

Abstract
Background
GSTM1 gene deletion is one of the most known copy number polymorphisms in human genome. It is most likely caused by homologous recombination between the repeats flanking the gene. However, taking into account that the deletion has no crucial effects on human well-being, and the ability of other GSTMs to compensate for the lack of GSTM1, a role for additional factors affecting GSTM1 deletion can be proposed. Our goal was to explore the relationships between GSTM1 deletion polymorphism and single nucleotide polymorphisms (SNPs) in the region of the GSTM cluster that includes GSTM2, GSTM3, GSTM4, and GSTM5 in addition to GSTM1.
Results
Real-time polymerase chain reaction was used to quantify the number of GSTM1 copies. Fourteen SNPs from the region were tested and their allelic patterns were compared in groups of Russian individuals subdivided according to their GSTM1 deletion genotypes. Linkage disequilibrium-based haplotype analysis showed substantial differences of haplotype frequencies between the groups, especially between individuals with homozygous GSTM1 −/− and +/+ genotypes. Exploration of the results of phasing of GSTM1 and SNP genotypes revealed unequal segregation of GSTM1 + and − alleles at different haplotypes.
Conclusions
The observed differences in haplotype patterns suggest the potential role of genetic context in GSTM1 deletion frequency (appearance) and in the determination of the deletion-related effects.
Keywords: DNA polymorphism, GSTM1 deletion, GSTM1 copy number polymorphism, Single nucleotide polymorphism, Haplotype
Background
Glutathione-S-transferases (GSTs) are a group of enzymes that play an important role in the metabolism and detoxification of a wide range of exo- and endogenous compounds. Functionally, GSTs act by conjugating electrophilic centers of the compounds with molecules of reduced glutathione. The reaction products are generally less reactive and readily excreted [1, 2]. Although many GST substrates can be potentially harmful to cellular macromolecules, some GST genes, particularly GSTT1 and GSTM1, are homozygously deleted in a high percentage (15 to 60 %) of individuals in various human populations [3]. GSTT1 and GSTM1 copy number variations are most likely caused by homologous recombination between the repeats flanking the both genes [4, 5]. However, the reasons for the high frequency of the deletions across different human populations are not well understood, especially if considered in the context of their associations with a higher risk for the development of a variety of pathophysiological conditions in individuals carrying deletion phenotypes [6]. A possible explanation could be the capability of other GSTs to compensate, at least in part, for the absence of GSTT1- and GSTM1-related enzyme activities under normal conditions [1]. Recent studies [7, 8] showed distinct connections between the levels of GST enzymatic activity and the activity of genes belonging to the same class (i.e., GSTT2B and GSTM2), evidently suggesting a role for local genetic context in the deletion frequency (deletion appearance).
In the current study, we investigated relationships between GSTM1 deletion polymorphism and SNP-based haplotypes in the region of the GSTM cluster that includes GSTM2, GSTM3, GSTM4, and GSTM5 in addition to GSTM1. The obtained results demonstrated substantial differences in haplotype distribution between groups of individuals subdivided according to GSTM1 deletion genotypes.
Methods
Blood samples were obtained with informed consent from Russian donors from three locations in the European part of Russia (Andreapolsky District of the Tver Oblast, n = 96; Muromsky District of the Vladimir Oblast, n = 96; and Kursky and Oktyabrsky districts of the Kursk Oblast, n = 93). The ethnicity of the donors was determined by an interview. To be included, individuals had to be unrelated and represent the native ethnic group in the regions studied (i.e., they belonged to at least the third generation living in a particular geographic region). The interview protocol and informed-consent form were approved by the Ethic Commission of Institute of Molecular Genetics of the Russian Academy of Sciences.
The DNA was isolated from peripheral leukocytes of the blood with a standard technique using proteinase K treatment and phenol–chloroform extraction [9].
