

Abstract
Background
The clinical utility of primaquine (PQ), used as a racemic mixture of two enantiomers, is limited due to metabolism-linked hemolytic toxicity in individuals with genetic deficiency in glucose-6-phosphate dehydrogenase. The current study investigated differential metabolism of PQ enantiomers in light of the suggestions that toxicity and efficacy might be largely enantioselective.
Methods
Stable isotope 13C-labelled primaquine and its two enantiomers (+)-PQ, (−)-PQ were separately incubated with cryopreserved human hepatocytes. Time-tracked substrate depletion and metabolite production were monitored via UHPLC–MS/MS.
Results
The initial half-life of 217 and 65 min; elimination rate constants (λ) of 0.19 and 0.64 h−1; intrinsic clearance (Clint) of 2.55 and 8.49 (µL/min)/million cells, which when up-scaled yielded Clint of 6.49 and 21.6 (mL/min)/kg body mass was obtained respectively for (+)- and (−)-PQ. The extrapolation of in vitro intrinsic clearance to in vivo human hepatic blood clearance, performed using the well-stirred liver model, showed that the rate of hepatic clearance of (+)-PQ was only 45 % that of (−)-PQ. Two major primary routes of metabolism were observed—oxidative deamination of the terminal amine and hydroxylations on the quinoline moiety of PQ. The major deaminated metabolite, carboxyprimaquine (CPQ) was preferentially generated from the (−)-PQ. Other deaminated metabolites including PQ terminal alcohol (m/z 261), a cyclized side chain derivative from the aldehyde (m/z 241), cyclized carboxylic acid derivative (m/z 257), a quinone-imine product of hydroxylated CPQ (m/z 289), CPQ glucuronide (m/z 451) and the glucuronide of PQ alcohol (m/z 437) were all preferentially generated from the (−)-PQ. The major quinoline oxidation product (m/z 274) was preferentially generated from (+)-PQ. In addition to the products of the two metabolic pathways, two other major metabolites were observed: a prominent glycosylated conjugate of PQ on the terminal amine (m/z 422), peaking by 30 min and preferentially generated by (+)-PQ; and the carbamoyl glucuronide of PQ (m/z 480) exclusively generated from (+)-PQ.
Conclusion
Metabolism of PQ showed enantioselectivity. These findings may provide important information in establishing clinical differences in PQ enantiomers.
Keywords: Enantioselectivity, Hepatocytes, Malaria, Metabolism, Primaquine enantiomers, Primaquine metabolites
Background
The sustained utility and continued interest in primaquine (PQ), even more than six decades after its introduction into clinical medicine, underscores its unique therapeutic indications [1]. As a prototype 8-aminoquinoline, PQ is the only licensed option to treat the relapsing liver stages (hypnozoites) of Plasmodium vivax, in addition to its use in the prophylaxis of all forms of human malaria [2–5]. PQ is also active against mature, infective Plasmodium falciparum gametocytes, and there is thus current high interest in its use as a gametocytocidal drug for blocking transmission of P. falciparum. In areas of emerging drug resistance, primaquine has shown effectiveness against and in the prevention of the spread of artemisinin-resistant P. falciparum strains [6]. The major drawback in the clinical utility of primaquine however, is its haemolytic toxicity in individuals with genetic deficiency in glucose-6-phosphate dehydrogenase (G6PD) [4, 5].
Many attempts have been made to improve the therapeutic index of PQ and other 8-aminoquinolines [2]. PQ is a chiral drug currently used as a racemic mixture, approximating a 50:50 ratio of (+)-(S)- and (−)-(R)-enantiomers. It has been suggested that the stereo-configuration of PQ side chain may dramatically impact its metabolism, toxicity, and anti-malarial activity. More than three decades ago, Schmidt et al. suggested the exploration of the differential anti-malarial efficacy and toxicity profiles of the individual PQ enantiomers for possible improvement of the therapeutic index of PQ [7]. However, the lack of an economical stereospecific method to prepare individual enantiomers or an effective practical procedure to resolve the racemate into enantiomers delayed further studies on individual PQ enantiomers.
An efficient method for the resolution of racemic PQ mixture into individual enantiomers and findings on enantioselective activity and toxicity profiles in several animal models were recently reported [8, 9]. From these studies, it was observed that (+)-(PQ) was more active than (−)-PQ in suppressive and prophylactic anti-malarial assays as well as Pneumocystis carinii pneumonia efficacy assay in mice [8]. However, for radical curative assay in monkeys, previous studies have shown that both PQ enantiomers were equally effective [7, 9]. (+)-PQ caused higher acute toxicity than (−)-PQ in beagle dogs and mouse models. In monkeys, reversible liver damage was observed at higher than therapeutic (−)-PQ doses. In three different animal models (beagle dogs, humanized mouse model and monkeys) (+)-PQ caused higher methaemoglobinemia than (−)-PQ. The observed disparity of activity and toxicity profiles of PQ enantiomers in different animal models could be attributed to their differential pharmacokinetics and metabolism. Based on these results, it was suggested that (−)-(R)-PQ enantiomer may have a better safety margin than the currently used racemate and proposed a “phase I” type human study to compare pharmacokinetics and tolerability study of PQ enantiomers [7, 9].
It had long been speculated that PQ needed metabolic activation due to its lack of in vitro activity and toxicity, and the observed delayed onset of action in animal models. PQ treatment failures were reported in subjects with impaired cytochrome P450 2D6 (CYP2D6) function [10]. Subsequent studies using a CYP2D knock-out mouse model found evidence that CYP2D metabolism was a prerequisite for the causal prophylactic activity of PQ [11]. Differential metabolism of PQ enantiomers in vitro by recombinant human CYP2D6 was previously reported [12]. The principal CYP2D6-dependent metabolic pathway of PQ was found to involve the hydroxylation of the quinoline ring yielding 2-, 3-, and 4-hydroxyprimaquine, PQ-5,6-orthoquinone and dihydroxyprimaquine as the major metabolites. PQ-5,6-orthoquinone appeared to be the oxidative degradation product of 5-OH-PQ. Synthetic 5-OH-PQ spontaneously undergoes rapid oxidation sequentially to PQ-5,8-quinone-imine and PQ-5,6-orthoquinone [13]. Both quinone-imine and orthoquinone can generate reactive oxygen species (ROS) through redox cycling, hence are possible candidates as the active metabolites of PQ. ROS have shown to be responsible for both antiparasitic activity and haemotoxity. The rate and extent of the generation of these metabolites differ significantly with individual PQ enantiomers [12]. Primaquine-5,6-orthoquinone was twice as abundant from CYP2D6-catalyzed metabolism of (+)-PQ metabolism compared to (−)-PQ. The rate of metabolism in vitro with recombinant human CYP2D6 was significantly higher for (+)-PQ compared to (−)-PQ.
