

Abstract
Background
Single-nucleotide polymorphisms (SNPs) have become the genetic markers of choice in various genetic, ecological, and evolutionary studies. Genotyping-by-sequencing (GBS) is a next-generation-sequencing based method that takes advantage of reduced representation to enable high-throughput genotyping using a large number of SNP markers.
Results
In the present study, the distribution of non-redundant SNPs in the parents of 12 rice recombination line populations was evaluated through GBS. A total of 45 Gigabites of nucleotide sequences conservatively provided satisfactory genotyping of rice SNPs. By assembling to the genomes of reference genomes of japonica Nipponbare, we detected 22,682 polymorphic SNPs that may be utilized for QTL/gene mapping with the Recombinant Inbred Lines (RIL) populations derived from these parental lines. Meanwhile, we identified polymorphic SNPs with large effects on protein-coding and miRNA genes. To validate the effect of the polymorphic SNPs, we further investigated a SNP (chr4:28,894,757) at the miRNA binding site in the 3′-UTR region of the locus Os4g48460, which is associated with rice seed size. Os4g48460 encodes a putative cytochrome P450, CYP704A3. Direct degradation of the 3′-UTR of the CYP704A3 gene by a miRNA (osa-miRf10422-akr) was validated by in planta mRNA degradation assay. We also showed that rice seeds of longer lengths may be produced by downregulating CYP704A3 via RNAi.
Conclusions
Our study has identified the genome-wide SNPs by GBS of the parental varieties of RIL populations and identified CYP704A3, a miRNA-regulated gene that is responsible for rice seed length.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-016-0779-3) contains supplementary material, which is available to authorized users.
Keywords: SNP, miRNA, Genetic diversity, Seed size, Rice
Background
Rice is the first crop plant from which a high-quality reference genome sequence from a single variety has been produced. Single nucleotide polymorphisms (SNPs) may be functionally responsible for specific traits or phenotypes, or they may be informative in tracing the evolutionary history of a species or the pedigree of a variety. SNPs are rapidly replacing simple sequence repeats (SSRs) because these are more abundant, stable, amenable to automation, efficient, and increasingly cost-effective [1].
SNPs have become the genetic marker of choice in the analysis of partially or completely sequenced genomes due to its ubiquity in the genome [2]. In rice, the primary sequencing data that led to the first whole-genome SNP discovery was derived from the draft sequences of the japonica cultivar Nipponbare and indica cultivar 93–11 [3, 4]. The SNP pools were mainly limited to two varieties. Another SNP discovery set from the OryzaSNP project has identified approximately 160,000 high-quality SNPs and has provided more insights by detecting informative SNPs across 20 diverse rice varieties [5]. Next-generation sequencing at 19× coverage across 517 rice varieties has identified over 3.6 million SNPs, of which 167,000 SNPs were located within coding regions [6]. Recently, rice re-sequencing projects have been conducted for various germplasm [7–9].
The decreasing cost, along with the rapid advancement of next-generation sequencing technology and related bioinformatics resources, has facilitated the large-scale discovery of SNPs in various plant species. Genotyping-by-sequencing (GBS) is a next-generation-sequencing based method that takes advantage of reduced representation to enable high-throughput genotyping of a large number of SNP markers. GBS has been applied in SNP genotyping for quantitative trait loci (QTL) mapping and gene identification in rice [10–12]. The TASSEL-GBS pipeline successfully fulfills the following key design criteria: (1) Ability to run on the modest computing resources, including desktop or laptop machines with only 8–16 GB of RAM; (2) Scalability from small to extremely large studies, in which hundreds of thousands or even millions of SNPs can be scored; (3) Applicability in a fast-breeding context, in which rapid genotyping is required due to the high frequency of tissue collection [13].
A total of 12 parental lines were selected for GBS because of their specific characteristics, including grain quality in rice breeding. Guichao2 and IR24 are two varieties with indica rice characters such as seed type and disease resistance [14]. Guichao2 was of interest because of its wide range of adaptability, high yield potential, and could be planted as an early, or middle, or late variety. However, this particular variety was later excluded because of bad grain quality. Koshihikari and Asominori are varieties from Japan, and USSR5 is a variety from Russia that shows typical japonica rice characters such as good quality, but is susceptible to rice strip virus disease (RSVD) [15]. Sasanishiki is a high-yielding indica cultivar [16]. Habataki has very high yield potential and a short, stiff stem, but is susceptible to cold during booting and early growth [17]. Kasalath harbors resistance genes against standard differential blast isolates from the Philippines and Japan [18]. Nanjing35 has superior grain yield, but poor appearance. N22 possesses QTL/genes related to dormancy [19]. RILs and NILs derived from these parental varieties are currently under investigation for genes controlling agronomic traits; therefore the SNP database will be very useful for gene mapping and isolation by using high-density SNP markers. The number of recombination events and the marker density of parental varieties determine the resolution of gene mapping. By using the cost-effective GBS approach, we have detected SNPs among the parents of 12 rice recombination line populations.
The P450s in biosynthetic pathways play critical roles in the synthesis of lignins, pigments, defense compounds, fatty acids, hormones, and signaling molecules [20]. CYP704 shows higher homology with CYP86, CYP94, CYP96 compared to the other P450s, and these are all non A-type P450s belonging to CYP86 [21]. CYP704B2 is involved in anther cutin biosynthesis and pollen exine formation in rice [22]. CYP704A3 is a member of the CYP family, and most of its family members are located in the ER [20], including CYP78A13, which influences grain size and yield in rice [23]. We herein report SNPs for the parental varieties of mapping populations and a miRNA regulated gene, CYP704A3, underlying rice seed size.