To subdivide the individuals according to GSTM1 deletion polymorphism, the results of two genotyping methods were used. The first method was based on the simultaneous amplification of a site on GSTM1 with a region of another gene used as an internal control. The method makes it possible to identify individuals with a homozygous deletion genotype (i.e., they have no GSTM1 copies at all; GSTM1 −/− genotype). This approach was used in our previous study, and data for individuals with a GSTM1 −/− genotype were satisfactorily obtained [10]. Individuals with one or two copies of the GSTM1 gene are not distinguished by this method. To differentiate between individuals with one (i.e., heterozygotes; GSTM1 −/+) or two (i.e., normal homozygotes; GSTM +/+) copies of GSTM1, quantitative real-time PCR was used. It was conducted using a TaqMan (5′-nuclease) assay system with signal from a GSTM1-specific probe that was normalized to the signal from a reference autosomal β-2-microglobulin gene (B2M). Primers and probes used to amplify the GSTM1 and B2M regions are presented in Table 1.
Table 1.
Primers and probes used in the study
| Gene | Sequencea |
|---|---|
| B2M | Forward primer: 5′-TTGTTTCACTGTCCTGAGGACTATTTAT-3′ |
| Reverse primer: 5′-ATGTTACTCTGTCAATGTTCTCCACAT-3' | |
| Probe: 5′-ROX-CTCTAACATGATAACCCTCAC-BHQ2-3′ | |
| GSTM1 | Forward primer: 5'-CTGAGCCCTGCTCGGTTTAG-3' |
| Reverse primer: 5'-ATGGGCATGGTGCTGGTT-3' | |
| Probe: 5'-FAM-CTGTCTGCGGAATC-BHQ1-3' |
PCR was performed in 25 μL of 1× PCR buffer, containing 2.5 mM MgCl2, 200 μM each of dNTP, 20 pM each of GSTM1 and B2M primers, 1.25 units of Hot-Rescue Taq DNA polymerase (Syntol, Moscow, Russia), 10 pM and 5 pM of GSTM1- and B2M-specific probes, respectively, and 10–20 ng of genomic DNA. Thermal cycling and fluorescence intensity measurement were conducted using a StepOnePlus Real-Time PCR System (Applied Biosystems, Waltham, MA, USA). The samples were initially incubated for 10 min at 95 °C and then cycled 35 times at 95 °C for 20 s, followed by 60 °C for 60 s. All samples were tested in pentaplicate.
To quantify the number of GSTM1 copies a comparative Ct method was used [11]. The ratio (R) of the GSTM1 to B2M gene dosage was calculated using the formula R = 2−ΔΔCt, where ΔΔCt = (Ctcontrol_B2M – Ctcontrol_GSTM1) – (Ctsample_B2M – Ctsample_GSTM1). We used a control sample known to be heterozygous for GSTM1. Based on the observed variability, R values higher than 1.4 were interpreted as an indication that the sample carried two functional variants (two copies) of GSTM1. Ratios between 0.7 and 1.3 were considered attributable to samples with heterozygous deletions (containing one GSTM1 copy).
Population SNP genotypes were obtained from our previous study, in which they were generated using Illumina Human CNV370-Duo and Human 660 W-Quad chips [12]. The set of SNPs was chosen by considering the chromosomal region in which the genes of the GSTM family were located.
Data on individual GSTM1 deletion genotypes and SNP genotypes in other populations (i.e., CEU) were obtained from the database of the 1000 Genomes Project [13].
To explore patterns of genetic variation across GSTM cluster, a haplotype analysis was performed. Two approaches were used. The first was based on an analysis of haplotypes in the haplotype blocks (haploblocks). The haplotype blocks were defined using a block definition based on the linkage disequilibrium (LD) measure D′ and its confidence interval. The corresponding pairwise LD statistics between SNPs and the frequencies of haplotypes were estimated using Haploview software (version 4.2) [14]. Comparisons of haplotype frequencies between groups of individuals were performed using GraphPad InStat (version 3.00, GraphPad Software, San Diego, CA, USA). P < 0.05 was considered significant.
The second approach consisted of exploring the patterns of haplotypes at which the GSTM1 null and non-null allele(s) segregated in populations. To obtain such structural haplotypes, the GSTM1 and SNP genotypes of populations were phased together using the Beagle software package (version 4, release 1399) using default parameters [15]. Visualization of the sets of phased GSTM1 and SNP alleles was conducted using a custom R script kindly provided by Robert E. Handsaker (Broad Institute of MIT and Harvard, Cambridge, MA, USA) [16].
