

Abstract
Background
BRAF V600 mutant circulating cell-free tumor DNA (BRAF V600mut ctDNA) could serve as a specific biomarker in patients with BRAF V600 mutant melanoma. We analyzed the value of BRAF V600mut ctDNA from plasma as a monitoring tool for advanced melanoma patients treated with BRAF/MEK inhibitors.
Methods
Allele-specific quantitative PCR analysis for BRAF V600 E/E2/D/K/R/M mutations was performed on DNA extracted from plasma of patients with known BRAF V600 mutant melanoma who were treated with dabrafenib and trametinib.
Results
245 plasma samples from 36 patients were analyzed. In 16 patients the first plasma sample was obtained before the first dosing of dabrafenib/trametinib. At baseline, BRAF V600mut ctDNA was detected in 75 % of patients (n = 12/16). BRAF V600mut ctDNA decreased rapidly upon initiation of targeted therapy (p < 0.001) and became undetectable in 60 % of patients (n = 7/12) after 6 weeks of treatment. During treatment, disease progression (PD) was diagnosed in 27 of 36 patients. An increase of the BRAF V600mut ctDNA copy number and fraction, identified PD with a sensitivity of 70 % (n = 19/27) and a specificity of 100 %. An increase in the BRAF V600mut ctDNA fraction was detected prior to clinical PD in 44 % of cases (n = 12/27) and simultaneously with PD in 26 % of patients (n = 7/27).
Conclusions
Quantitative analysis of BRAF V600mut ctDNA in plasma has unique features as a monitoring tool during treatment with BRAF/MEK inhibitors. Its potential as an early predictor of acquired resistance deserves further evaluation.
Keywords: Melanoma, cfDNA, ctDNA, BRAF V600, Biomarkers, Dabrafenib, Trametinib
Background
The detection of mutations in circulating cell-free tumor DNA (ctDNA) is under investigation as a specific biomarker for the diagnosis and monitoring of patients with different cancer types [1–4]. Mutations in the BRAF gene at position V600 are detected in 40–50 % of cutaneous melanomas, and represent the most common oncogenic driver mutation in this disease [5]. Therefore, quantitative measurement of BRAF V600 mutant ctDNA in cell-free DNA (cfDNA) extracted from plasma could serve as a specific biomarker in this patient population [6, 7].
Treatment with a combination of a BRAF- and MEK inhibitor results in a high tumor response rate (64–69 %) and improves the survival of patients with BRAF V600 mutant melanoma [8–10]. Immune-checkpoint inhibition of either the CTLA-4 or PD-1 receptor can also improve the survival of patients with advanced melanoma, irrespective of the BRAF V600 mutation status [11–13]. Optimal sequencing of available treatment options for patients with BRAF V600 mutant melanoma has not been defined. Retrospective series have raised concern that ipilimumab may have lesser activity when applied after the emergence of resistance to BRAF-inhibitors [14, 15]. Alternatively, ipilimumab fails to improve the survival of patients with a life-expectancy of less than 3–4 months from initiating therapy, and durable complete responses have been reported on ipilimumab in patients who developed prior resistance to treatment with a BRAF-inhibitor [16]. Of concern is the high incidence of progression within the central nervous system (CNS) at first progression on BRAF-inhibitors, as metastases to the CNS often imply a poor prognosis and necessitate the use of corticotherapy, implying an unfavorable condition for initiating immunotherapy [17, 18]. As conventional clinical tools for assessment of early tumor progression lack sensitivity, many patients will be symptomatic at the time of progression or will experience rapid progression and deterioration in the few weeks that follow the diagnosis of progression [17, 19–21]. Therefore, more sensitive tools for monitoring of response and resistance to BRAF/MEK targeted therapy is of interest in order to optimize treatment of advanced BRAF V600 mutant melanoma. Furthermore, changes in the BRAF V600mut ctDNA concentration might be helpful for the interpretation of imaging results during immunotherapy where atypical tumor responses are more frequent [22, 23].
In this translational research study we analyze the value of longitudinal quantitative measurement of BRAF V600mut ctDNA from plasma as a therapeutic monitoring tool for patients with advanced BRAF V600 mutant melanoma treated with the BRAF/MEK inhibitors dabrafenib and trametinib.
Methods
This was an exploratory translational study with a primary objective of investigating longitudinal quantitative measurement of BRAF V600mut ctDNA in patients treated with a combination of a BRAF and a MEK inhibitor using the Idylla™ ctBRAF Mutation prototype assay on the Idylla™ system (Biocartis). The study was conducted at a single university hospital (UZ Brussel, academic study sponsor) in collaboration with Biocartis (Mechelen, Belgium).
Patients were eligible for plasma BRAF V600mut ctDNA monitoring when a BRAF V600 mutation had been detected in tumor tissue and were either treated or initiated treatment with dabrafenib and trametinib. Blood samples were prospectively collected after obtaining informed consent with an Ethical Committee approved document. Response evaluation to targeted therapy was performed every 2 months with standard imaging techniques [including 18-fluorodeoxyglucose positron emission tomography/computed tomography (FDG PET–CT), computed tomography (CT) of thorax and abdomen, magnetic resonance imaging (MRI) of the brain]. Plasma samples were collected together with routine blood collections (every 2 weeks during the first month of therapy; every month after the first month of therapy), until progressive disease (PD) was detected according to RECIST v1.1 [24].