In addition to the CYP-mediated oxidation of the quinoline ring, oxidative deamination of the terminal amine by monoamine oxidase A (MAO) has been identified as the other major metabolic pathway of PQ. Oxidation of PQ by MAO results in formation of an aldehyde metabolite, which is further oxidized or reduced to the stable metabolites namely, carboxyprimaqine (CPQ) and PQ-alcohol, respectively. CPQ had been identified as the major circulating human metabolite of racemic PQ [14, 15]. Pharmacokinetic studies of racemic PQ in mice, monkeys and humans have shown that CPQ rapidly appeared in the plasma and was predominantly the (−)-form [9, 15, 16]. The current study is aimed at characterizing the kinetics of metabolic profiles of the individual PQ enantiomers in human hepatocytes, where both the CYP and MAO pathways are present.
The primary aim of this study was therefore to characterize the metabolism of the individual enantiomers of PQ in human hepatocytes, while profiling the enantioselective metabolites generated. This is expected to provide projections of enantioselective metabolic profiles in human. To facilitate identification of metabolites a 1:1 mixture 13C-(C6)-labelled and normal 12C-PQ was used in this study.
Methods
HPLC-grade acetonitrile and methanol were purchased from Fisher Scientific (Fair Lawn, NJ, USA). Water for the HPLC mobile phase was purified using a Milipore Synergy Water Purification System (Milipore SAS, Molsheim, France). PQ diphosphate and formic acid were purchased from Sigma (St Louis, MO, USA).
Synthetic chemicals
13C(6)-labelled PQ was prepared [17] and resolved into (+)-(S)- and (−)-(R)- enantiomers as previously reported [8]. CPQ [18] and CPQ-lactam [19] were prepared using the procedures reported. Synthesis of 2-, 3-, and 4-OHPQ and PQ alcohol were reported earlier [12].
Synthesis of primaquine alcohol glucuronide
Primaquine alcohol-β-d-2,3,4-O-triacetylglucuronide methyl ester
Ag2CO3 (414 mg, 1.5 mM) was added potion wise for 2 h to a stirring mixture of acetobromo-α-d-glucuronic acid methyl ester (398 mg, 1 mM), and primaquine alcohol (390 mg, 1.5 mM) in dry toluene (10 mL) at 75 °C and the reaction was left overnight. The reaction mixture filtered through Celite, and the filtrate was evaporated to dryness The product was purified by column chromatography on silica gel with hexane:EtOAc (90:10) to give primaquine alcohol-O-β-d-2,3,4-O-triacetylglucuronide methyl ester (410 mg).
1H NMR δ (CDCl3): 1.08 (3H, d, J = 4.0 Hz), 1.49 (4H, m), 1.82 (3H,s), 1.84 (6H, s), 3.41 (4H, m), 3.53 (3H, s), 3.66 (3H, s), 4.27 (1H, d, J = 8.0 Hz), 4.95 (1H, d, J = 8.0 Hz), 5.05 (1H, dd, J = 6.0, 8.0 Hz), 5.35 (1H, dd, J = 6.0. 8.0 Hz), 6.10 (1H,s), 6.13 (1H, s), 6.21 (1H, d, J = 4.0 Hz), 7.07 (1H, dd, J = 6.0, 4.0 Hz), 7.70 (1H, d, J = 6.0 Hz), 8.29 (1H, d, J = 4.0 Hz); HRESIMS [M + H]+m/z 577.2423 (calculated for (C28H36N2O11 + H)+ 577.2397).
Primaquine alcohol-O-β-d-glucuronide
Primaquine alcohol-O-β-d-2,3,4-O-triacetylglucuronide methyl ester (350 mg) was dissolved in a mixture of MeOH/H2O (5:1)(10 mL) stirred at 0 °C, added ethyl di-isopropyl amine (2 mL) and continued stirring overnight at room temperature. Solvent was evaporated under vacuum and the residue was chromatographed on reverse phase silica (C-18) column with H2O:MeOH to give primaquine alcohol-O-β-d-glucuronic acid.
1H NMR δ (CD3OD): 8.67 (1H, brs), 8.44(1H, d, J = 6.0 Hz), 7.64 (1H, m), 6.48 (1H, s), 5.14 (1H, s), 4.52 (1H, d, J = 8.0 Hz), 3.88 (3H, s), 3.84 (1H, m), 3.68 (1H, m), 3.40 (1H, m), 3.34 (1H, m), 3.20 (2H,m), 1.80 (4H,m), 1.30 (3H, d, J = 6.0 Hz); HRESIMS [M + H]+m/z 437.1884 (calculated for (C21H28N2O8 + H)+ 437.1924).
Synthesis of carboxyprimaquine-β-d-glucuronide
Benzyl 1-O-levulinyl-d-glucuronate
N-Methyl mopholine (0.25 mL) was added to a mixture of benzyl d-glucuronate (426 mg, 1.5 mM), levulinic acid (185 mg, 1.6 mM) and HATU (380 mg, 1 mM) in anhydrous acetonitrile (10 mL) and the reaction mixture was stirred 2 h at ambient temperature. Reaction was quenched with Ambalite H+ resin, filtered and the residue was thoroughly washed with CH2Cl2. Combined organic extract was evaporated to dryness and chromtographed on silica column with CH2Cl2:MeOH (96:4) to give benzyl 1-O-levulinyl-d-glucuronate (550 mg).
1H NMR δ (CDCl3): 2.06 (3H, s), 2.72-2.48 (4H, m), 3.80-3.58 (4H, m), 4.02 (1H, d, J = 9.6 Hz), 5.15 (2H, s), 5.60 (1H,d, J = 7.6 Hz), 7.30 (5H, m).
Carboxyprimaquine-β-d-glucuronide benzyl ester
NaBH4 was added portion wise to a mixture of benzyl 1-O-levulinyl-d-glucuronate (275 mg, 0.72 mM) and 6-methoxy-8-aminoquinoline (128 mg, 0.8 mM) in glacial acetic acid (5 ml) at 20 °C under stirring while monitoring the reaction by TLC. Upon completion, the reaction was quenched with ice/water, neutralized with 10 % NaOH and extracted with CH2Cl2. The organic extract was dried on anhydrous Na2SO4, evaporated to dryness. The product was purified by chromatography on silica gel and elution with CH2Cl2/MeOH (5 %) to give carboxyprimaquine-β-d-glucuronide benzyl ester (260 mg).
Carboxyprimaquine-β-d-glucuronide
Carboxyprimaquine-β-d-glucuronide benzyl ester (210 mg) and 10 % Pd/C in EtOH (10 mL) was hydrogenated with H2 (20 psi) for 2 h. Reaction mixture was filtered through Celite purified by silica column chromatography with CH2Cl2:MeOH to give carboxyprimaquine-β-d-glucuronide (140 mg).