Results
In previous studies based on both whole-genome polymorphism analysis and testing SNPs in GSTA and GSTM clusters, high similarity between three Russian populations from the Central European part of Russia was demonstrated [12, 17]. To create a more effective sample, particularly in the context of generally under-represented GSTM1 +/+ genotype carriers, three Russian populations were combined. In total, 128 individuals with a GSTM1 −/− genotype, 121 individuals with a GSTM1 −/+ genotype, and 36 individuals with a GSTM1 +/+ genotype were detected in the sample. Genotype frequencies did not differ from those predicted by the Hardy–Weinberg rule (P = 0.45).
Fourteen SNPs determined as located in the region of the GSTM cluster were found among SNPs from the Illumina chip analyses and used in the current study. Figure 1 shows the LD between the SNPs, and the haploblocks inferred in the combined Russian sample. In total, four haploblocks were inferred in the chromosome region. We started our analysis from haplotypes of haploblock 2, comprising SNPs rs673151 and rs929166. These SNPs were the nearest to the region of the GSTM1 deletion. The frequencies of haplotypes CT and TT were maximal in the group of individuals with a GSTM1 +/+ genotype, while the third haplotype, CG, was the most frequent among the carriers of the GSTM1 −/− genotype. All three haplotypes had intermediate frequencies in the group of individuals with the GSTM1 −/+ genotype (Table 2). The same picture of haplotype distribution (i.e., intermediate values of haplotype frequencies in the group of individuals with the GSTM1 −/+ genotype) was observed for haploblocks 1, 3, and 4 (Table 2). As a consequence, pairwise comparisons of haplotypes showed the greatest differences between individuals with the GSTM1 −/− genotype and individuals with the GSTM1 +/+ genotype. Haplotype distributions in all four blocks were significantly different between these two groups (Table 3). A slightly lower number of significant differences were found between the groups with GSTM1 −/− and GSTM −/+ genotypes, and only one when the group with the GSTM1 +/+ genotype was compared with the group with the GSTM −/+ genotype. The same analysis was also carried out in the CEU population. The found correlations in haplotype distributions were similar to those observed in the Russian sample.
Fig. 1.
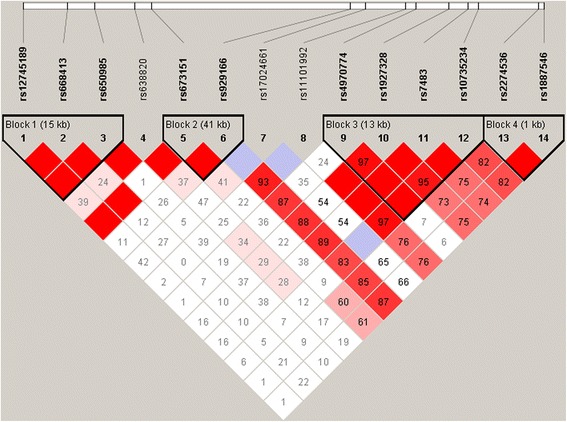
Linkage disequilibrium (LD) between SNPs in the region of the GSTM cluster in a combined Russian sample. A standard Haploview D′/LOD color scheme is used to demonstrate LD, with bright red for strong LD (LOD ≥ 2, D′ = 1), white for no LD (LOD < 2, D′ < 1), shades of pink/red for intermediate LD (LOD ≥ 2, D′ < 1), and blue for statistically ambiguous LD (LOD < 2, D′ = 1) [14]. Numbers in cells represent D′ values between pairs of SNPs (empty cells indicate that D′ = 1 between the corresponding SNPs). Black triangles indicate inferred haplotype blocks
Table 2.