Results
Sequencing and variation calling
We identified a total of 22,682 polymorphic SNPs in 12 parental varieties and Nipponbare in relation to the reference genome (Additional file 1: Table S1). To explore the genomic distribution of the patterns of DNA polymorphisms between the indica and japonica subspecies, the SNP count based on our sample was plotted along each chromosome (Fig. 1). SNP count (Fig. 1, solid line) was defined as the number of SNPs in a 200-kb interval. Non-random patterns of SNP distribution were observed, with highly different SNP frequencies detected on all chromosomes. These data support previous findings that polymorphisms in the rice genome (from the indica-japonica perspective) are non-randomly distributed [24]. Relatively low SNP polymorphisms were observed in the regions highlighted in green bars (Fig. 1), which could be due to stringent recombination restrictions, or lack of restriction enzyme sites or reads in these regions. More markers need to be developed in these low-density SNP regions for QTL/gene mapping.
Fig. 1.
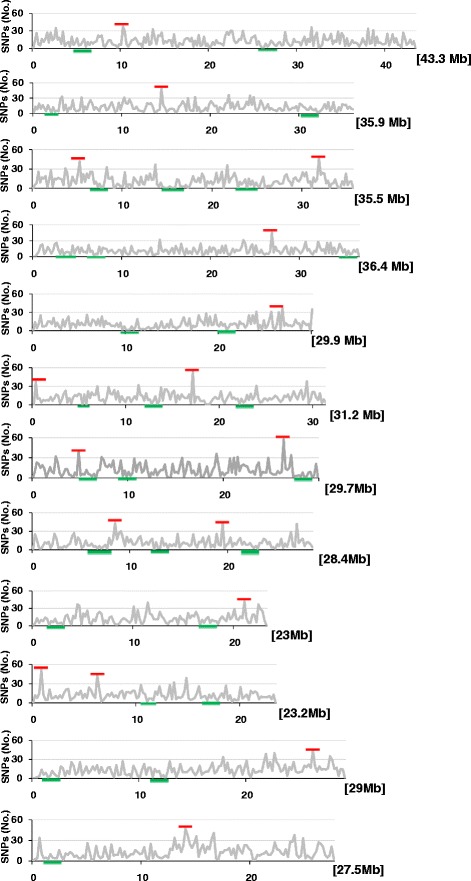
Distribution of SNPs in 12 chromosomes of different rice varieties. The x-axis represents the physical distance along each chromosome, which is split into 200-kb windows. The total size of each chromosome is shown in brackets. The y-axis indicates the number of SNPs. The regions with relatively high and low density are labeled in red and green bars, respectively
We aimed to detect SNPs in the difference rice varieties by using the TASSEL GBS pipeline. Furthermore, we validated 12 SNP genotypes (Additional file 2: Table S2) from 12 chromosomes, respectively, by using Sanger sequencing on the ABI3730xl DNA sequencher (ABI, CA, USA) with the BigDye V3.0 kit. The SNP database with a low rate of missing data will be useful for QTL detection in the populations derived from the 12 varieties.
The phylogenetic tree produced using the 22,682 SNPs revealed three distinct groups, with japonicas clustered into one group, and the other two groups together with aus and indica types (Fig. 2a). Three distinct groups were identified by principal component analysis with well-separated lines, corresponding to indica, aus, and japonica rice species (Fig. 2b). Consistent with the phenotypical classification, the varieties from China, Guichao2, Nanjing11 and Nanjing35 were grouped into the indica type, together with IR36 and IR24 from IRRI; the varieties from Japan, Sasanishiki, Koshihikari, Habataki, and Asominori were grouped into japonica, together with USSR5 from Russia; the varieties N22 and Kasalath from India were grouped into aus. Based on this classification, more SNP polymorphisms could be expected between the parental lines with distinct relationships. Thus, germplasm classification of the 12 rice varieties was properly conducted and the results were informative for further RIL or NIL construction.
Fig. 2.

Genetic structure and population differentiation in 13 rice parental varieties. a Unweighted pair group method with arithmetic mean (UPGMA) dendrogram based on 22,682 SNPs. b Principal component analysis for the entire set of RILs (n = 306)
We checked 12 SNP genotypes showing the same genotypes within indica and japonica subgroups by using Sanger sequencing on the ABI3730xl DNA sequencher (ABI, CA, USA) using the BigDye V3.0 kit. Sequences generated by each primer pair were aligned using Sequencher and SNPs were validated by visual inspection.
SNP annotations and large-effect SNPs
The SNPs were annotated and classified based on their location in intergenic regions, introns, 5′-UTR, 3′-UTR or exon (63.1, 8.5, 4.1, 4.8, and 19.4 %, respectively) (Table 1). We analyzed the SNPs with potentially large effects on gene expression and protein alterations.
Table 1.
Summary of SNPs in the 12 varieties
| All | Intergene | Intron | 5′-UTR | 3′-UTR | Exon | |
|---|---|---|---|---|---|---|
| chr1 | 2703 | 1545 | 228 | 120 | 217 | 690 |
| chr2 | 2205 | 1367 | 175 | 122 | 89 | 518 |
| chr3 | 2099 | 1281 | 212 | 79 | 100 | 499 |
| chr4 | 2033 | 1239 | 211 | 107 | 92 | 453 |
| chr5 | 1626 | 1010 | 154 | 67 | 81 | 354 |
| chr6 | 1927 | 1232 | 128 | 76 | 111 | 406 |
| chr7 | 1746 | 1092 | 148 | 60 | 67 | 408 |
| chr8 | 1613 | 1057 | 148 | 83 | 49 | 292 |
| chr9 | 1463 | 986 | 133 | 70 | 38 | 254 |
| chr10 | 1471 | 994 | 131 | 35 | 72 | 287 |
| chr11 | 1972 | 1213 | 148 | 63 | 119 | 490 |
| chr12 | 1824 | 1303 | 117 | 42 | 65 | 327 |
| All | 22682 | 14319 | 1933 | 924 | 1100 | 4978 |
The mutant of the cytochrome P450 gene CYP724B1 accumulates large amounts of mRNA; furthermore, its seed is shorter than that of the wide-type [25]. In the present study, we detected an A/T polymorphism between Asominori and IR24 that was associated with short- and long-grain phenotypes respectively. The SNP in the fourth exon (chr4:28,894,497) is predicted to result in the amino acid alteration of Aspartic acid to Valine. These findings suggest that CYP704A3 might be associated with grain length. Thus, we decided to further investigate a protein-altering A/T polymorphism located in the exon of the cytochrome P450 gene, CYP704A3 (LOC_Os04g48460).