1H NMR δ (CD3OD): 8.48 (1H, brs), 8.02 (1H, d, J = 8.0 Hz), 7.35 (1H, m), 6.38 (1H, d, J = 7.6 Hz), 5.51 (1H, m), 3.91 (1H, m), 3.84 (3H,s), 3.68 (1H, m), 3.46 (1H, m), 3.39 (1H, m), 3.30 (1H, m), 2.54 (2H, m), 1.90 (2H, m), 1.25 (3H, d, J = 6.4 Hz): 13C NMR δ (CD3OD): 19.1, 30.2, 30.8, 54.3, 71.6, 72.1, 75.5, 76.0, 94.0, 97.4, 121.5, 130.1, 135.1, 143.8, 144.5, 159.5, 172.4: HRESIMS [M + H]+m/z 451.1776 (calculated for (C21H26N2O9 + H)+ 451.1717).
Synthesis of (S)-primaquine-N-carbamoyl-β-d-glucuronide
(S)-primaquine-N-carbamoyl-2,3,4-O-triacetyl-β-d-glucuronide methyl ester
A solution of 4-dimethylamino pyridine (112 mg, 0.92 mM) in anhydrous CH3CN (5 mL) was added dropwise to a solution of di-tert-butyl dicarbonate (1.2 g, 5.5 mM) in anhydrous CH3CN (5 mL) at 0 °C under stirring. After 10 min, a solution of (+)-(S)-primaquine free amine (1.2 g, 4.6 mM) in anhydrous CH3CN (5 mL) was added dropwise and the reaction mixture was stirred for further 30 min at 0 °C. Solvent was evaporated under reduced pressure and the resulting dark yellow oily residue was dissolved in toluene (5 mL), cooled to 0 °C, treated dropwise with a solution of methyl 2,3,4-tri-O-acetyl-d-glucuronnte (1.8 g, 5.5 mM) in toluene (5 mL) followed by triethylamine (1.2 mL) and stirred for 2 h at 0 °C and another 2 h at room temperature. Solvent was evaporated under reduced pressure and the dark brown residue was chromatographed on silica column and eluted with ethyl acetate/hexane (40:60) to give (S)-primaquine-N-carbamoyl -2,3,4-O-triacetyl-β-d-glucuronide methyl ester (1.80 g).
1H NMR δ (CDCl3): 1.25 (3H, d, J = 6.8 Hz), 1.60 (4H, m), 1.99 (9H, brs), 3.15 (2H, m), 3.58 (1H, m), 3.66 (3H, s), 3.85(3H, s), 4.12 (1H, d, J = 10, Hz), 5.31-5.09 (4H, m), 5.67 (1H, d, J = 8.4 Hz), 5.94 (1H, brd, J = 8.4 Hz), 6.23 (1H, s), 6.30 (1H, s), 7.27 (1H, dd, J = 8.4, 7.6 Hz), 7.88 (1H, d, J = 8.4 Hz), 8.49 (1H, d, J = 4.4 Hz); HRESIMS [M + H]+m/z 620.2451 (calculated for (C29H37N3O12 + H)+ 620.2455).
(S)-primaquine-N-carbamoyl-β-d-glucuronide
Ethyldiisopropyl amine (6 mL) was added to a solution of primaquine-N-carbamoyl-2,3,4-O-triacetyl-β-d-glucuronide methyl ester (700 mg, 1.12 mM) in MeOH (30 mL) and H2O (6 mL) at 0 °C while stirring. After 10 min at 0 °C, cooling bath was removed and stirring was continued overnight. Solvent was removed under reduced pressure and the residue was dissolved in MeOH (2 mL), introduced to a Sephadex column (4 × 30 cm) and eluted with methanol (0.5 mL/min) to give (S)-primaquine-N-carbamoyl-β-d-glucuronide (284 mg) as a pale yellow powder.
1H NMR δ (D6-DMSO): 1.18 (3H, d, J = 6.4 Hz), 1.60 (4H, m), 3.33-2.99 (7H, m), 3.66 (3H, s), 3.59 (2H, m), 3.80 (3H, s), 5.16 (1H, d, J = 8.4 Hz), 6.11 (1H, d, J = 8.4 Hz), 6.24 (1H, d, J = 2.0 Hz), 6.44 (1H, d, J = 2.0 Hz), 7.40 (1H, dd, J = 8.0, 8.0 Hz), 8.05 (1H, d, J = 8.4 Hz), 8.51 (1H, d, J = 3.6 Hz); 13C NMR δ (D6-DMSO): 20.6, 26.6, 33.8, 47.5, 55.4, 72.3, 72.8, 74.8, 77.1, 92.1, 95.4, 96.6, 122.6, 130.0, 135.0, 135.3, 145.1, 144.7, 155.6, 159.4, 173.1; HRESIMS [M + H]+m/z 480.1954 (calculated for (C22H29N3O9 + H)+ 480.1982).
Hepatocytes
Pooled mixed-gender cryopreserved metabolism-qualified (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4, UGT2B7, UDP-glucuronosyltransferase) primary human hepatocytes (catalog number 454504) sourced from free donors (who were free of HIV, HBV, HCV and other liver-related diseases) were purchased from Corning Life Sciences (Woburn MA, USA) and stored in liquid nitrogen until use.
Hepatocytes incubations and viability measurements
The cryopreserved hepatocytes were thawed and the cells were suspended in recovery medium (Corning Life Sciences). According to the supplier’s instructions, the suspended cells were centrifuged (300g, 5 min), the recovery medium discarded and the cells were re-suspended in the plating medium. Viability of the suspended hepatocytes was computationally determined based on cell counts using a Bio-Rad automated cell counter (Hercules, CA, USA). The cell density was adjusted to approximately 1X106 viable cells/mL in the plating media.
PQ, racemic and enantiomers, prepared as a 1:1 mixture of 12-C and 13-C-labelled compounds, were separately incubated at a final concentration of 20 µM in the cell-containing plating media using the 48-well plate at 37 °C under a humidified atmosphere of 95 % air and 5 % CO2 in an Eppendorf incubator (Hauppauge, NY, USA) attached with a shaker set at 75 rpm. A separate control incubation of PQ and the two enantiomers in the cell-free hepatocyte media was performed to detect any non-enzymatically generated product. Metabolic reactions were halted by the addition of two volumes of ice-cold methanol containing 0.5 µg/mL 6-d3-methoxyprimaquine as an internal standard at predetermined time intervals. This mixture was vortexed and kept at −20 °C for at least 4 h before further analysis. Parallel incubation of substrate-free hepatocytes was performed. At each time point, aliquots were taken for cell viability measurement.