Blood samples were collected in 10 mL EDTA tubes and immediately centrifuged at a relative centrifugal force of 1410.63×g, during 15 min at room temperature. Plasma was separated and stored in 1-mL aliquots at −80 °C. Silica-based extraction of DNA and subsequent allele-specific quantitative PCR (qPCR) to detect the BRAF wild-type gene and the G1798>A and T1799>A changes in the BRAF gene were performed with Idylla™ (Biocartis) on 1 mL of the stored plasma. The G1798>A change is present in patients with V600 K, V600R, and V600 M mutations, whereas the T1799>A change is present in patients with V600E, V600 K, V600E2, and V600D mutations. A linear correlation between Cq values reported by prototype Idylla assays and digital droplet PCR was previously established, allowing precise and sensitive quantification of mutant ctDNA fragments in plasma down to 3 mutant copies per PCR reaction with an analytical sensitivity of 0.01 %. The investigators that performed the DNA extraction and the subsequent qPCR were blinded for the clinical patient information. Quantitative PCR results were expressed as median and 25th/75th percentiles of the BRAF V600mut ctDNA copy number per milliliter, and the BRAF V600mut ctDNA fraction to the total amount of cfDNA. The evolution of the BRAF V600mut ctDNA copy number and fraction, were analyzed in relation to the clinical diagnosis of PD. Progression-free survival (PFS) was measured from the time of first administration of targeted therapy, to the time of PD or death, and was analyzed using the Kaplan–Meier method (SPSS software, IBM). For comparison of PFS between different groups, the log-rank test was used. We applied the Wilcoxon signed-rank test to compare changes in the concentrations during treatment. Fisher’s exact test was used to compare categorical variables. A 2-tailed p value ≤0.05 was considered to be statistically significant. Statistical analysis was performed with SPSS statistics 22.
To assess the early evolution of the BRAF V600mut ctDNA concentration during treatment with dabrafenib and trametinib, five samples were collected during the first 24 h of treatment in two additional patients treated “in hospital”. In these two patients, all plasma samples were analyzed in duplicate. The first sample of 1 mL was analyzed with the Idylla™ system using a fully automated cartridge that contains all reagents for the above mentioned extraction of DNA followed by qPCR detection and reporting of the results. In the second sample of 1 mL, DNA was extracted using the QIAamp Circulating Nucleic Acid Kit (Qiagen), followed by droplet digital PCR (ddPCR, Bio-rad) for detection of BRAF V600Emut ctDNA. Each ddPCR analysis was performed in triplicate. Five μL of eluate was added to each ddPCR reaction containing 11.5 μL ddPCR Supermix for probes (Bio-Rad) and 3 μL of BRAF V600E and wild-type assay (dHsaCP2000027, dHsaCP2000028, Bio-Rad), adjusted to a total volume of 23 μL. The reaction was then compartmentalized into ~15,000 droplets per sample in a QX-100 droplet generator (Bio-Rad) according to the manufacturer’s instructions. Emulsified PCR reactions were run in 96-well format on a Bio-rad CFX96 Touch Real-Time PCR Detection System according to the manufacturer’s instructions. Plates were subsequently read on a Bio-Rad QX-100 droplet reader using QuantaSoft software (Bio-Rad). No-template controls were included in each run and the BRAF V600E copies per sample were quantified using the Quantasoft software.
Results
Patients and procurement of plasma samples
From February 2014 to October 2015, 245 plasma samples from 36 patients with advanced melanoma were collected. Clinical baseline patient characteristics are summarized in Table 1. In 16 patients the first plasma sample was obtained before the first dosing of dabrafenib (150 mg BID) and trametinib (2 mg QD). In 20 patients the first sample was obtained after initiation of targeted therapy.
Table 1.
Patient baseline characteristics (n = 36)
| Variable | Overall |
|---|---|
| Age | 52 (12) |
| Sex | |
| Male | 12 (33 %) |
| Female | 24 (67 %) |
| Stage | |
| IVa | 4 (11 %) |
| IVb | 2 (6 %) |
| IVc | 30 (83 %) |
| Primary site | |
| Cutaneous | 28 (78 %) |
| Acral | 4 (11 %) |
| Mucosal | 0 (0 %) |
| Unknown | 4 (11 %) |
| Treatment | |
| Dabrafenib + trametinib | 34 (94 %) |
| Dabrafenib | 2 (6 %) |
| Vemurafenib | 0 (0 %) |
| Re-challenge dabrafenib + trametiniba | 8 (22 %) |
| First plasma sample | |
| Before initiation of targeted therapy | 16 (44 %) |
| After initiation of targeted therapy | 20 (56 %) |
Data are mean (SD) or n (%).
Baseline characteristics at the moment of collection of the first plasma sample
aEight patients were treated with dabrafenib and trametinib after documentation of disease progression at least 12 weeks following the last day of dosing of a BRAF-inhibitor containing treatment regimen (ClinicalTrials.gov Identifier:NCT02296996)
Early changes in BRAF V600 mutant ctDNA after treatment initiation with dabrafenib/trametinib
In 75 % of patients (n = 12/16) BRAF V600mut ctDNA was detected before initiation of dabrafenib and trametinib (median fraction 16.5 %; Q1–Q3 9.8–25 %—median copy number 571 copies/mL; Q1–Q3 79–1610 copies/mL) (Fig. 1a). In 25 % of patients (n = 4/16) no BRAF V600mut ctDNA was detected before initiation of dabrafenib and trametinib.