We sequenced this gene that exhibited a total of four SNPs, with two present in the exon and two within the 3′-UTR. A G/A SNP (chr4:28,894,757) in the 3′-UTR was detected in the binding region between CYP704A3 and its miRNA gene, osa-miRf10422-akr (Fig. 3a).
Fig. 3.
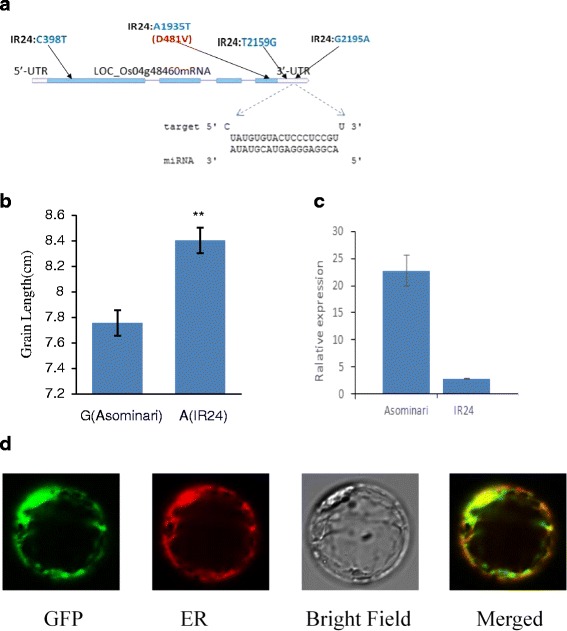
osa-miRf10422-akr precursors and its putative target gene. a CYP704A3 is a putative target of osa-miRf10422-akr. CYP704A3 structure and mutation sites are labeled as SNPs (blue) and the changed amino acid residues (red). b Significant differences in grain length between SNP genotypes of the CYP704A3 gene. **indicates a significance at P < 0.01. c qRT-PCR analysis shows the expression of CYP704A3 in maturing seeds. Asominori and IR24 were analyzed in terms of the expression of the CYP704A3 transcript. Seeds with longer lengths have lower levels of relative expression, similar to the other members of subfamily CYP450 such as CYP724B1, of which its mRNA accumulates at higher levels in CYP724B1 mutants with shorter seed compared to that in the wide-type. d Subcellular localization of the CYP704A3Protein. GFP signals of the CYP704A3-GFP fusion proteins localized in the endoplasmic reticulum of rice protoplasts
We also observed the effects of CYP704A3 on grain length in a population consisting 184 rice landraces by developing a SNP marker for G/A genotyping the CYP704A3 gene. The average seed length of plants with the IR24 allele (8.40 ± 0.1 mm) was longer than that harboring the Asominori allele (7.76 ± 0.09 mm) (P < 0.01, Fig. 3b). Because of the positive seed length/SNP correlation and the critical position of SNP in the binding region, we deduced that the SNP mutation was crucial to grain size.
Seeds with CYP704A3 of IR24 allele genotype were longer than that of Asominori allele (Fig. 3b). We further compared the expression levels of CYP704A3 in maturing seeds of IR24 and Asominori with long and short seeds respectively. The seeds of IR24 with longer size had relative lower expression of CYP704A3 (Fig. 3d).
To verify the subcellular location of CYP704A3, we constructed a CYP704A3-GFP fusion expression vector, and then transformed the recombinant expression vector into rice protoplasts. Confocal microscopy showed that the green fluorescent signals of CYP704A3-GFP co-localized with the autofluorescence signals of the ER (Fig. 3f).
To further confirm the function of CYP704A3, CYP704A3-RNAi transgenic rice plants were generated. Several independently transformed plants showed a reduction in the level of expression of the CYP704A3 gene (Fig. 4a). Three of these were selected to represent the relative expression of CYP704A3 in relation to the transgenic negative control RNAi-4 (Fig. 4b). The reduced expression of CYP704A3 caused various degrees of elongation in grain length (Fig. 4c). Significant difference (P ≤ 0.01) in grain length (Fig. 4d) and length/width ratio (P ≤ 0.01) (Fig. 4d) among the wild-type, RNAi-4, and OsCYP704A3-RNAi transgenic plants were observed. These results further indicated that CYP704A3 negatively regulates grain length in rice.
Fig. 4.