In preparation for HPLC–MS analysis, samples were centrifuged (10,000g, 10 min) and clear supernatants were dried using a Speedvac. The dried samples were re-suspended in 150 µL methanol, centrifuged and clear supernatants were transferred to HPLC sample vials for analysis.
Detection, identification and quantification of metabolites
The analytical method for simultaneous analysis of primaquine and its metabolites using the Ultra-High Performance Liquid Chromatography-Mass Spectrometry (UHPLC-QToF-MS), as earlier developed and reported was employed in this study [16]. Typically, total separation and elution of the analytes was achieved within 10 min retention time, using the Acquity UPLC™, BEH Shield RP18 column (100 mm × 2.1 mm I.D, 1.7 µm) equipped with an LC-18 guard column (Vanguard 2.1 × 5 mm, Waters Corp., Milford, MA, USA) on an ACQUITY UPLC system (Waters Corporation, Milford, MA, USA) to which a conditioned auto-sampler (at 20 °C) was attached. Details of the chromatography set-up are published [16].
Metabolites in the accurate mass data were found using the Metabolynx® software. The data were searched using predicted metabolite mass, mass defects, isotope, and fragmentation patterns. Each sample was subjected to data acquisition in full scan and data-dependent positive MS/MS, targeted MS/MS (ESI positive ionization mode) and high-resolution MS (HRMS) modes using the Waters ACQUITY™ XEVO QTOF Mass Spectrometer (Waters Corporation, Manchester, UK) connected to the UHPLC system via an electrospray ionization (ESI) interface. Metabolites were distinguished from artifacts and unrelated products by the presence of twin peaks with a mass difference of six, arising from the six labels in the 1:1 mixture of 12-C and 13-C-labelled PQ. The Identification of each metabolite was assisted by its HRMS data, which were used to calculate their elemental compositions. The full scan mass data were screened and filtered using Waters MetaboLynx XS software. The qualitative metabolite identification was performed using this software package.
In vitro elimination calculations
Initial half-life (t1/2) and elimination rate constants (λ = ln2/t1/2) of PQ in hepatocyte incubates were calculated by log-linear regression of PQ concentrations profiled against time using data from the sampling points of the 20 µM PQ incubates [20]. The intrinsic clearance in vitro (CLint in vitro) was calculated from λ and the cell density in the respective incubation (number of viable hepatocytes per mL at time zero) and scaled up to the intrinsic clearance in vivo (CLint in vivo) using the human liver mass (25.7 g/kg body mass) and the hepatocellularity (number of hepatocytes per gram of liver = 99 millions cells/g liver) [21]: CLint in vitro = λ/cell density. CLint in vivo = CLint in vitro × liver mass × heptocellularity. From CLint in vivo and the hepatic blood flow [Q (20.7 mL/min)/kg body mass], the hepatic metabolic blood clearance (CLh,b) was predicted using the well-stirred model: CLh,b = (CLint in vivo × Q)/(CLint in vivo + Q) [21].
Since this estimation was for comparisons between enantiomers only, no corrections were made for the free fractions in vitro or in vivo—i.e. they were assumed to be identical [21].
Statistical analysis
Intra- and inter-day variations in the data obtained from replicated experiments were assessed for significance. Replicate agreements were within 5 % variations. All results were statistically analyzed for significance by Student’s t test using GraphPad Prism (GraphPad Software, San Diego, CA).
Results
PQ was incubated in vitro with human hepatocytes (1 million cells/mL) at a final concentration of 20 µM. Substrate concentrations, as initially titrated, showed optimal metabolism at 20 µM (Km = 37 µM). Although this is higher than expected peak plasma concentrations in humans as we earlier reported (<2 µM) [15], it approximates expected liver concentrations of PQ, which we observed to be—at therapeutic doses about 20 times higher than plasma in our studies in mice [22]. The viable cell counts in the drug-free incubation at the predetermined time-points were similar to those of the PQ-containing incubates, suggesting the absence of PQ-induced direct hepatocellular toxicity (Fig. 1a). Hepatocyte viability at the 2 h termination time was 88 % of the starting value, an indication that the integrity of the hepatocytes was not compromised through the incubation period.
Fig. 1.
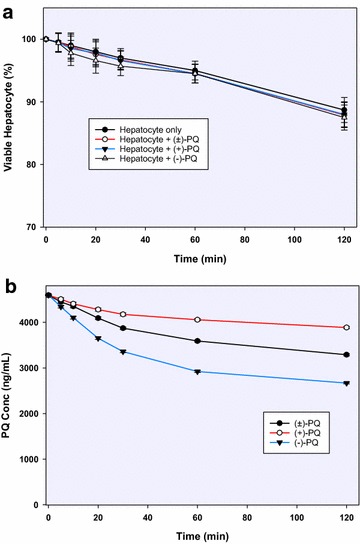
a Hepatocyte viability time course determined through cell counts in the presence and absence of (±)-primaquine and its (+)- and (−)-enantiomers; b differential depletion of the 20 µM racemic (±)-primaquine and its (+)-, and (−)-enantiomers in primary human hepatocytes (1 million cells/mL) after 2 h incubation. Each point represents values mean ±SD (n = 4)
Intrinsic metabolic clearance from hepatocytes
Substrate depletion and metabolite production were monitored via UHPLC-MS/MS. By 2 h, 30, 16 and 42 % of PQ, (+)-PQ and (−)-PQ, respectively, were depleted (Fig. 1b).
The initial half-life, calculated by log-linear regression of PQ concentrations profiled against time using data from the sampling points of the 20 µM incubates, was 217 and 65 min for (+)- and (−)-PQ, with elimination rate constants (ʎ) of 0.19 and 0.64 h−1 respectively. The in vitro intrinsic clearance, calculated from ʎ and the cell density in the respective incubations, was 2.55 and 8.49 (µL/min)/million cells, which when upscaled in vivo based on known human hepatocellularity (hepatocytes/g liver) and liver mass (g/kg body mass) yielded 6.49 and 21.6 (mL/min)/kg body mass respectively for (+)- and (−) PQ. Extrapolation to in vivo human hepatic blood clearance was performed using the well-stirred liver model, which showed that the rate of hepatic clearance of (+)-PQ was only 30 % that of (−) PQ (Table 1).
Table 1.