Haplotype frequencies in groups of individuals subdivided according to GSTM1 deletion polymorphism
| Number of haploblock | Haplotypes | Individuals with GSTM1 −/− genotype (2 N = 256) | Individuals with GSTM1 −/+ genotype (2 N = 242) | Individuals with GSTM1 +/+ genotype (2 N = 72) |
|---|---|---|---|---|
| 1 | TCT | 0.390 | 0.512 | 0.597 |
| CAT | 0.508 | 0.380 | 0.250 | |
| CCT | 0.028 | 0.069 | 0.125 | |
| CCC | 0.075 | 0.039 | 0.028 | |
| 2 | CT | 0.583 | 0.705 | 0.736 |
| CG | 0.406 | 0.237 | 0.139 | |
| TT | 0.012 | 0.055 | 0.125 | |
| 3 | AACG | 0.390 | 0.392 | 0.319 |
| CGTA | 0.374 | 0.254 | 0.167 | |
| AACA | 0.154 | 0.188 | 0.306 | |
| CACA | 0.079 | 0.158 | 0.208 | |
| CGCA | 0.004 | 0 | 0 | |
| AGCG | 0 | 0.008 | 0 | |
| 4 | GT | 0.217 | 0.343 | 0.542 |
| AG | 0.776 | 0.657 | 0.458 | |
| GG | 0.008 | 0 | 0 |
Table 3.
Statistics (P-values) of intergroup comparisons of haplotype frequenciesa
| Number of haploblock | Individuals with GSTM1 −/− genotype vs. Individuals with GSTM1 +/+ genotype | Individuals with GSTM1 −/− genotype vs. Individuals with GSTM1 −/+ genotype | Individuals with GSTM1 +/+ genotype vs. Individuals with GSTM1 −/+ genotype |
|---|---|---|---|
| 1 | ‹0.0001 | 0.0060 | 0.9512 |
| 2 | ‹0.0001 | ‹0.0001 | 0.0391 |
| 3 | ‹0.0001 | 0.0038 | 0.0700 |
| 4 | ‹0.0001 | 0.0022 | 0.0024 |
asignificant differences are in bold
The output of processing phased Russian genotypes generally supported the results of the haploblock-based analysis. The output was expressed as an unequal occurrence of GSTM1 alleles on different haplotypes (Fig. 2). Understanding the paucity of the SNP set tested, we attempted to increase the resolution by imputing additional genotypes. However, the additional genotypes did not markedly influence the pattern of haplotypes inferred, although the total number of SNPs increased to 49. This might be because none of the new SNPs were closer to the GSTM1 deletion than the two aforementioned SNPs, rs673151 and rs929166, which could be the result of earlier and crucial branching. The relevance of earlier branching was supported by the data from the processing of a set of 356 phased SNPs with a MAF ≥ 0.01 in the CEU sample, in which some SNPs were located at some hundreds of base pairs from the deletion (Fig. 3). Furthermore, in the resulting plot, nonrandom occurrence of particular GSTM1 alleles at different haplotypes was more evident (i.e., the lower left part of the plot was occupied exclusively by haplotypes with a GSTM1 null allele) (Fig. 3).
Fig. 2.
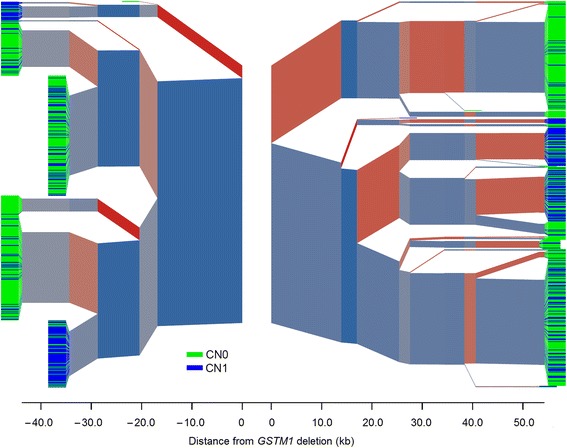
GSTM1 copy number distribution and haplotype structure of the GSTM cluster genomic region in a combined Russian sample. SNP haplotypes in the region of the GSTM cluster are shown. The gap in the center of the plot indicates the edges of the GSTM1 deletion. The branch points represent SNPs at which flanking haplotypes diverge because of mutation or recombination. The thickness of the branches corresponds to the frequency of haplotypes. Blue to red color intensity indicates the allele frequency of individual SNPs used to define the haplotypes, where the major allele is bluer, and the minor allele is redder. The color of the “leaves” at the ends of the branches indicates the GSTM1 state of the chromosomes: green, no copies of GSTM1 (deleted gene; CN0); blue, one functional copy of GSTM1 (CN1)
Fig. 3.