Fig. 1.
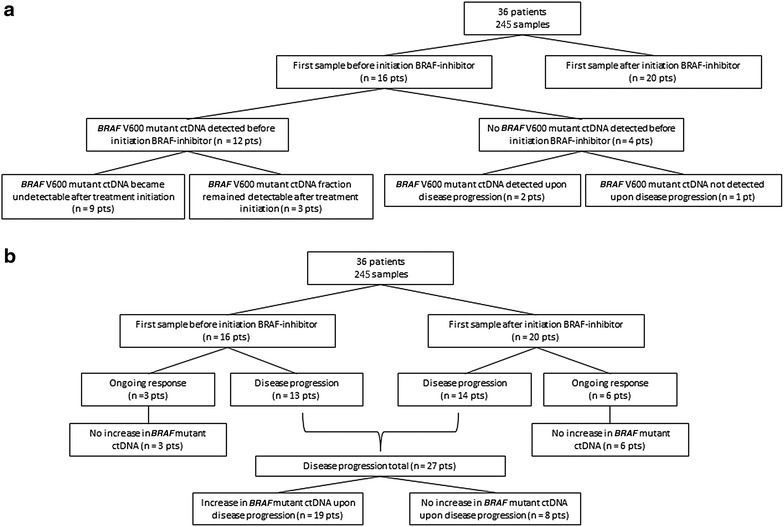
a Flow diagram showing subgroups of patients according to the detection of BRAF V600mut ctDNA after treatment initiation. b Flow diagram showing subgroups of patients according to the evolution of BRAF V600mut ctDNA in relation to disease progression. Pts patients
To assess changes in the BRAF V600mut ctDNA concentration during the first hours after treatment initiation, five samples were collected in the first 24 h of treatment in 2 patients treated ‘‘in hospital’’ (Fig. 2). In both patients the BRAF V600mut ctDNA copy number decreased significantly within 5 days after treatment initiation. In one patient only a transient increase in the BRAF V600mut ctDNA copy number was observed within the first 24 h on therapy (Fig. 2b, d).
Fig. 2.
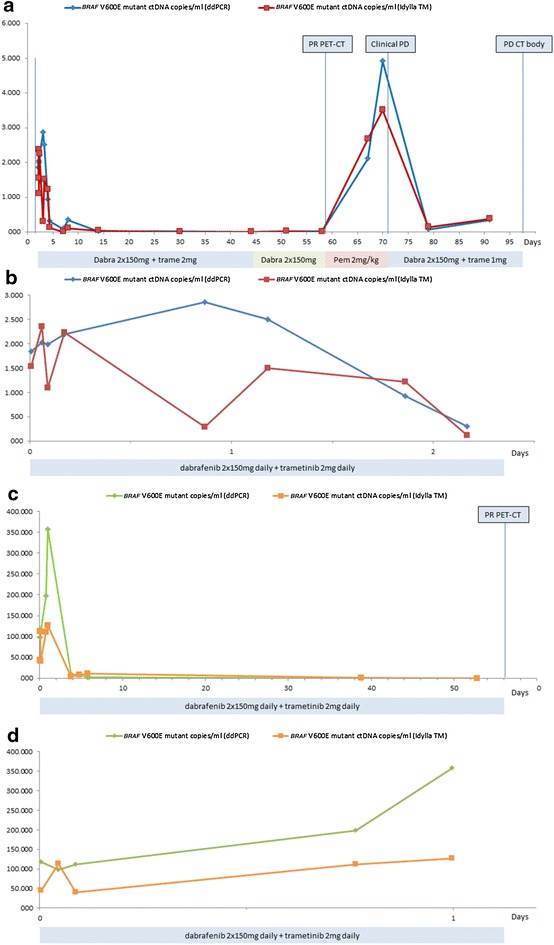
Serial measurement of BRAF V600mut ctDNA in plasma of 2 patients (a, b = patient 1; c, d = patient 2) with advanced melanoma during targeted therapy. The y-axis represents the BRAF V600mut ctDNA copy number per millilitre. Results obtained with the Idylla™ system (Biocartis) and digital PCR (Bio-rad) are shown. a Treatment with dabrafenib and trametinib (dabra 2 × 150 mg + trame 2 mg) was initiated after obtaining the baseline plasma sample. Within 1 week the BRAF V600mut ctDNA copy number dropped significantly. After switching targeted therapy to pembrolizumab (Pem 2 mg/kg) due to side effects on the moment of best response (PR PET–CT), an increase of the BRAF v600mut ctDNA copy number was detected within 9 days. After reintroducing dabrafenib and trametinib, due to rapid clinical disease progression (Clinical PD), the BRAF V600mut ctDNA copy number dropped again. b The evolution of the BRA F V600mut ctDNA copy number in patient 1 during the first 2 days of treatment. c Treatment with dabrafenib and trametinib (dabra 2 × 150 mg + trame 2 mg) was initiated after obtaining the baseline plasma sample. Within 1 week the BRAF V600mut ctDNA copy number dropped significantly. d The evolution of the BRAF V600mut ctDNA copy number in patient 2 during the first day of treatment
During treatment, the BRAF V600mut ctDNA fraction and copy number decreased significantly compared to baseline in all 12 patients (p < 0.01, Fig. 3), and became undetectable (n = 7) or <1 % (n = 5) after a median of 13 days (range 6–40 days). At the time when no ctDNA could be measured any more, none of the patients had obtained a complete radiological remission (e.g. Fig. 4a, e). In 3 patients, BRAF V600mut ctDNA remained detectable from the initiation of targeted therapy until PD was diagnosed (e.g. Fig. 4b). Median PFS was not significantly longer for patients in whom BRAF V600mut ctDNA became undetectable 1 month after the initiation of targeted therapy, as compared to patients in whom ctDNA remained detectable during the first month of therapy (p = 0.56). Progression free survival was significantly shorter for patients in whom BRAF V600mut ctDNA remained detectable throughout the treatment with targeted therapy compared to patients in whom BRAF V600mut ctDNA became undetectable (p < 0.001) (Fig. 5).