The intrinsic clearance of PQ and its enantiomers in human hepatocytes
| (±)-PQ | (+)-PQ | (−)-PQ | |
|---|---|---|---|
| Time interval (min) for calculation of elimination rate constant | 0–60 | 0–60 | 0–60 |
| λ (h−1)a | 0.34 | 0.19 | 0.64 |
| t½ (h)b | 2.03 | 3.62 | 1.09 |
| t½ (min) | 121.8 | 217 | 65 |
| CLint in vitro [(mL/h)/million cellsc] | 0.273 | 0.153 | 0.509 |
| CLint in vitro [(µL/min)/million cells] | 4.55 | 2.55 | 8.49 |
| CLint in vivo [(L/h)/kg body mass]d | 0.69 | 0.39 | 1.30 |
| CLint in vivo [(mL/min)/kg body mass] | 11.58 | 6.49 | 21.60 |
| CLh,b [(L/h)/kg body mass]e | 0.45 | 0.30 | 0.63 |
| CLh,b [(mL/min)/kg body mass] | 7 | 5 | 11 |
| CLh,b (% hepatic blood flow)f | 36 | 24 | 51 |
λ elimination rate constant, Clint intrinsic clearance, CL h,b hepatic blood clearance
aλ is taken as the -slope of the linear portion of the log-linear regression (0-60 min)
bHalf-lives computed from
c
d . Hepatocellularity is given as 99 million cells/g liver; average human liver weight is 25.7 g/kg body mass
e Qh hepatic blood flow (well-stirred liver model) is given as 20.7 (mL/min)/kg body mass
fQh (mL/min/kg body weight): 55 (rat); 30.9 (dog); 43.6 (monkey); 20.7 (human)
Differential kinetics and profile of metabolites generated from PQ enantiomers
From the metabolite profile, two major routes of metabolism were observed as earlier reported: oxidative deamination of the side chain terminal amine; and hydroxylations on the quinoline moiety [16]. The metabolites generated through both the pathways were further metabolized by phase II conjugation pathways (Fig. 2). The comparative concentrations of the metabolites generated at 1 h post-incubation is presented in Fig. 3.
Fig. 2.
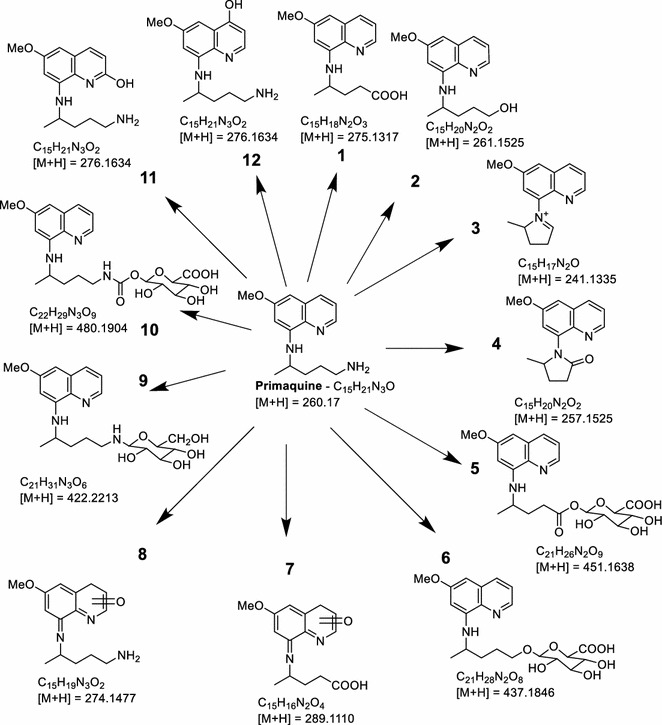
Putative identities and predicted structures of primaquine metabolites generated in human hepatocytes, as determined through MS/MS fragmentation, twin peak detection on the UHPLC chromatogram and prediction by Waters’ Metabolynx® software package
Fig. 3.
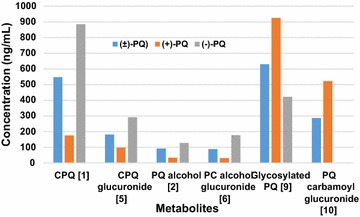
The relative concentrations of the major metabolites generated by primaquine and its enantiomers at 1 h incubation time point in primary human hepatocytes
The major deaminated metabolite, CPQ (Fig. 2—1) (75 % of which was generated within 30 min), was preferentially generated from the (−)-PQ, when compared to (+) PQ (about 8:1 at early time points) (Fig. 4a). This metabolite has been shown earlier to be the major product of PQ biotransformation circulating in the plasma of mice [23], monkeys [9] and humans [15]. It is formed through the oxidative deamination of the 8-N-aminoalkyl side chain of PQ to an intermediate PQ aldehyde which is further converted to the carboxylic acid by an aldehyde dehydrogenase. The PQ aldehyde formed from PQ may also undergo a reduction reaction to form the primaquine alcohol (m/z 261; Fig. 2—2), or cyclization of the side chain through the loss of a water molecule (m/z 241, Fig. 2—3). Further metabolism of CPQ involves the elimination of water and cyclization to the 8-amino group forming a lactam product (m/z 257, Fig. 2—4). CPQ and the PQ alcohol undergo conjugation reactions with glucuronides (m/z 451 and 437 respectively; Fig. 2—5, 6) is also formed from hydroxylated CPQ. These metabolites of the oxidative deamination of the amino side chains were preferentially generated from the (−)-PQ compared to (+)-PQ (Fig. 5a, b, c).
Fig. 4.
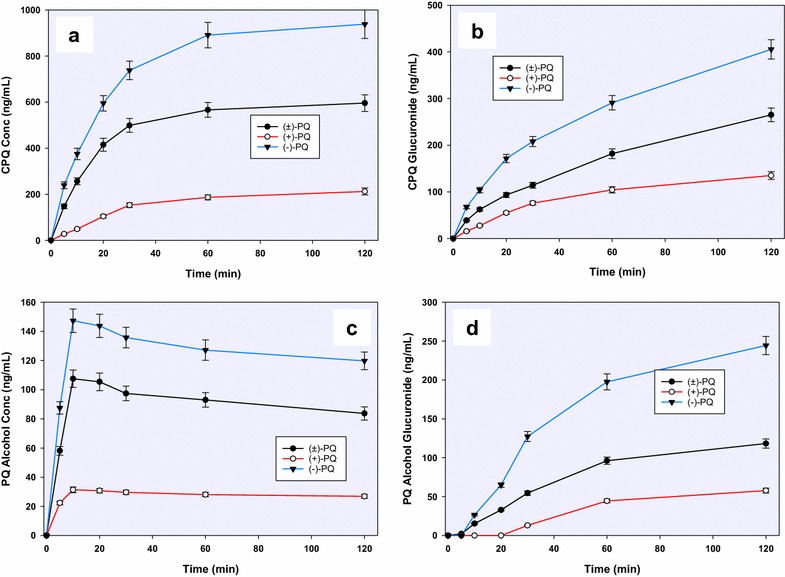
A time-course analysis of the in vitro generation of a carboxyprimaquine (1), b primaquine alcohol (2) and c, d their glucuronide conjugates (5, 6), from (+)-, (−)- and (±)-primaquine in human hepatocytes. Each point represents value mean ±SD of four observations
Fig. 5.