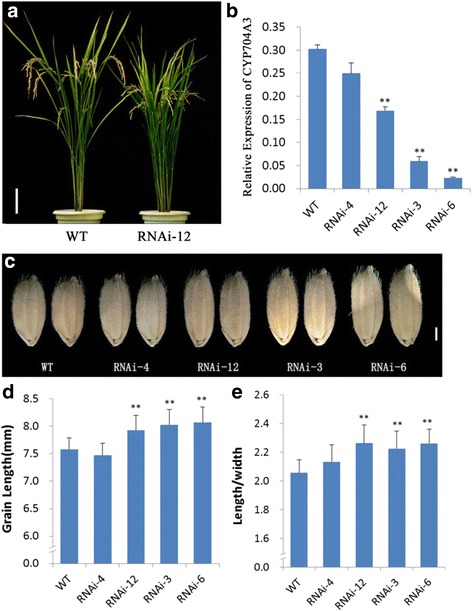
CYP704A3-RNAi transgenic plantsproduced longer grains. a The morphology of wild-type and RNAi-12 transgenic plants. Scale bar: 10 mm. b Relative expression levels of CYP704A3 mRNA of the wild-type RNAi-4 (as a transgenic negative control) and T1CYP704A3-RNAi transgenic plants which were determined by qRT-PCR. c Grains of the wild-type, RNAi-4 (as a transgenic negative control), RNAi-12, RNAi-3 and RNAi-6. Scale bar: 1 mm. d Grain length (n = 100). e Grain length to width ratio (n = 100)
Interaction between osa-miRf10422-akr and the CYP704A3 target gene
To validate that CYP704A3 is regulated by osa-miRf10422-akr, a schematic representation of the reporters and the effectors (Fig. 5a) was used in this assay. To examine the ability of plant expression vectors to produce osa-miRf10422-akr miRNAs in vivo, an Agrobacterium strain harboring pCAMBIA1300-35S:osa-miRf10422-akr or the control pCAMBIA 1300 vector (35S) was infiltrated into N. benthamiana leaves, together with the reporter gene EGFP which was fused with the 3′-UTR of the rice CYP704A3 gene, which contained the putative miRNA target. When the effecter recognizes the miRNA target in the reporter construct, the mRNA level of EGFP and the fluorescence of EGFP are downregulated. The fluorescence of the agroinfiltrated leaves was taken at 2 dpi under UV illumination. Fluorescence imaging showed that EGFP and osa-miRf10422-akr were co-expressed (Fig. 5b) and together with miRNA target region in the 3′-UTR of the CYP704A3 gene (Fig. 5c). Total RNA was extracted at 3 dpi, and quantitative EGFP mRNA analysis using the average measurements of three leaves utilized in each infiltration treatment (Fig. 5d). As expected, osa-miRf10422-akr expression affected the CYP704A3 gene expression both at the transcriptional and protein levels. These findings therefore indicate that osa-miRf10422-akr participates in seed size determination by directly regulating CYP704A3.
Fig. 5.
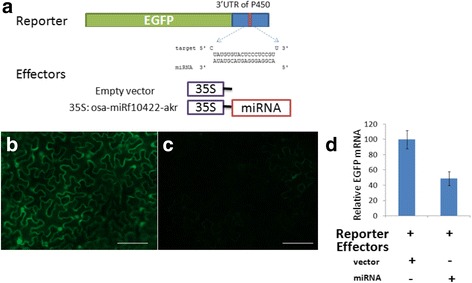
The effects of osa-miRf10422-akrexpression on the accumulation of the CYP704A3 gene. The schematic representation of the reporters and the effectors used in this assay is shown in (a). GFP fluorescence images of the co-expression of osa-miRf10422-akr with the reporter gene EGFP, which was fused with the empty vector control (b) and miRNA target region in the 3′-UTR of the CYP704A3 gene (c). Fluorescence imaging analysis of the agroinfiltrated leaves at 2 dpi under UV illumination. Quantitation of EGFP mRNA as averaged from three leaves from each infiltration treatment (d)
The CYP704A3 target gene underwent selection for seed size improvement
To test whether the CYP704A3 target gene and the osa-miRf10422-akr had undergone selection within the parental varieties, we calculated the linkage disequilibrium (LD) of the two genes and its flanking regions on both sides using our GBS data. LD analysis of the two genomic regions, namely, osa-miRf10422-akr and its putative target gene, CYP704A3, of these parental varieties, revealed different patterns of LD blocks (Fig. 6). The r2-values for the miRNA osa-miRf10422-akr did not show any increase in these parental varieties that are commonly used in rice breeding. No LD blocks were detected in the osa-miRf10422-akr region (~24,872 kb) on chromosome 3 (Fig. 6a). In contrast, the r2-values of the CYP704A3 gene increased in these parental varieties. A strong LD block was detected in the target gene LOC_Os04g48460 on chromosome 4 (Fig. 6b). The detection of a strong LD block in CYP704A3 compared to that in the miRNA gene indicated that the CYP704A3 target gene may have undergone selection within these parental rice varieties, although additional investigations should be performed.
Fig. 6.
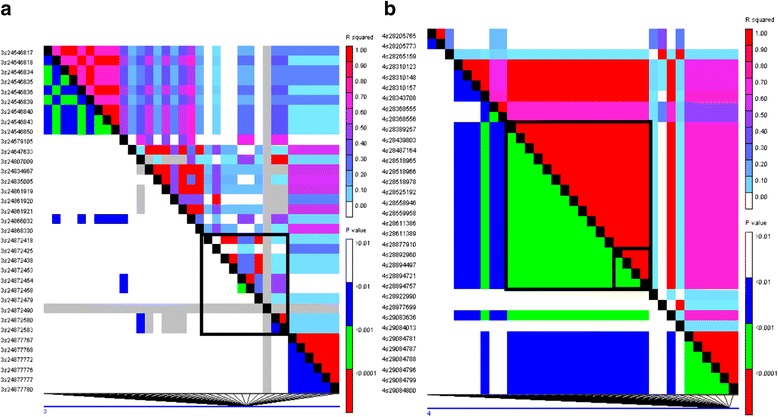
Patterns of LD blocks in two genomic regions of osa-miRf10422-akr precursors and its putative target gene, LOC_Os04g48460. a No LD blocks in the osa-miRf10422-akr region (~24,872Kb) on chromosome 3 were detected. b LD block in the big black block encompassing the LOC_Os04g48460 region (~28,892 kb in small black block) on chromosome 4. Red and white spots indicate strong (r 2 = 1) and weak (r 2 = 0) LD, respectively
Discussion and conclusions
De novo genome sequencing or re-sequencing generates the gigabytes of data that need to be analyzed at a very large scale. On the other hand, smaller subsets of SNP data may be utilized for trait-marker analysis and genomic selection for breeding selection. By simplifying genomic data by using GBS, linkage and linkage disequilibrium may be cost-effectively analyzed, and deep candidate gene re-sequencing may be conducted during targeted SNP detection [26]. The genome can be significantly simplified by restriction enzyme digestion. The SNPs can be detected at restriction-associated sites. A higher number of short-read alignments at regions of interest may help in more precisely resolving the real allele frequency of mutant alleles in bulked DNA [26]. Therefore, GBS may be employed for further gene fine-mapping and allele analysis.