Fig. 3.
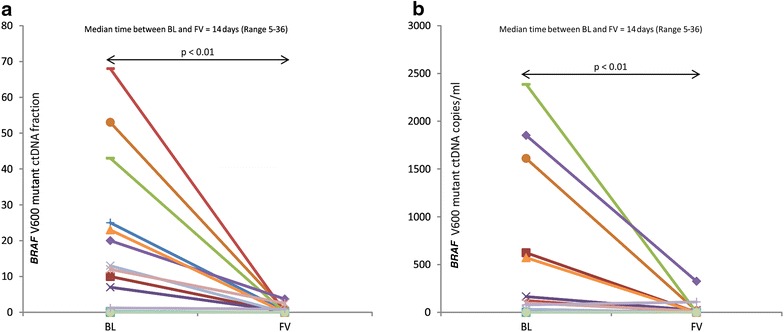
a Individual evolutions in the BRAF V600mut ctDNA fraction from initiation of treatment with dabrafenib (150 mg BID) and trametinib (2 mg QD), at baseline (BL) to the first visit (FV). b Individual evolutions in the BRAF V600mut ctDNA copy number from initiation of treatment with dabrafenib (150 mg BID) and trametinib (2 mg QD) at baseline (BL) to the first visit (FV)
Fig. 4.
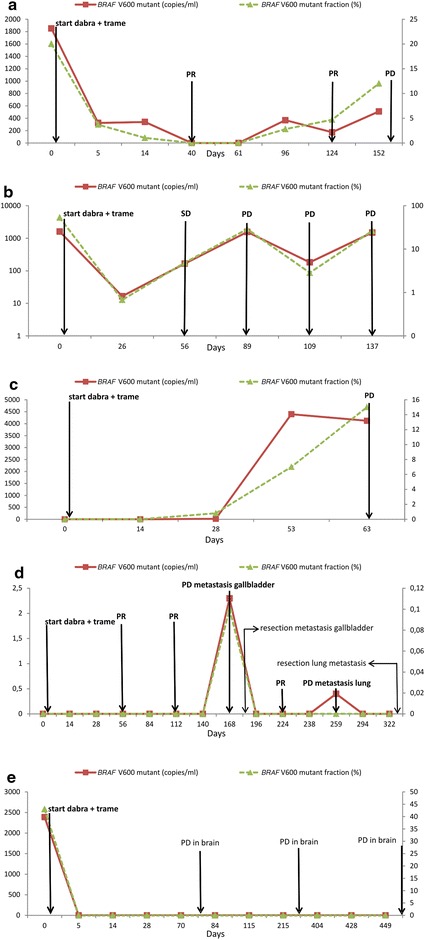
Serial measurement of BRAF V600mut ctDNA from plasma in 5 patients (a–e) with advanced melanoma during targeted therapy. Treatment with dabrafenib (dabra, 150 mg BID) and trametinib (trame, 2 mg QD) was initiated after obtaining the baseline plasma sample. The left y-axis represents the BRAF V600mut ctDNA copy number per milliliter (solid line), the right y-axis represents the BRAF V600mut ctDNA fraction to the total amount of cfDNA (dashed line). SD, PR and PD respectively denote stable disease, partial response and progressive disease according to RECIST v1.1. a The BRAF V600mut ctDNA copy number and fraction dropped after treatment initiation. After 40 days, no BRAF V600mut ctDNA could be detected anymore, and CT body showed a PR. The BRAF V600mut ctDNA fraction reappeared after 96 days and increased on day 124, although an ongoing PR was reported on PET–CT. PET–CT showed PD, 50 days after the reappearance of BRAF V600mut ctDNA in plasma. b BRAF V600mut ctDNA remained detectable from treatment initiation until PD was detected after 89 days. c At baseline, no BRAF V600mut ctDNA was detected in plasma. BRAF V600mut ctDNA appeared after 28 days, 35 days prior to the detection of PD on CT body. d At baseline, no BRAF V600mut ctDNA was detected in plasma. After 168 days, PD was detected in a gallbladder metastasis, and BRAF V600mut ctDNA was detected concomitantly. The only other lesion, a lung metastasis, had remained strictly stable. After resection of the gallbladder metastasis, the BRAF V600mut ctDNA fraction could not be detected until PD occurred in the remaining lung metastasis. e Five days after treatment initiation, BRAF V600mut ctDNA could not be detected anymore. Brain MRI showed a new millimetric brain lesion on the first response evaluation, while PET–CT showed clear regression of all liver and lung metastases. Subsequent MRI’s showed ongoing slow PD in the brain, while liver and lung lesions showed ongoing PR. No BRAF V600mut ctDNA was detected during PD in this patient
Fig. 5.