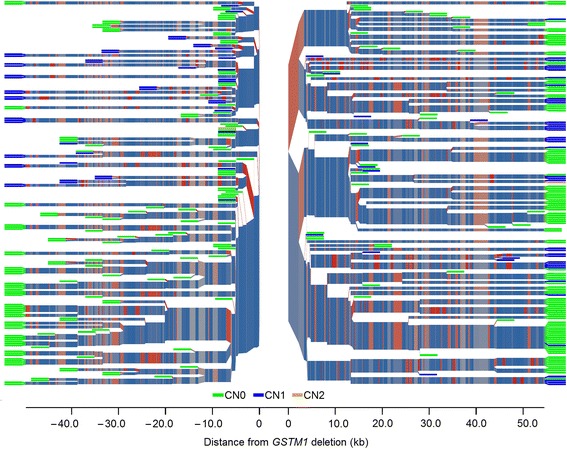
GSTM1 copy number distribution and haplotype structure of the GSTM cluster genomic region in the CEU population. The plot has been generated based on the data on the phase state of the alleles of GSTM1 and 356 SNPs (MAF ≥ 0.01). One individual had two copies of GSTM1 on one of his chromosomes (CN2), which is indicated as an orange “leaf” in the right-hand part of the plot. Other designations are the same as in Fig. 2
Discussion
The GSTM1 homozygous deletion is very common in human populations, varying between 16 and 40 % in Africans (sub-Saharan Africa), 42 and 55 % in Europeans, and 42 and 65 % in East Asian populations [3]. The frequency of the deletion suggests that GSTM1 has not been subjected to strong environmental selection pressure during evolution, and thus it might have resulted from an ancestral deletion that was widely spread across populations because of a founder effect.
An alternative explanation can be proposed in the context of the molecular structure of the GSTM1 region. Xu and coauthors [4] found that GSTM1 in GSTM1 +/+ individuals was flanked by two highly homologous 4.2 kb regions. By contrast, individuals with a completely deleted GSTM1 had only one segment that was identical to the regions in GSTM1 +/+ individuals. Such a structural organization of the region (i.e., existence of two highly homologous repeats) allows for the possibility of nonallelic homologous recombination in the chromosomal area, resulting in gene deletion [18], particularly the GSTM1 deletion, which in principle might occur independently on multiple occasions. The results of our intergroup haplotype comparisons support the hypothesis that the deletion might occur at different haplotypes because there were no differences in haplotype spectra between the groups. At the same time, substantial differences in the frequencies of the haplotypes were found. The greatest differences were observed between groups of individuals with homozygous GSTM1 −/− and +/+ genotypes, which differed in haplotype frequency in all four haploblocks inferred. We hypothesized that the differences may show the potential influence of genomic context on the occurrence of the deletion. The assumption was supported by the results of exploring structural haplotypes in the region of the GSTM1 deletion, where unequal occurrence of GSTM1 − and + alleles at different SNP haplotypes was demonstrated. In what way could the context affect the deletion frequency (appearance)? It is unlikely that individual SNPs could affect recombination if they were not in a recombination hotspot [19]. However, taking into account the data on the possibility of functional compensation of GSTM1 −/− by GSTM2 [8], one can suggest that the correlations found may reflect the existence of functional associations between GSTM1 and allelic variants of other genes of the GSTM cluster. Such associations can explain both why GSTM1 has not been subjected to strong environmental selection and the high frequency of GSTM1 deletion. The associations seem to be also relevant to the conflicting results reported in studies that correlated GSTM1 with risk of cancer and other diseases [6, 20–24] in which the absence of GSTM1-related enzymatic activity could be masked by catalytic activities of other GSTMs and resulted in no or reduced impact of GSTM1 deletion on disease risk. Finally, taken in the context of the results of associative studies, our findings highlight the necessity of parallel examination of allelic status of functionally and structurally related members of the gene family.
