013;8:2404–2411. doi: 10.1021/cb4004942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Long DE, Karmakar P, Wall KA, Sucheck SJ. Synthesis of alpha-L-rhamnosyl ceramide and evaluation of its binding with anti-rhamnose antibodies. Bioorg Med Chem. 2014;22:5279–89. doi: 10.1016/j.bmc.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sheridan RT, Hudon J, Hank JA, Sondel PM, Kiessling LL. Rhamnose glycoconjugates for the recruitment of endogenous anti-carbohydrate antibodies to tumor cells. Chembiochem. 2014;15:1393–8. doi: 10.1002/cbic.201402019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood. 2012;119:5640–5649. doi: 10.1182/blood-2012-01-380121. [DOI] [PubMed] [Google Scholar]
- 56.Ninkovic T, Kinarsky L, Engelmann K, Pisarev V, Sherman S, Finn OJ, Hanisch FG. Identification of O-glycosylated decapeptides within the MUC1 repeat domain as potential MHC class I (A2) binding epitopes. Mol Immunol. 2009;47:131–140. doi: 10.1016/j.molimm.2008.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ninkovic T, Hanisch FG. O-glycosylated human MUC1 repeats are processed in vitro by immunoproteasomes. J Immunol. 2007;179:2380–2388. doi: 10.4049/jimmunol.179.4.2380. [DOI] [PubMed] [Google Scholar]
- 58.Apostolopoulos V, Yuriev E, Ramsland PA, Halton J, Osinski C, Li W, Plebanski M, Paulsen H, McKenzie IFC. A glycopeptide in complex with MHC class I uses the GalNAc residue as an anchor. Proc Natl Acad Sci U S A. 2003;100:15029–34. doi: 10.1073/pnas.2432220100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ryan SO, Turner MS, Gariepy J, Finn OJ. Tumor antigen epitopes interpreted by the immune system as self or abnormal-self differentially affect cancer vaccine responses. Cancer Res. 2010;70:5788–96. doi: 10.1158/0008-5472.CAN-09-4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ryan SO, Vlad AM, Islam K, Gariepy J, Finn OJ. Tumor-associated MUC1 glycopeptide epitopes are not subject to self-tolerance and improve responses to MUC1 peptide epitopes in MUC1 transgenic mice. Biol Chem. 2009;390:611–8. doi: 10.1515/BC.2009.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lakshminarayanan V, Thompson P, Wolfert MA, Buskas T, Bradley JM, Pathangey LB, Madsen CS, Cohen PA, Gendler SJ, Boons GJ. Immune recognition of tumor-associated mucin MUC1 is achieved by a fully synthetic aberrantly glycosylated MUC1 tripartite vaccine. Proc Natl Acad Sci U S A. 2012;109:261–6. doi: 10.1073/pnas.1115166109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jackson DC, O’Brien-Simpson N, Ede NJ, Brown LE. Free radical induced polymerization of synthetic peptides into polymeric immunogens. Vaccine. 1997;15:1697–1705. doi: 10.1016/s0264-410x(97)00085-6. [DOI] [PubMed] [Google Scholar]
- 63.Wiesmuller KH, Jung G, Hess G. Novel low-molecular-weight synthetic vaccine against foot-and-mouth disease containing a potent B-cell and macrophage activator. Vaccine. 1989;7:29–33. doi: 10.1016/0264-410x(89)90007-8. [DOI] [PubMed] [Google Scholar]
- 64.Deres K, Schild H, Wiesmuller KH, Jung G, Rammensee HG. In vivo priming of virus-specific cytotoxic T lymphocytes with synthetic lipopeptide vaccine. Nature. 1989;342:561–4. doi: 10.1038/342561a0. [DOI] [PubMed] [Google Scholar]
- 65.Roth A, Espuelas S, Thumann C, Frisch B, Schuber F. Synthesis of thiol-reactive lipopeptide adjuvants. Incorporation into liposomes and study of their mitogenic effect on mouse splenocytes. Bioconj Chem. 2004;15:541–553. doi: 10.1021/bc034184t. [DOI] [PubMed] [Google Scholar]
- 66.Bessler W, Baier W, Hoffmann P, Heinevetter L, Wiesmüller K, Jung G. Bacterial lipopeptides constitute efficient novel immunogens and adjuvants in parenteral and oral immunization. Behring Institute Mitteilungen. 1997:390–399. [PubMed] [Google Scholar]
- 67.Braun V. Covalent lipoprotein from the outer membrane of Escherichia coli. Biochim Biophys Acta. 1975;415:335–377. doi: 10.1016/0304-4157(75)90013-1. [DOI] [PubMed] [Google Scholar]
- 68.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 69.Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between Toll-like receptors. Proc Natl Acad Sci U S A. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cai H, Sun ZY, Chen MS, Zhao YF, Kunz H, Li YM. Synthetic multivalent glycopeptide-lipopeptide antitumor vaccines: impact of the cluster effect on the killing of tumor cells. Angew Chem Int Ed Engl. 2014;53:1699–703. doi: 10.1002/anie.201308875. [DOI] [PubMed] [Google Scholar]
- 71.Matzinger P. The JAM test - a simple assay for DNA fragmentation and cell-Death. J Immunol Methods. 1991;145:185–192. doi: 10.1016/0022-1759(91)90325-a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.