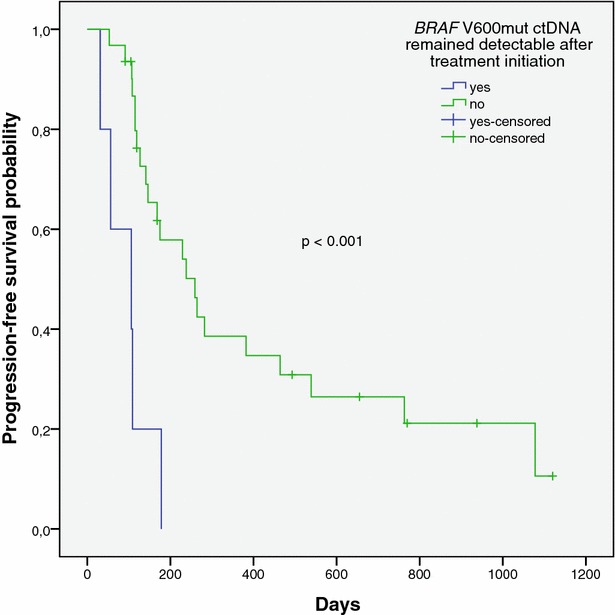
Kaplan–Meier plot representing the progression-free survival probability for patients with advanced melanoma in whom BRAF V600mut ctDNA became undetectable after initiation of a BRAF inhibitor containing treatment regimen (solid line), and for patients in whom BRAF V600mut ctDNA remained detectable during follow-up (dashed line)
Correlation between BRAF V600 mutant ctDNA levels and disease progression
During the course of plasma BRAF V600mut ctDNA monitoring, clinical PD was diagnosed in 27 of 36 patients after a median of 111 days [95 % CI 98–124] (Figs. 1b, 6). An increase of the BRAF V600mut ctDNA copy number and fraction was diagnosed in 19 of 27 (70 %) patients with PD and in none of the patients with an ongoing response, resulting in a sensitivity of 70 % and a specificity of 100 % (Kappa 0.54 [0.26–0.82]).
Fig. 6.
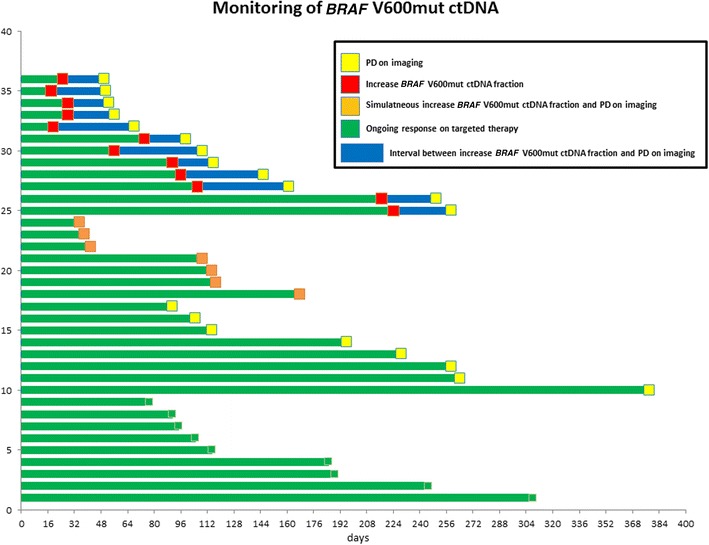
Swimmer plot showing the interval between increase of the BRAF V600mut ctDNA fraction and progressive disease according to RECIST v1.1 on imaging (PD), for all 36 patients patients from start of ctDNA monitoring. In 12 patients, an increase in the BRAF V600mut ctDNA fraction was detected prior to PD. In 7 patients, an increase of the BRAF V600mut ctDNA fraction was detected simultaneously with PD. In 8 patients, no increase in the BRAF V600mut ctDNA fraction was detected prior to or simultaneously with PD. None of the patients with an ongoing response showed an increase of the BRAF V600mut ctDNA fraction
In 30 % (n = 8/27) of patients, no BRAF V600mut ctDNA was detected at the time of clinical PD. Of these eight patients, three showed PD in the brain (2 asymptomatic, e.g. Fig. 4e) and five showed slow PD in a single subcutaneous, bone, muscle or lung lesion.
An increase in the BRAF V600mut ctDNA fraction was detected prior to the clinical diagnosis of PD in 12 out of 27 (44 %) patients and simultaneously with PD in 7 out of 27 (26 %) patients. The first increase in the BRAF V600mut ctDNA fraction, after the BRAF V600mut ctDNA fraction became undetectable or was decreasing, was detected after a median of 74 days [95 % CI 19–130] from the collection of the first plasma sample. The median interval between the first increase in the BRAF V600mut ctDNA fraction and PD was 25 days [95 % CI 1–48 days]. A first increase in the BRAF V600mut ctDNA fraction or copy number predicted PD within the following month in 63 % of patients (n = 12/19; p < 0.001) and within the following 2 months in 100 % of patients (n = 19/19; p < 0.001).