Conclusions
In summary, we have reported the results of exploring the haplotype structure in the GSTM cluster region in relation to GSTM1 deletion polymorphism. By using both haploblock-based and extended phased haplotypes, substantial differences in haplotype distribution were observed when they were correlated with the GSTM1 genotypes and alleles. The results suggest the potential role of genetic context in GSTM1 deletion frequency (appearance), and in the determination of deletion-related effects.
Acknowledgements
The authors are grateful to Robert E. Handsaker for a custom R script and for productive discussion of the results obtained. This study was supported by grants from the Program “Molecular and Cell Biology” of the Russian Academy of Sciences, the Federal Support of Leading Scientific Schools and the Russian Foundation for Basic Research.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AVK, PAS and SAL conceived and designed the study, INF and TVT carried out genotyping experiments, AVK performed the statistical analysis and drafted the manuscript, AMA participated in the statistical analysis. SAL helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Andrey V. Khrunin, Email: khrunin@img.ras.ru
Irina N. Filippova, Email: ireha_08@mail.ru
Aydar M. Aliev, Email: aidaraliev@gmail.com
Tat’yana V. Tupitsina, Email: tanya_tupitsina@mail.ru
Petr A. Slominsky, Email: slomin@img.ras.ru
Svetlana A. Limborska, Email: limbor@img.ras.ru
References
- 1.Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 2.Oakley A. Glutathione transferases: a structural perspective. Drug Metab Rev. 2011;43:138–51. doi: 10.3109/03602532.2011.558093. [DOI] [PubMed] [Google Scholar]
- 3.Saitou M, Ishida T. Distributions of the GSTM1 and GSTT1 null genotypes worldwide are characterized by latitudinal clines. Asian Pac J Cancer Prev. 2015;16:355–61. doi: 10.7314/APJCP.2015.16.1.355. [DOI] [PubMed] [Google Scholar]
- 4.Xu S, Wang Y, Roe B, Pearson WR. Characterization of the human class Mu glutathione S-transferase gene cluster and the GSTM1 deletion. J Biol Chem. 1998;273:3517–27. doi: 10.1074/jbc.273.6.3517. [DOI] [PubMed] [Google Scholar]
- 5.Sprenger R, Schlagenhaufer R, Kerb R, Bruhn C, Brockmöller J, Roots I, Brinkmann U. Characterization of the glutathione S-transferase GSTT1 deletion: discrimination of all genotypes by polymerase chain reaction indicates a trimodular genotype-phenotype correlation. Pharmacogenetics. 2000;10:557–65. [DOI] [PubMed]
- 6.Bolt HM, Thier R. Relevance of the deletion polymorphisms of the glutathione S-transferases GSTT1 and GSTM1 in pharmacology and toxicology. Curr Drug Metab. 2006;7:613–28. doi: 10.2174/138920006778017786. [DOI] [PubMed] [Google Scholar]
- 7.Zhao Y, Marotta M, Eichler EE, Eng C, Tanaka H. Linkage disequilibrium between two high-frequency deletion polymorphisms: implications for association studies involving the glutathione-S transferase (GST) genes. PLoS Genet. 2009;5:e1000472. doi: 10.1371/journal.pgen.1000472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhattacharjee P, Paul S, Banerjee M, Patra D, Banerjee P, Ghoshal N, Bandyopadhyay A, Giri AK. Functional compensation of glutathione S-transferase M1 (GSTM1) null by another GST superfamily member, GSTM2. Sci Rep. 2013;3:2704. [DOI] [PMC free article] [PubMed]
- 9.Milligan BG. Total DNA, isolation. In: Hoelzel AR, editor. Molecular genetic analysis of populations. London: Oxford University Press; 1998. pp. 29–60. [Google Scholar]
- 10.Khrunin AV, Khokhrin DV, Limborskaia SA. Glutathione-S-transferase gene polymorphism in Russian populations of European Russia. Genetika. 2008;44:1429–34. [PubMed] [Google Scholar]
- 11.Shadrina MI, Semenova EV, Slominsky PA, Bagyeva GH, Illarioshkin SN, Ivanova-Smolenskaia II, Limborska SA. Effective quantitative real-time polymerase chain reaction analysis of the parkin gene (PARK2) exon 1-12 dosage. BMC Med Genet. 2007;8:6. [DOI] [PMC free article] [PubMed]
- 12.Khrunin AV, Khokhrin DV, Filippova IN, Esko T, Nelis M, Bebyakova NA, et al. A genome-wide analysis of populations from European Russia reveals a new pole of genetic diversity in northern Europe. PLoS One. 2013;8:e58552. [DOI] [PMC free article] [PubMed]
- 13.1000 Genomes: A Deep Catalog of Human Genetic Variation http://www.1000genomes.org. Accessed 25 June 2015.