A time-course analysis of a cyclized PQ aldehyde (m/z 241) (3), b cyclized carboxyprimaquine (m/z 257) (4), c hydroxylated CPQ quinone-imine (m/z 289) (7) and d hydroxyprimaquine quinone-imine (m/z 274) (8) differentially generated from (+)-, (−)- and (±)-primaquine in vitro in human hepatocytes. Each point represents value mean ±SD of four observations
A major product of the quinoline ring oxidation identified was the one that corresponds to an m/z 274, formed by hydroxylation of PQ (Figs. 2—8) and subsequent dehydrogenation to the quinone-imine. This ring oxidation product was preferentially formed from (+)-PQ compared to (−)-PQ (2:1) (Fig. 5d). Site of hydroxylation on the quinolone ring could not be established unequivocally at present, but based on other studies, hydroxylation of the 5-position is unlikely due to instability of this hydroxylation product which is rapidly converted to PQ-5,6 orthoquinone. Three other metabolites corresponding to m/z 274 were also observed, which were exclusively generated by (+)-PQ.
In addition to the ring hydroxylations and side-chain terminal amine oxidative deamination pathways, two other metabolites were identified, which were generated through conjugation directly on the PQ side chain terminal amine. A prominent conjugate (m/z 422; Fig. 2—9), identified as a glycosylated PQ, was generated. Formation of this metabolite showed a biphasic increase, peaking by 30 min. This metabolite was preferentially generated from (+)-PQ rather than (−)-PQ (Fig. 6a). This glycosylated conjugate of PQ was also formed non-enzymatically in the cell-free media (Fig. 6b). A linearly accumulating metabolite (m/z 480; Fig. 2—10) over the 2 h time course was identified as an N-carbamoyl glucuronide of PQ, and was exclusively generated from (+)-PQ (Fig. 6c).
Fig. 6.
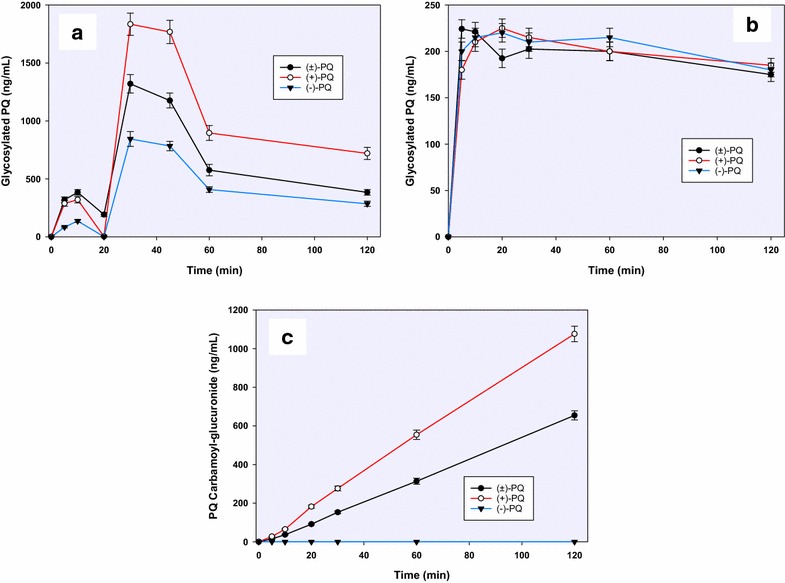
A time-course analysis of a glycosylated primaquine (9) generated through the activity of human hepatocytes; b non-enzymatic generation of primaquine-glucose conjugates observed following the incubation of primaquine and its metabolites in cell-free hepatocyte media and; c primaquine carbamoyl-glucuronide (10) differentially generated from (+)-, (−)-and (±)-primaquine in vitro in human hepatocytes. Each point represents value mean ±SD of four observations
Other metabolites were detected but with too minute peak areas to profile. These include trace amounts of 2- and 4-hydroxyprimaquine (Fig. 2—11 and 12), which were exclusively generated from the (+)-PQ. Figure 2 presents the putative structural identities of the metabolites of PQ in human hepatocytes.
Discussion
Pharmacological and toxicological activities of PQ have been attributed to the metabolites of the drug, presumably generated through CYP-mediated pathways [24, 25]. Until recently, understanding regarding the nature of metabolites and pathways involved in formation of these metabolites has been incomplete. Application of stable 13C isotope labeling of PQ, development of sensitive UHPLC-MS/MS methods for analysis and availability of synthetic metabolites have provided the tools necessary for more accurate identification and quantification of PQ metabolites [16]. However, the situation has been further complicated by the reports regarding differential pharmacological, toxicological and pharmacokinetics profile of enantiomers of PQ [7–9, 15]. Therefore, it is important to get a more complete picture of the metabolism and pharmacokinetics of PQ especially the differential qualitative, quantitative and kinetic profiles of the metabolites generated from individual PQ enantiomers. In the current study, the enantiomers of PQ have been shown to have differential metabolic and pharmacokinetic profiles in human hepatocytes in vitro.
The results presented herein have predicted that the initial half-life (as well as other pharmacokinetic parameters derived therefrom) of (+)-PQ was more than three times longer than that that of (−)-PQ. MAO and CYP are known to be involved in the phase I hepatic metabolism of PQ [26]. A majority of the metabolites identified in this study appeared to be generated through MAO-mediated oxidative deamination of PQ. CPQ, the product of the sequential oxidative deamination of the 8-N-aminoalkyl side chain by MAO and oxidation of the resulting PQ-aldehyde was the most abundant primary metabolite of PQ and was preferentially generated from the (−)-PQ (about 8 times higher than in (+)-PQ at the early time points). These results support our previous data on in vivo pharmacokinetics of PQ enantiomers in rodents, primates and humans as well in vitro results with freshly isolated human hepatocytes [9, 15]. It is noteworthy that all the metabolites formed via the oxidative deamination of the 8-N-aminoalkyl side chain were consistently more abundant from the (−)-PQ than (+)-PQ. A recent pharmacokinetic study in human [15] showed that CPQ appeared readily in plasma after the administration of racemic primaquine and at peak, its concentration was 15 times higher from (−)-PQ than from (+)-PQ. In fact, in some human subjects, the observed CPQ was almost exclusively of the minus enantiomer [15]. Thus, reactions on the amino side-chain appeared to be the major clearance pathway of (−)-PQ.
Only two ring-hydroxylated PQ metabolites attributable to CYP pathway were present in substantial amounts. They were identified by accurate mass as quinone-imine metabolites of hydroxylated PQ and CPQ (m/z 274 and 289 respectively). Traces of other hydroxylated metabolites were observed, including the 2-OH and 4-OH-PQ, and three other quinone imines (m/z 274). The position of the carbonyl group of these metabolites could not be determined by mass analysis. Stability of the keto-imines however, suggests that the keto group is unlikely to be present at C-5 of the quinoline ring, as for this metabolite, a rapid degradation with demethylation of the 6-methoxy is observed.