When BRAF V600mut ctDNA could not be detected in plasma (n = 173 samples), PD did not occur in the following month in 86 %, and not in the following 2 months in 76 % of cases. Out of the four patients with no detectable BRAF V600 ctDNA at baseline, three patients showed PD during follow-up, and in two of these patients BRAF V600mut ctDNA was detected simultaneously with or before prior to PD on imaging. (Fig. 4c, d).
Discussion
In this study, BRAF V600mut ctDNA was detected at baseline in 75 % of stage IV melanoma patients with a known BRAF V600 mutation. This is in line with two other studies using PCR based methods, where BRAF V600mut ctDNA was detected in plasma of 73–84 % of BRAF V600 mutant melanoma patients [6, 25]. These results are also in line with an overall detection rate of 77 % in 4 clinical studies with 746 stage IV BRAF V600 mutant melanoma patients using BEAMing (beads, emulsions, amplification, and magnetics analysis) after cfDNA extraction from plasma [26–30]. Moreover, one study showed a 100 % agreement between digital PCR, qPCR and BEAMing, suggesting that the suboptimal detection rate of BRAF V600 mutant DNA in plasma in advanced melanoma is the result of a variable amount of BRAF V600mut ctDNA in advanced melanoma patients, rather than an variability in the sensitivity of different test-platforms [25]. We observed that after initiating BRAF/MEK inhibitor treatment, BRAF V600mut ctDNA can drop below the detection limit, notwithstanding the absence of radiological complete remission. This phenomenon of rapid decrease in the concentration of BRAF V600mut ctDNA has been reported before and has been attributed to the destruction of tumor cells and a subsequent rapid clearance of ctDNA [6, 31–33]. Our combined observations of (1) decrease of the BRAF V600mut ctDNA concentration within days after treatment initiation, (2) of early increase during disease progression and (3) of early increase after discontinuation of targeted therapy, are suggestive of a correlation between ctDNA levels and the proliferation of melanoma cells. Therefore BRAF V600mut ctDNA seems to reflect the BRAF V600mut-dependent proliferative tumor burden, and not tumor mass as evidenced by CT-imaging. From this perspective, ctDNA could be attributed to increased cell death during melanoma cell proliferation or might be actively secreted or passively released by living cells, rather than by apoptotic or necrotic cells [34]. This latter hypothesis is supported by several preclinical arguments. Spontaneous active DNA release or apoptosis have been put forward as the main source of ctDNA, because plasma DNA often presents a ladder pattern, typical for active cleaving, when subjected to electrophoresis [32, 35]. Several arguments support the hypothesis that DNA is primarily released by living cancer cells rather than by apoptotic cancer cells: (1) DNA concentration increases in normal lymphocyte cultures following stimulation with phytohemagglutinin, lipopolysaccharide or antigen; (2) in a leukemic cell-line malignant cells released newly synthesized DNA; (3) cancer cell DNA concentration in cell culture supernatant increases with cell proliferation when few apoptotic or necrotic cells are present; and (4) circulating exosomal BRAF V600E mutant DNA was isolated from SK-MEL-28 melanoma-bearing mice [32, 33, 36, 37].
In our study population, PFS was significantly better for the patients in whom BRAF V600mut ctDNA became undetectable after the initiation of targeted therapy. When the BRAF V600mut ctDNA fraction increased during treatment, clinical PD always followed within 2 months. Moreover, in 44 % of cases an increase in the mutant fraction preceded PD. We recently reported that reappearance of BRAF V600mut ctDNA in plasma cfDNA can precede changes on PET–CT in patients under BRAF/MEK inhibitor treatment [34]. In this study an increase in the BRAF V600mut ctDNA fraction preceded PD with a median interval of 25 days. However, plasma samples were collected on a monthly basis, whereas imaging was performed every 2 months. In a study on ctDNA monitoring in metastatic breast cancer, increasing levels of ctDNA appeared on average 5 months before the establishment of PD by means of imaging [3].
An increase in the BRAF V600mut ctDNA fraction during BRAF/MEK targeted therapy could potentially serve as a trigger for early evaluation with imaging techniques, and allow for a timely switch to immunotherapy or other appropriate therapeutic interventions (e.g. stereotactic radiotherapy to brain metastases). Early detection of progression is important in metastatic melanoma, because of the aggressive course of the disease as exemplified by the observation that the central nervous system is a frequent site (30 %) of first progression under BRAF inhibitor therapy [17]. Moreover, potential second line therapeutic options such as ipilimumab and, to a lesser extent anti-PD-1 therapies, are associated with a latency of their anti-tumor effect. Therefore, the possibility of offering immunotherapy may be compromised if PD is only detected on imaging or when clinical symptoms are present [19, 20].
Conclusions
Our results indicate that quantitative analysis of BRAF V600mut ctDNA in plasma holds promise as a monitoring tool in BRAF V600 mutant melanoma during treatment with BRAF/MEK targeted therapy. Prospective trials are needed to assess if an add-on strategy or switch to immunotherapy upon early detection of therapy resistance through ctDNA evolution, adds value to the current treatment strategies. Finally we provide clinical arguments suggesting that an important fraction of BRAF V600mut ctDNA correlates with the BRAF V600mut-dependent proliferative tumor burden.