SNP markers of CYP704A3 gene were developed for genotyping a population consisting of 184 landraces, and significant mean differences in the t-test suggested that the gene may play roles in grain development. In rice, at least five proteins have been identified through quantitative genetic studies of seed size. Except for the identified novel positive regulator, which is the putative serine carboxypeptidase encoded by GS5 [27], four other proteins are required to limit final seed size and weight [28–31]. In the present study, we observed the upregulation of the CYP704A3 gene in the short seed variety Asominori, compared to the long seed variety IR24. This result was in agreement with previous findings on the P450 gene CYP724B1 [25]. CYP724B1 mRNA accumulated at higher levels in d11 mutants with shorter seeds compared to the wild-type plants [25].
In addition to the cytochrome P450 gene, CYP724B1, in rice that controls seed growth, a gene encoding the ortholog of KLUH, SlKLUH, a P450 enzyme of the CYP78A subfamily in tomato (Solanumly copersicum), was reported to affect fruit mass and size [32]. Here, we present evidences indicating that another rice P450 gene, CYP704A3, was associated with seed size. Interestingly, CYP704A3 was found and validated to be a target gene of osa-miRf10422-akr. The mechanisms on whether and how the interaction between osa-miRf10422-akr and CYP704A3 controls seed length require further investigation.
In conclusion, we detected a total of 22,682 DNA polymorphisms by high-throughput GBS of mapped reads by assembling these using the reference genomes of japonica Nipponbare. Detection of genome-wide DNA polymorphisms by high-throughput GBS enabled us to identify sequence diversity derived from rice differentiation and genomic locations that were related to traits of agronomic importance. We identified polymorphic SNPs in the rice cytochrome P450 gene, CYP704A3, which was targeted by a miRNA gene, osa-miRf10422-akr, and associated with seed size.
Methods
Plant materials
A total of 184 rice accessions were used as study material. The seeds of all accessions were collected, stored and supplied by the State Key Laboratory of Crop Genetics and Germplasm Enhancement of Nanjing Agricultural University, Jiangsu, China. The 184 landraces are collected from eight geographic regions. East China had the most entries, accounting for 27.7 % of the study material, followed by Southwest China (21.2 %), South China (14.7 %), North China (7.6 %), Middle China (8.2 %), Northeast China (5.4 %), Northwest (4.4 %), and Southeast Asia (10.9 %). This study population, including the 12 landraces, was used in the association analysis of seed size. In the past decades, these accessions have been widely used as parents in plant breeding. The 184 accessions were planted from May to November in 2013 and 2014 at the Tuqiao Experimental Farm of Nanjing Agricultural University. For the field experiments, the accessions were grown in a randomized complete block design using two replicates. The space was 20 cm between rows and 17 cm between individuals, with standard agronomic management.
Sample preparation and sequencing
Genomic DNA was extracted from the 12 rice accessions, including varieties originating from China (Guichao2, Nanjing11, and Nanjing35), Japan (Sasanishiki, Koshihikari, Habataki, and Asominori), India (N22 and Kasalath), and IRRI (IR36 and IR24), and Russia (USSR5). DNA was extracted from leaf tissues using the DNeasy Plant Mini Kit (Qiagen, Germany).
Sequencing library preparation and sequencing
RAD sequencing is one of several strategies recently developed to improve short-read sequencing by reducing their complexity [33]. RAD sequencing reduces genome complexity by resequencing only the stretches of DNA adjacent to recognition sites of a chosen restriction endonuclease and has been proven to be a powerful tool for genetic analysis [34]. The RAD library of the 12 varieties was prepared for single end-sequencing according to Baird et al. [23] with some modifications. Briefly, barcodes were 6-bp long, being at least two mutational steps separated from each other. A total of 2 μg genomic DNA from each inbred was digested for 1 h at 37 °C in a 50-μl reaction with 50 U of EcoRI (New England Biolabs). The RAD library was sequenced on an Ion Torrent PGM and Illumina Hiseq2500. The raw reads that were of high quality were used for the analysis of genetic variations in the 12 accessions.
Mapping of reads
A large number of reads were assembled based on the genomic sequences of the japonica rice cultivars Nipponbare using TMAP3.6. SNPs were detected by comparison alignment using the Nipponbare sequences as reference. Parameters were set as default to classify whether mismatches were sequencing errors or genomic variations.
SNP detection and analysis
Reads were separated by barcode and trimmed at the 3′ ends. The RAD tags at the RAD clusters were screened for SNPs and InDels using Oryza sativa L. cv. Nipponbare (http://rapdb.dna.affrc.go.jp/download/irgsp1.html) as reference. SNPs of each sample were collected using the TASSEL pipeline [27]. Filtering and imputation procedures were performed to call the first 64-bp of the high quality reads with default parameters in the pipeline. A phylogenetic tree was produced using the 22,682 SNPs to show the relationships among the 13 landraces. The SNP cladogram-tree dataset was generated using the neighbor-joining method as provided in TASSEL [35]. Three distinct groups were identified by principal component analysis using TASSEL [35]. LD of the two genes and its flanking regions on both sides was calculated using our GBS data using TASSEL [35].
A dCAPs marker was developed for the SNP in the binding region between CYP704A3 and its miRNA gene, osa-miRf10422-akr. The average seed length of plants with T identical to IR24 was compared to the A that was identical to Asominori using the t-test, using a significance level of P < 0.01.