In previous studies with recombinant human CYP2D6, substantial depletions of PQ enantiomers (50 and 30 % for (+)- and (−)-PQ, respectively) were observed during a 2 h incubation period, and CYP2D6 preferentially converted (+)-PQ to its PQ-5,6-orthoquinone and 2-hydroxyprimaquine [12]. Meanwhile, in the current study, only trace amounts of primary ring oxidative products were detected with hepatocyte incubation, all of which were exclusively generated from the (+)-PQ. The low quantities of the primary ring hydroxylation products, especially those derived from the 5-OH PQ, which has been deemed an important active metabolite [25] could possibly be due to rapid degradation or conjugation with other products. Further studies are required to resolve this issue.
The formation of the phase II conjugation reaction metabolite, identified as an N-carbamoyl glucuronide of PQ accounted for most of the (+)-PQ consumption. Identity of N-carbamoyl glucuronide of PQ was confirmed by comparison of its HPLC retention time and MS/MS fragmentation properties with the synthetic standard. It accumulated linearly over time and was the major product after prolonged incubation of PQ with human hepatocytes. The formation of this carbamoyl glucuronide of PQ as a putative metabolite in the incubation mixture of racemic PQ with pooled human hepatocytes was previously reported [26]. In the current studies, this appears to be the quantitatively predominant metabolite of (+)-PQ in the human hepatocytes, and we have also observed it in plasma and urine of human subjects receiving primaquine (manuscript in preparation). Such N-carbamoyl glucuronides are unusual, but have been reported as minor products for other drugs containing primary and secondary amines [27, 28]. Incubation of these drugs in a high CO2 environment with UDP-glucuronyltransferases can readily yield the carbamoyl glucuronides. It is, however, somewhat surprising that in the hepatocytes, this product is enantioselectively formed.
Another unexpected metabolite observed was an apparent glucose conjugate of PQ. The biphasic kinetics of formation of PQ-glucose conjugate suggests that formation of this metabolite may also depend on other metabolic reactions in the hepatocytes. It would be interesting to further investigate the pathway(s) involved in formation of this metabolite. The earlier small peak of formation of PQ-glucose conjugate may not be enzyme-dependent, since formation of this conjugate could be observed even in cell-free control incubations.
As observed against time of incubation, metabolite profiles showed different shapes, suggesting variation in kinetics and disposition. The interplay of factors including multiple enzyme involvement, dependence of one metabolite on the formation of the other, possible interactions among the metabolites might be responsible for the varying kinetics of formation of the different metabolites.
Conclusion
The enantiomers of PQ showed significant variation in in vitro pharmacokinetics as well as metabolite generation in human hepatocytes. The in vitro clearance of (−)-PQ is three times faster than that of (+)-PQ in hepatocytes. Trace amounts of ring oxidation products were observed with (+)-PQ while (−)-PQ was preferentially metabolized by oxidative deamination of the terminal amine on the N-8 alkyl amino side-chain. Overall, the ring oxidation products appeared to be formed in limited amounts. A phase II conjugation reaction metabolite N-carbamoyl glucuronide of PQ was identified as a major metabolite, which was exclusively formed from (+)-PQ. Another PQ conjugate formed by apparent N-glucosylation was preferentially formed with (+)-PQ. These findings enhance the understanding of the pathways of PQ metabolism, and further inform the enantioselectivity of its metabolic profiles.
Authors’ contributions
LAW, BLT, GAR, NPDN, JDM developed the overall project direction; LAW, SRM, PSF and BLT conceptualized the study; PSF, LAW and BLT designed the experiments; NPDN and HMTBH synthesized primaquine and its metabolites, PSF and SIK guided and performed the enzyme incubation studies and the sample preparation; PSF, BA, IAK and YW performed the LC-MS analysis; PSF, LAW, JDM, MAE, BLT performed data analysis and result interpretation. All authors contributed to the writing and the approval of the final version of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This study was supported by the Bill and Melinda Gates Foundation Phase I Grand Challenge Explorations Award (OPP53288) and the US Army Medical Research and Materiel Command Awards Nos. W81XWH-10-2-0059 and W81XWH-13-2-0026 to the University of Mississippi. The authors thank Dr. David Jollow, Medical University of South Carolina, for his continuing input and advice and help in data interpretations.
This material has been reviewed by the Walter Reed Army Institute of Research. There is no objection to its presentation and/or publication. The opinions or assertions contained herein are the private views of the authors, and are not to be construed as official, or as reflecting true views of the Department of the Army or the Department of Defense.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Pius S. Fasinu, Phone: +1-662-915-1005, Email: psfasinu@olemiss.edu
Bharathi Avula, Email: bavula@olemiss.edu.
Babu L. Tekwani, Email: btekwani@olemiss.edu
N. P. Dhammika Nanayakkara, Email: dhammika@olemiss.edu.
Yan-Hong Wang, Email: wangyh@olemiss.edu.
H. M. T. Bandara Herath, Email: bherath@olemiss.edu
James D. McChesney, Email: jdmcchesney@yahoo.com
Gregory A. Reichard, Email: gregory.a.reichard2.ctr@mail.mil
Sean R. Marcsisin, Email: marcsisin.s@gmail.com
Mahmoud A. Elsohly, Email: melsohly@olemiss.edu
Shabana I. Khan, Email: skhan@olemiss.edu
Ikhlas A. Khan, Email: ikhan@olemiss.edu
Larry A. Walker, Email: lwalker@olemiss.edu
References
- 1.Ashley EA, Recht J, White NJ. Primaquine: the risks and the benefits. Malar J. 2014;13:418. doi: 10.1186/1475-2875-13-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tekwani BL, Walker LA. 8-Aminoquinolines: future role as antiprotozoal drugs. Curr Opin Infect Dis. 2006;19:623–631. doi: 10.1097/QCO.0b013e328010b848. [DOI] [PubMed] [Google Scholar]
- 3.Hill DR, Baird JK, Parise ME, Lewis LS, Ryan ET, Magill AJ. Primaquine: report from CDC expert meeting on malaria chemoprophylaxis. Am J Trop Med Hyg. 2006;75:402–415. [PubMed] [Google Scholar]
- 4.Vale N, Moreira R, Gomes P. Primaquine revisited six decades after its discovery. Eur J Med Chem. 2009;44:937–953. doi: 10.1016/j.ejmech.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 5.Fernando D, Rodrigo C, Rajapakse S. Primaquine in vivax malaria: an update and review on management issues. Malar J. 2011;10:351. doi: 10.1186/1475-2875-10-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.WHO. Global Plan for Artemisinin Resistance Containment (GPARC). http://www.wpro.who.int/mvp/artemisinin_resistance_containment_2011.pdf Accessed 27 Nov 2015.