Authors’ contributions
Study concepts: MS, BN. Study design: MS, BN. Data acquisition: MS, GM, SVDH, YJ, BJacobs. Quality control of data and algorithms: MS, AB. Data analysis and interpretation: MS, BN. Statistical analysis: MS, RB. Manuscript preparation: MS. Manuscript editing: MS, IC, BN. Manuscript review: MS, IC, SW, TS, BN, YJ. All authors read and approved the final manucript.
Competing interests
The following authors are employed by Biocartis: Geert Meersseman, Geert Maertens, Sari Van Den Herrewegen, Bart Jacobs. There is no potential competing interests for the other authors.
Consent for publication
We have obtained consent to publish from the participants.
Ethics approval and consent to participate
An ethics committee approved informed consent file was signed (Ethics committee: Commissie Medische Ethiek UZ Brussel).
Funding
Prof. Bart Neyns received funding from the Flemish government (“Kom Op Tegen Kanker”) for this research project. Max Schreuer received a doctoral (PhD) grant from the Flemish government (“Kom Op Tegen Kanker”).
Abbreviations
- BRAF V600mut ctDNA
BRAF V600 mutant circulating cell-free tumor DNA
- ctDNA
circulating cell-free tumor DNA
- cfDNA
circulating cell-free DNA
- CNS
central nervous system
- FDG PET–CT
fluorodeoxyglucose positron emission tomography/computed tomography
- CT
computed tomography
- MRI
magnetic resonance imaging
- PD
progressive disease
- qPCR
allele-specific quantitative PCR
- PFS
progression-free survival
- BID
bis in die (twice daily)
- QD
quaque die (once daily)
- BEAMing
beads, emulsions, amplification, and magnetics analysis
- CR
complete remission
Contributor Information
Max Schreuer, Phone: + 32 2 474 93 91, Email: max.schreuer@uzbrussel.be.
Geert Meersseman, Email: gmeersseman@biocartis.com.
Sari Van Den Herrewegen, Email: svandenherrewegen@biocartis.com.
Yanina Jansen, Email: yanina.jansen@uzbrussel.be.
Ines Chevolet, Email: ines.chevolet@uzgent.be.
Ambre Bott, Email: ambre.bott@gmail.com.
Sofie Wilgenhof, Email: sofie.wilgenhof@vub.ac.be.
Teofila Seremet, Email: teofila.caplanusi@erasme.ulb.ac.be.
Bart Jacobs, Email: bjacobs@biocartis.com.
Ronald Buyl, Email: rbuyl@vub.ac.be.
Geert Maertens, Email: gmaertens@biocartis.com.
Bart Neyns, Email: bart.neyns@uzbrussel.be.
References
- 1.Gonzalez-Masia JA, Garcia-Olmo D, Garcia-Olmo DC. Circulating nucleic acids in plasma and serum (CNAPS): applications in oncology. Oncotargets Ther. 2013;6:819–832. doi: 10.2147/OTT.S44668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bettegowda C, Sausen M, Leary R, Kinde I, Agrawal N, Bartlett B, et al. Detection of circulating tumor DNA in early and late stage human malignancies. Cancer Res. 2014; 74(19). [DOI] [PMC free article] [PubMed]
- 3.Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368(13):1199–1209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 4.Oxnard GR, Paweletz CP, Kuang YA, Mach SL, O’Connell A, Messineo MM, et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 2014;20(6):1698–1705. doi: 10.1158/1078-0432.CCR-13-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell. 2012;150(2):251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanmamed MF, Fernandez-Landazuri S, Rodriguez C, Zarate R, Lozano MD, Zubiri L, et al. Quantitative cell-free circulating BRAFV600E mutation analysis by use of droplet digital PCR in the follow-up of patients with melanoma being treated with BRAF inhibitors. Clin Chem. 2015;61(1):297–304. doi: 10.1373/clinchem.2014.230235. [DOI] [PubMed] [Google Scholar]
- 7.Chang-Hao Tsao S, Weiss J, Hudson C, Christophi C, Cebon J, Behren A, et al. Monitoring response to therapy in melanoma by quantifying circulating tumour DNA with droplet digital PCR for BRAF and NRAS mutations. Sci Rep. 2015;5. [DOI] [PMC free article] [PubMed]
- 8.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–39. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 9.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015;386:444–451. doi: 10.1016/S0140-6736(15)60898-4. [DOI] [PubMed] [Google Scholar]
- 10.Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–1876. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 11.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robert C, Thomas L, Bondarenko I, O’Day S, Garbe C, Weber J, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 13.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372(26):2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 14.Ascierto PA, Simeone E, Sileni VC, Del Vecchio M, Marchetti P, Cappellini GC, et al. Sequential treatment with ipilimumab and BRAF inhibitors in patients with metastatic melanoma: data from the Italian cohort of the ipilimumab expanded access program. Cancer Invest. 2014;32(4):144–149. doi: 10.3109/07357907.2014.885984. [DOI] [PubMed] [Google Scholar]
- 15.Ackerman A, Klein O, McDermott DF, Wang W, Ibrahim N, Lawrence DP, et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014;120(11):1695–1701. doi: 10.1002/cncr.28620. [DOI] [PubMed] [Google Scholar]