Polymorphism verification and dCAPs marker genotyping
Twelve polymorphic RAD clusters that were located in the 12 chromosomes were randomly selected. Primers were designed to flank the entire RAD cluster. The target sequence was amplified in all 12 varieties. The PCR conditions were as follows: 94 °C for 3 min; followed 35 cycles of 94 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s; 72 °C for 5 min, and then held at 4 °C. Sanger sequencing of PCR products was conducted using an ABI3730xl DNA sequencer, following standard protocols. Primers for CYP704A3 were as follows: Forward 5′-CAAGGGCGGCGCTGGTCTATT-3′ and Reverse 5′-ATTTTCCTTTGGTTATGTTTTGTA-3′.
Real-time PCR and subcellular localization of the CYP704A3 protein
Total RNA was isolated from maturing seeds using a plant RNA purification reagent (Invitrogen). Synthesis of cDNA and real-time PCR were performed as described elsewhere [36]. The rice Actin gene was selected as endogenous reference. PCR specificity was examined by 3 % agarose gel electrophoresis using 5 μL of each reaction to check the right product length and to make sure that no primer dimers or non-specific amplicons were generated. The primers for real-time PCR were as follows: Forward 5′-GTCGCCTTGTCGCTGCTGCTAC-3′ and Reverse 5′-CGGGCGGATACCTGCGTTTCT-3′ for the CYP704A3 gene.
For the subcellular localization of the CYP704A3 protein in rice protoplasts, the coding sequence of the CYP704A3 gene was amplified and inserted into the BglII/NotI sites of the PA7 vector to form a translational fusion with the C-terminus of the GFP. The transient expression constructs were transformed into rice protoplasts as described elsewhere [37]. The fluorescence of GFP was observed using a confocal laser scanning microscope (Leica TCS SP5). The primers used in subcellular localization assays were as follows: Forward: 5′-ATGGACGAGCTGTACAGATCTATGGAGTCGCCGCT-3′ and Reverse: 5′-GAACTGCAGCCGGGCGGCCGCTCACCGGGCCAATG-3′.
Rice transformation
To validate the function of CYP704A3, a CYP704A3-RNAi vector was constructed and introduced to wild-type plants. To construct the RNAi vector, a 316-bp fragment within the CDS was amplified using the forward primer, 5′-GGGGTACCTCCGGCGGCGAAGG-3′ and reverse primer, 5′-CGAGCTCTTGCTCTCTGCTCATCTG-3′ with the KpnI and SacI enzyme digestion sites, and the reverse sequence was amplified using the forward primer, 5′- GGTACGTATCCGGCGGCGAAGG-3′ and reverse primer, 5′-AACTGCAGTTGCTCTCTGCTCATCTG-3′ with the SnaBI and PstI enzyme digestion sites.
The rice plants examined under natural field conditions were grown in normal rice growing seasons at the Experimental Station of Nanjing Agricultural University, Nanjing, China. Seeds were planted in a seedbed in mid-May and transplanted to the field in mid-June 2015. Field management, including irrigation, fertilizer application, and pest control, was essentially performed using standard agricultural practices. Harvested paddy rice was air-dried and stored at room temperature prior to testing. Fully filled grains from each plant were randomly chosen (n = 100) for grain size evaluation.
Quantitative EGFP fluorescence and miRNA analysis
GFP fluorescence imaging of the coexpression of osa-miRf10422-akr with the reporter gene EGFP fused with the empty vector control and the miRNA target region in 3′-UTR of the CYP704A3 gene was performed. The fluorescence of the agroinfiltrated leaves was examined at 2 dpi under UV illumination. Quantitative EGFP mRNA of three leaves from each infiltration treatment was also analyzed.
Availability of data and materials
The data sets supporting the results of this article are included within the article and its additional files. All the sequencing data produced in this study have been deposited in NCBI Short Read Archive (http://www.ncbi.nlm.nih.gov/sra/) and can be accessed under the SRA accession numbers: SRR3307074, SRR3307908, SRR3308144, SRR3308415, SRR3308416, SRR3308417, SRR3308419, SRR3308421, SRR3308737, SRR3310108, SRR3310109, SRR3310110, SRR3310115, SRR3310157, SRR3310158. SRR3310111, SRR3310156, and SRR3308736.
Acknowledgments
The Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD, 010809001), and the Fundamental Research Funds for the Central Universities (KYRC201208, KYZ201202-6), and National Key Technology Support Program project (2015BAD01B02-7), China supported this study. CM was supported by the “Shuangchuang”, Project (2012), and “Innovation Team Core Member” (2013) 140, Jiangsu Province, China. JY (Jian Ye) was supported by the State Key Laboratory of Plant Genomics, Institute of Microbiology, Chinese Academy of Sciences. The funding agencies had no role in the study design, data collection and analysis, decision to publish, or manuscript preparation. We thank Dr Yunlu Tian for his help in data collection and management in field experiments.
Abbreviations
- 3′-UTR
3′-untranslated region
- EGFP
enhanced green fluorescent protein
- ER
endoplasmic reticulum
- GBS
genotyping-by-sequencing
- LD
linkage disequilibrium
- QTL
quantitative trait loci
- RAD
restriction-site-associated DNA
- RIL
recombinant inbred line
- SNP
single-nucleotide polymorphism
Additional files
22,682 polymorphic SNPs among the 12 parental varieties and Nipponbare as the reference genome. (XLSX 1756 kb)
Primers for 12 SNP genotypes from the 12 chromosomes respectively, using Sanger sequencing on the ABI3730xl DNA sequencher (ABI, CA, USA). (PDF 95 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CW conceived and performed the experiments, and wrote the manuscript; WT performed the GBS experiments and analyzed the data; TW and J Ye performed the miRNA and transgenic experiments; J Ye analyzed the data and wrote the manuscript; JS, YJ, J Yu, JT and GC performed the field experiments; all authors read and approved the final manuscript.
Contributor Information
Weijie Tang, Email: 2014201050@njau.edu.cn.
Tingting Wu, Email: 2014101123@njau.edu.cn.
Jian Ye, Email: jianye@im.ac.cn.
Juan Sun, Email: 15150532132@163.com.
Yue Jiang, Email: 2013101101@njau.edu.cn.