- 14.Barrett J, Fry B, Maller J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 15.Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing data inference for whole genome association studies using localized haplotype clustering. Am J Hum Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Handsaker RE, Van Doren V, Berman JR, Genovese G, Kashin S, Boettger LM, McCarroll SA. Large multiallelic copy number variations in humans. Nat Genet. 2015;47:296–303. [DOI] [PMC free article] [PubMed]
- 17.Filippova IN, Khrunin AV, Limborska SA. Analysis of DNA variations in GSTA and GSTM gene clusters based on the results of genome-wide data from three Russian populations taken as an example. BMC Genet. 2012;13:89. doi: 10.1186/1471-2156-13-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu W, Zhang F, Lupski JR. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1:4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Myers S, Spencer CC, Auton A, Bottolo L, Freeman C, Donnelly P, McVean G. The distribution and causes of meiotic recombination in the human genome. Biochem Soc Trans. 2006;34:526–30. [DOI] [PubMed]
- 20.Pietro G, Magno LA, Rios-Santos F. Glutathione S-transferases: an overview in cancer research. Expert Opin Drug Metab Toxicol. 2010;6:153–70. doi: 10.1517/17425250903427980. [DOI] [PubMed] [Google Scholar]
- 21.Carlsten C, Sagoo GS, Frodsham AJ, Burke W, Higgins JP. Glutathione S-transferase M1 (GSTM1) polymorphisms and lung cancer: a literature-based systematic HuGE review and meta-analysis. Am J Epidemiol. 2008;167:759–74. doi: 10.1093/aje/kwm383. [DOI] [PubMed] [Google Scholar]
- 22.Economopoulos KP, Sergentanis TN. GSTM1, GSTT1, GSTP1, GSTA1 and colorectal cancer risk: a comprehensive meta-analysis. Eur J Cancer. 2010;46:1617–31. doi: 10.1016/j.ejca.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 23.Minelli C, Granell R, Newson R, Rose-Zerilli MJ, Torrent M, Ring SM, et al. Glutathione-S-transferase genes and asthma phenotypes: a Human Genome Epidemiology (HuGE) systematic review and meta-analysis including unpublished data. Int J Epidemiol. 2010;39:539–62. doi: 10.1093/ije/dyp337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu H, Ma HF, Chen YK. Association between GSTM1 polymorphisms and lung cancer: an updated meta-analysis. Genet Mol Res. 2015;14:1385–92. doi: 10.4238/2015.February.13.17. [DOI] [PubMed] [Google Scholar]
- 25.Covault J, Abreu C, Kranzler H, Oncken C. Quantitative real-time PCR for gene dosage determinations in microdeletion genotypes. Biotechniques. 2003;35:594–597. doi: 10.2144/03353dd02. [DOI] [PubMed] [Google Scholar]
- 26.Nørskov MS, Frikke-Schmidt R, Loft S, Tybjaerg-Hansen A. High-throughput genotyping of copy number variation in glutathione S-transferases M1 and T1 using real-time PCR in 20,687 individuals. Clin Biochem. 2009;42:201–209. doi: 10.1016/j.clinbiochem.2008.10.020. [DOI] [PubMed] [Google Scholar]