- 7.Schmidt LH, Alexander S, Allen L, Rasco J. Comparison of the curative antimalarial activities and toxicities of primaquine and its d and l isomers. Antimicrob Agents Chemother. 1977;12:51–60. doi: 10.1128/AAC.12.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nanayakkara ND, Tekwani BL, Herath HB, Sahu R, Gettayacamin M, Tungtaeng A, et al. Scalable preparation and differential pharmacologic and toxicologic profiles of primaquine enantiomers. Antimicrob Agents Chemother. 2014;58:4737–4744. doi: 10.1128/AAC.02674-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saunders D, Vanachayangkul P, Imerbsin R, Khemawoot P, Siripokasupkul R, Tekwani BL, et al. Pharmacokinetics and pharmacodynamics of (+)-primaquine and (−)-primaquine enantiomers in rhesus macaques (Macaca mulatta) Antimicrob Agents Chemother. 2014;58:7283–7291. doi: 10.1128/AAC.02576-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bennett JW, Pybus BS, Yadava A, Tosh D, Sousa JC, McCarthy WF, et al. Primaquine failure and cytochrome P-450 2D6 in Plasmodium vivax malaria. N Engl J Med. 2013;369:1381–1382. doi: 10.1056/NEJMc1301936. [DOI] [PubMed] [Google Scholar]
- 11.Pybus BS, Marcsisin SR, Jin X, Deye G, Sousa JC, Li Q, et al. The metabolism of primaquine to its active metabolite is dependent on CYP 2D6. Malar J. 2013;12:212. doi: 10.1186/1475-2875-12-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fasinu PS, Tekwani BL, Nanayakkara NP, Avula B, Herath HM, Wang YH, et al. Enantioselective metabolism of primaquine by human CYP2D6. Malar J. 2014;13:507. doi: 10.1186/1475-2875-13-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allahyari R, Strother A, Fraser IM, Verbiscar AJ. Synthesis of certain hydroxy analogues of the antimalarial drug primaquine and their in vitro methemoglobin-producing and glutathione-depleting activity in human erythrocytes. J Med Chem. 1984;27:407–410. doi: 10.1021/jm00369a031. [DOI] [PubMed] [Google Scholar]
- 14.Mihaly GW, Ward SA, Edwards G, Orme ML, Breckenridge AM. Pharmacokinetics of primaquine in man: identification of the carboxylic acid derivative as a major plasma metabolite. Br J Clin Pharmacol. 1984;17:441–446. doi: 10.1111/j.1365-2125.1984.tb02369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tekwani BL, Avula B, Sahu R, Chaurasiya ND, Khan SI, Jain S, et al. Enantioselective pharmacokinetics of primaquine in healthy human volunteers. Drug Metab Dispos. 2015;43:571–577. doi: 10.1124/dmd.114.061127. [DOI] [PubMed] [Google Scholar]
- 16.Avula B, Tekwani BL, Chaurasiya ND, Nanayakkara NP, Wang YH, Khan SI, et al. Profiling primaquine metabolites in primary human hepatocytes using UHPLC-QTOF-MS with 13C stable isotope labeling. J Mass Spectrom. 2013;48:276–285. doi: 10.1002/jms.3122. [DOI] [PubMed] [Google Scholar]
- 17.Herath HM, McChesney JD, Walker LA, Nanayakkara NP. Synthesis of [13C6] primaquine. J Labelled Comp Radiopharm. 2013;56:341–343. doi: 10.1002/jlcr.3039. [DOI] [PubMed] [Google Scholar]
- 18.McChesney JD, Sarangan S. Synthesis of 8-(3-carboxy-l-methyl-propylammo)-6-methoxyquinoline: a newly characterized primaquine metabolite. Pharm Res. 1984;1:96–98. doi: 10.1023/A:1016311616719. [DOI] [PubMed] [Google Scholar]
- 19.Hufford CD, Clark AM, Quinones IN, Baker JK, McChesney JD. Microbial metabolism studies on the major microbial and mammalian metabolite of primaquine. J Pharm Sci. 1983;72:92–94. doi: 10.1002/jps.2600720124. [DOI] [PubMed] [Google Scholar]
- 20.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos. 1999;27:1350–1359. [PubMed] [Google Scholar]
- 21.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093–1095. doi: 10.1023/A:1018943613122. [DOI] [PubMed] [Google Scholar]
- 22.Potter BM, Xie LH, Vuong C, Zhang J, Zhang P, Duan D, et al. Differential CYP 2D6 metabolism alters primaquine pharmacokinetics. Antimicrob Agents Chemother. 2015;59:2380–2387. doi: 10.1128/AAC.00015-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Avula B, Khan SI, Tekwani BL, Nanayakkara NPD, McChesney JD, Walker LA, et al. Analysis of primaquine and its metabolite carboxyprimaquine in biological samples: enantiomeric separation, method validation and quantification. Biomed Chromatogr. 2011;25:1010–1017. doi: 10.1002/bmc.1557. [DOI] [PubMed] [Google Scholar]
- 24.Ganesan S, Chaurasiya ND, Sahu R, Walker LA, Tekwani BL. Understanding the mechanisms for metabolism-linked hemolytic toxicity of primaquine against glucose 6-phosphate dehydrogenase deficient human erythrocytes: evaluation of eryptotic pathway. Toxicology. 2012;294:54–60. doi: 10.1016/j.tox.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 25.Potter BM, Xie LH, Vuong C, Zhang J, Zhang P, Duan D, et al. Differential CYP 2D6 metabolism alters primaquine pharmacokinetics. Antimicrob Agents Chemother. 2015;59:2380–2387. doi: 10.1128/AAC.00015-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pybus BS, Sousa JC, Jin X, Ferguson JA, Christian RE, Barnhart R, et al. CYP450 phenotyping and accurate mass identification of metabolites of the 8-aminoquinoline, anti-malarial drug primaquine. Malar J. 2012;11:259. doi: 10.1186/1475-2875-11-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Obach RS, Reed-Hagen AE, Krueger SS, Obach BJ, O’Connell TN, Zandi KS, et al. Metabolism and disposition of varenicline, a selective α4β2 acetylcholine receptor partial agonist, in vivo and in vitro. Drug Metab Dispos. 2006;34:121–130. doi: 10.1124/dmd.105.006767. [DOI] [PubMed] [Google Scholar]
- 28.Sadeque AJ, Usmani KA, Palamar S, Cerny MA, Chen WG. Identification of human UDP-glucuronosyltransferases involved in N-carbamoyl glucuronidation of lorcaserin. Drug Metab Dispos. 2012;40:772–778. doi: 10.1124/dmd.111.043448. [DOI] [PubMed] [Google Scholar]