Jun Yu, Email: 2015101124@njau.edu.cn.
Jianpeng Tang, Email: 2013101132@njau.edu.cn.
Gaoming Chen, Email: 2014101124@njau.edu.cn.
Chunming Wang, Email: wangchm@njau.edu.cn.
Jianmin Wan, Email: wanjianmin@caas.cn.
References
- 1.McCouch SR, Zhao K, Wright M, Tung C-W, Ebana K, Thomson M, Reynolds A, Wang D, DeClerck G, Ali ML. Development of genome-wide SNP assays for rice. Breed Sci. 2010;60(5):524–535. doi: 10.1270/jsbbs.60.524. [DOI] [Google Scholar]
- 2.Kumar S, Banks TW, Cloutier S. SNP discovery through next-generation sequencing and its applications. Int J Plant Genomics. 2012;2012:831460. doi: 10.1155/2012/831460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feltus FA, Wan J, Schulze SR, Estill JC, Jiang N, Paterson AH. An SNP resource for rice genetics and breeding based on subspecies indica and japonica genome alignments. Genome Res. 2004;14(9):1812–1819. doi: 10.1101/gr.2479404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen Y-J, Jiang H, Jin J-P, Zhang Z-B, Xi B, He Y-Y, Wang G, Wang C, Qian L, Li X. Development of genome-wide DNA polymorphism database for map-based cloning of rice genes. Plant Physiol. 2004;135(3):1198–1205. doi: 10.1104/pp.103.038463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McNally KL, Childs KL, Bohnert R, Davidson RM, Zhao K, Ulat VJ, Zeller G, Clark RM, Hoen DR, Bureau TE. Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc Natl Acad Sci. 2009;106(30):12273–12278. doi: 10.1073/pnas.0900992106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, Li C, Zhu C, Lu T, Zhang Z. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet. 2010;42(11):961–967. doi: 10.1038/ng.695. [DOI] [PubMed] [Google Scholar]
- 7.Xu X, Liu X, Ge S, Jensen JD, Hu F, Li X, Dong Y, Gutenkunst RN, Fang L, Huang L. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat Biotechnol. 2012;30(1):105–111. doi: 10.1038/nbt.2050. [DOI] [PubMed] [Google Scholar]
- 8.Alexandrov N, Tai S, Wang W, Mansueto L, Palis K, Fuentes RR, Ulat VJ, Chebotarov D, Zhang G, Li Z. SNP-Seek database of SNPs derived from 3000 rice genomes. Nucleic Acids Res. 2015;43(D1):D1023–D1027. doi: 10.1093/nar/gku1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duitama J, Silva A, Sanabria Y, Cruz DF, Quintero C, Ballen C, Lorieux M, Scheffler B, Farmer A, Torres E. Whole genome sequencing of elite rice cultivars as a comprehensive information resource for marker assisted selection. PLoS One. 2015;10(4):e0124617. doi: 10.1371/journal.pone.0124617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arbelaez JD, Moreno LT, Singh N, Tung C-W, Maron LG, Ospina Y, Martinez CP, Grenier C, Lorieux M, McCouch S. Development and GBS-genotyping of introgression lines (ILs) using two wild species of rice, O. meridionalis and O. rufipogon, in a common recurrent parent, O. sativa cv. Curinga. Mol Breed. 2015;35(2):1–18. doi: 10.1007/s11032-015-0276-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spindel J, Wright M, Chen C, Cobb J, Gage J, Harrington S, Lorieux M, Ahmadi N, McCouch S. Bridging the genotyping gap: using genotyping by sequencing (GBS) to add high-density SNP markers and new value to traditional bi-parental mapping and breeding populations. Theor Appl Genet. 2013;126(11):2699–2716. doi: 10.1007/s00122-013-2166-x. [DOI] [PubMed] [Google Scholar]
- 12.Thomson MJ. High-throughput SNP genotyping to accelerate crop improvement. Plant Breed Biotechnol. 2014;2(3):195–212. doi: 10.9787/PBB.2014.2.3.195. [DOI] [Google Scholar]
- 13.Deschamps S, Llaca V, May GD. Genotyping-by-sequencing in plants. Biology. 2012;1(3):460–483. doi: 10.3390/biology1030460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun D, Jiang L, Zhang Y. Detection of QTL associated with rice stripe resistance in cultivar IR24. Acta Agron Sin. 2007;33:25–30. [Google Scholar]
- 15.Wang B, Jiang L, Zhang Y, Zhang W, Wang M, Cheng X, Liu X, Zhai H, Wan J. QTL mapping for resistance to strip virus disease in rice. Plant Breed. 2011;130(3):321–327. doi: 10.1111/j.1439-0523.2010.01796.x. [DOI] [Google Scholar]
- 16.Ando T, Yamamoto T, Shimizu T, Ma XF, Shomura A, Takeuchi Y, Lin SY, Yano M. Genetic dissection and pyramiding of quantitative traits for panicle architecture by using chromosomal segment substitution lines in rice. Theor Appl Genet. 2008;116(6):881–890. doi: 10.1007/s00122-008-0722-6. [DOI] [PubMed] [Google Scholar]
- 17.Ashikari M, Sakakibara H, Lin S, Yamamoto T, Takashi T, Nishimura A, Angeles ER, Qian Q, Kitano H, Matsuoka M. Cytokinin oxidase regulates rice grain production. Science. 2005;309(5735):741–745. doi: 10.1126/science.1113373. [DOI] [PubMed] [Google Scholar]
- 18.Ebitani T, Hayashi N, Omoteno M, Ozaki H, Yano M, Morikawa M, Angeles ER, Qian Q, Kitano H, Matsuoka M. Characterization of Pi13, a blast resistance gene that maps to chromosome 6 in indica rice (Oryza sativa L. variety, Kasalath). Breed Sci. 2011;61(3):251–9.