- 16.Schreuer M, Chevolet I, Jansen Y, Seremet T, Wilgenhof S, Lienard D, et al. Objective responses can be obtained by CTLA-4 inhibition in metastatic melanoma after BRAF inhibitor failure. Melanoma Res. 2015;25(1):68–74. doi: 10.1097/CMR.0000000000000131. [DOI] [PubMed] [Google Scholar]
- 17.Peuvrel L, Saint-Jean M, Quereux G, Brocard A, Khammari A, Knol AC, et al. Incidence and characteristics of melanoma brain metastases developing during treatment with vemurafenib. J Neurooncol. 2014;120(1):147–154. doi: 10.1007/s11060-014-1533-z. [DOI] [PubMed] [Google Scholar]
- 18.Margolin KA, Di Giacomo AM, Maio M. Brain metastasis in melanoma: clinical activity of CTLA-4 antibody therapy. Semin Oncol. 2010;37(5):468–472. doi: 10.1053/j.seminoncol.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 19.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 20.Prieto PA, Yang JC, Sherry RM, Hughes MS, Kammula US, White DE, et al. CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res. 2012;18(7):2039–2047. doi: 10.1158/1078-0432.CCR-11-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abusaif S, Jradi Z, Held L, Pflugfelder A, Weide B, Meier F, et al. S100B and lactate dehydrogenase as response and progression markers during treatment with vemurafenib in patients with advanced melanoma. Melanoma Res. 2013;23(5):396–401. doi: 10.1097/CMR.0b013e3283650741. [DOI] [PubMed] [Google Scholar]
- 22.Wolchok JD, Hoos A, O’Day S, Weber JS, Hamid O, Lebbe C, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15(23):7412–7420. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 23.Du Four S, Wilgenhof S, Duerinck J, Michotte A, Van Binst A, De Ridder M, et al. Radiation necrosis of the brain in melanoma patients successfully treated with ipilimumab, three case studies. Eur J Cancer. 2012;48(16):3045–3051. doi: 10.1016/j.ejca.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 24.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45(2):228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 25.Janku F HH, Claes B, Falchook GS, Fu S, Tsimberidou AM, et al. Rapid, automated BRAF mutation testing of cell-free DNA from plasma of patients with advanced cancers using the novel Idylla platform. Proceedings of the 106th Annual Meeting of the American Association for Cancer Research (#2413), 4/2015. 2015.
- 26.Santiago-Walker AGB, Mazumdar J, Casey M, Haney P, O’Hagan A, et al. Correlation of BRAF mutation status in cfDNA and tumor and association with clinical outcome across four BRAFi and MEKi clinical trials. Pigment Cell Melanoma Res. 2013;26(6):997. [Google Scholar]
- 27.Long GV, Trefzer U, Davies MA, Kefford RF, Ascierto PA, Chapman PB, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087–1095. doi: 10.1016/S1470-2045(12)70431-X. [DOI] [PubMed] [Google Scholar]
- 28.Ascierto PA, Minor D, Ribas A, Lebbe C, O’Hagan A, Arya N, et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol. 2013;31(26):3205–3211. doi: 10.1200/JCO.2013.49.8691. [DOI] [PubMed] [Google Scholar]
- 29.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated Melanoma. N Engl J Med. 2012;367(2):107–114. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 30.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 31.Pupilli C, Pinzani P, Salvianti F, Fibbi B, Rossi M, Petrone L, et al. Circulating BRAF(V600E) in the diagnosis and follow-up of differentiated papillary thyroid carcinoma. J Clin Endocrinol Metab. 2013;98(8):3359–3365. doi: 10.1210/jc.2013-1072. [DOI] [PubMed] [Google Scholar]
- 32.Stroun M, Lyautey J, Lederrey C, Olson-Sand A, Anker P. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin Chim Acta. 2001;313(1–2):139–142. doi: 10.1016/S0009-8981(01)00665-9. [DOI] [PubMed] [Google Scholar]
- 33.Van der Vaart M, Pretorius PJ. The origin of circulating free DNA. Clin Chem. 2007;53(12):2215. doi: 10.1373/clinchem.2007.092734. [DOI] [PubMed] [Google Scholar]
- 34.Schreuer M, Van Den Herrewegen S, Jansen Y, Seremet T, Bott A, Chevolet I, et al. Applications for quantitative measurement of BRAF V600 mutant cell-free tumor DNA in the plasma of patients with metastatic melanoma. Melanoma Res. 2015 Dec 3. [Epub ahead of print]. [DOI] [PubMed]
- 35.Giacona MB, Ruben GC, Iczkowski KA, Roos TB, Porter DM, Sorenson GD. Cell-free DNA in human blood plasma: length measurements in patients with pancreatic cancer and healthy controls. Pancreas. 1998;17(1):89–97. doi: 10.1097/00006676-199807000-00012. [DOI] [PubMed] [Google Scholar]
- 36.Chen Z, Fadiel A, Naftolin F, Eichenbaum KD, Xia Y. Circulation DNA: biological implications for cancer metastasis and immunology. Med Hypotheses. 2005;65(5):956–961. doi: 10.1016/j.mehy.2005.04.042. [DOI] [PubMed] [Google Scholar]
- 37.Thakur B, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014;24(6):766–769. doi: 10.1038/cr.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]