- 19.Xie K, Jiang L, Lu B, Yang C, Li L, Liu X, Zhang L, Zhao Z, Wan J. Identification of QTLs for seed dormancy in rice (Oryza sativa L.) Plant Breed. 2011;130(3):328–332. doi: 10.1111/j.1439-0523.2010.01829.x. [DOI] [Google Scholar]
- 20.Schuler MA, Werck-Reichhart D. Functional genomics of P450s. Annu Rev Plant Biol. 2003;54(1):629–667. doi: 10.1146/annurev.arplant.54.031902.134840. [DOI] [PubMed] [Google Scholar]
- 21.Nelson DR, Schuler MA, Paquette SM, Werck-Reichhart D, Bak S. Comparative genomics of rice and Arabidopsis. Analysis of 727 cytochrome P450 genes and pseudogenes from a monocot and a dicot. Plant Physiol. 2004;135(2):756–772. doi: 10.1104/pp.104.039826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Pinot F, Sauveplane V, Werck-Reichhart D, Diehl P, Schreiber L, Franke R, Zhang P, Chen L, Gao Y. Cytochrome P450 family member CYP704B2 catalyzes the ω-hydroxylation of fatty acids and is required for anther cutin biosynthesis and pollen exine formation in rice. Plant Cell. 2010;22(1):173–190. doi: 10.1105/tpc.109.070326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu F, Fang J, Ou S, Gao S, Zhang F, Du L, Xiao Y, Wang H, Sun X, Chu J. Variations in CYP78A13 coding region influence grain size and yield in rice. Plant Cell Environ. 2015;38(4):800–811. doi: 10.1111/pce.12452. [DOI] [PubMed] [Google Scholar]
- 24.Rowe H, Renaut S, Guggisberg A. RAD in the realm of next‐generation sequencing technologies. Mol Ecol. 2011;20(17):3499–3502. doi: 10.1111/j.1365-294x.2011.05197.x. [DOI] [PubMed] [Google Scholar]
- 25.Tanabe S, Ashikari M, Fujioka S, Takatsuto S, Yoshida S, Yano M, Yoshimura A, Kitano H, Matsuoka M, Fujisawa Y. A novel cytochrome P450 is implicated in brassinosteroid biosynthesis via the characterization of a rice dwarf mutant, dwarf11, with reduced seed length. Plant Cell. 2005;17(3):776–790. doi: 10.1105/tpc.104.024950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartwig B, James GV, Konrad K, Schneeberger K, Turck F. Fast Isogenic Mapping-by-Sequencing of Ethyl Methanesulfonate-Induced Mutant Bulks. Plant Physiol. 2012;160(2):591–600. doi: 10.1104/pp.112.200311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mascher M, Wu S, Amand PS, Stein N, Poland J. Application of genotyping-by-sequencing on semiconductor sequencing platforms: a comparison of genetic and reference-based marker ordering in barley. PLoS One. 2013;8(10):e76925. doi: 10.1371/journal.pone.0076925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shomura A, Izawa T, Ebana K, Ebitani T, Kanegae H, Konishi S, Yano M. Deletion in a gene associated with grain size increased yields during rice domestication. Nat Genet. 2008;40(8):1023–1028. doi: 10.1038/ng.169. [DOI] [PubMed] [Google Scholar]
- 29.Fan C, Xing Y, Mao H, Lu T, Han B, Xu C, Li X, Zhang Q. GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor Appl Genet. 2006;112(6):1164–1171. doi: 10.1007/s00122-006-0218-1. [DOI] [PubMed] [Google Scholar]
- 30.Song X-J, Huang W, Shi M, Zhu M-Z, Lin H-X. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet. 2007;39(5):623–630. doi: 10.1038/ng2014. [DOI] [PubMed] [Google Scholar]
- 31.Weng J, Gu S, Wan X, Gao H, Guo T, Su N, Lei C, Zhang X, Cheng Z, Guo X. Isolation and initial characterization of GW5, a major QTL associated with rice grain width and weight. Cell Res. 2008;18(12):1199–1209. doi: 10.1038/cr.2008.307. [DOI] [PubMed] [Google Scholar]
- 32.Chakrabarti M, Zhang N, Sauvage C, Munos S, Blanca J, Canizares J, Diez MJ, Schneider R, Mazourek M, McClead J. A cytochrome P450 regulates a domestication trait in cultivated tomato. Proc Natl Acad Sci U S A. 2013;110:17125–17130. doi: 10.1073/pnas.1307313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andolfatto P, Davison D, Erezyilmaz D, Hu TT, Mast J, Sunayama-Morita T, Stern DL. Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Res. 2011;21(4):610–617. doi: 10.1101/gr.115402.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet. 2011;12(7):499–510. doi: 10.1038/nrg3012. [DOI] [PubMed] [Google Scholar]
- 35.Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23(19):2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- 36.Wang C, Ye J, Tang W, Liu Z, Zhu C, Wang M, Wan J. Loop Nucleotide Polymorphism in a Putative miRNA Precursor Associated with Seed Length in Rice (Oryza sativa L.) Int J Biol Sci. 2013;9(6):578–586. doi: 10.7150/ijbs.6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen S, Tao L, Zeng L, Vega-Sanchez ME, Umemura K, Wang GL. A highly efficient transient protoplast system for analyzing defence gene expression and protein–protein interactions in rice. Mol Plant Pathol. 2006;7(5):417–427. doi: 10.1111/j.1364-3703.2006.00346.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sets supporting the results of this article are included within the article and its additional files. All the sequencing data produced in this study have been deposited in NCBI Short Read Archive (http://www.ncbi.nlm.nih.gov/sra/) and can be accessed under the SRA accession numbers: SRR3307074, SRR3307908, SRR3308144, SRR3308415, SRR3308416, SRR3308417, SRR3308419, SRR3308421, SRR3308737, SRR3310108, SRR3310109, SRR3310110, SRR3310115, SRR3310157, SRR3310158. SRR3310111, SRR3310156, and SRR3308736.