

Abstract
Very little is known about lipid function during wound healing, and much less during impaired healing. Such understanding will help identify what roles lipid signaling plays in the development of impaired/chronic wounds. We took a lipidomics approach to study the alterations in lipid profile in the LIGHT−/− mouse model of impaired healing which has characteristics that resemble those of impaired/chronic wounds in humans, including high levels of oxidative stress, excess inflammation, increased extracellular matrix degradation and blood vessels with fibrin cuffs. The latter suggests excess coagulation and potentially increased platelet aggregation. We show here that in these impaired wounds there is an imbalance in the arachidonic acid (AA) derived eicosonoids that mediate or modulate inflammatory reactions and platelet aggregation. In the LIGHT−/− impaired wounds there is a significant increase in enzymatically derived breakdown products of AA. We found that early after injury there was a significant increase in the eicosanoids 11-, 12-, and 15-hydroxyeicosa-tetranoic acid, and the proinflammatory leukotrienes (LTD4 and LTE) and prostaglandins (PGE2 and PGF2α). Some of these eicosanoids also promote platelet aggregation. This led us to examine the levels of other eicosanoids known to be involved in the latter process. We found that thromboxane (TXA2/B2), and prostacyclins 6kPGF1α are elevated shortly after wounding and in some cases during healing. To determine whether they have an impact in platelet aggregation and hemostasis, we tested LIGHT−/− mouse wounds for these two parameters and found that, indeed, platelet aggregation and hemostasis are enhanced in these mice when compared with the control C57BL/6 mice. Understanding lipid signaling in impaired wounds can potentially lead to development of new therapeutics or in using existing nonsteroidal anti-inflammatory agents to help correct the course of healing.
Acute wounds that do not follow a concerted and overlapping set of repair processes, become impaired and may enter a state of chronicity.1 Deciphering the etiology of impaired and chronic wounds has remained one of the biggest challenges in addressing healing outcomes of problematic wounds. Hallmarks of impaired and chronic wounds include increased oxidative stress, deregulated levels of growth factors, imbalance in cytokines and chemokines, sustained inflammation, leaky blood vessels, and uncontrolled function of proteases.2 Although therapies have been developed to correct the course of impaired healing and have been successful in varying degrees in animal models of impaired healing, their results in human clinical trials have been limited due to the multifactorial imbalance in the wound microenvironment.3
Lipids are an integral part of skin structure and function, and have been shown to be involved in the pathogenesis of several diseases including psoriasis, atopic dermatitis, and disorders arising from exposure to ultraviolet radiation (UVR).4 The study of individual lipids and their regulation relevant to acute wound healing has been studied for the past four decades.5–10 However, evaluation of lipids using lipidomics approaches has only recently been established. Lipidomics is a branch of metabolomics dedicated to the systematic identification and quantification of an extensive assortment of lipids in cells, organs and extracellular fluids to correlate them to disease states.11,12 The use of liquid chromatography-mass spectrometry (LC-MS) allows us to measure various lipids quantitatively at the same time. Not only does lipidomics hold promise to further our knowledge of the underlying mechanisms to chronic wound development and progression, it also opens new avenues of risk assessment and evaluation of targeted therapeutics in a personalized and timely manner.13
Arachidonic acid (AA), the precursor for a large number of signaling lipids, is a polyunsaturated fatty acid present in phospholipids of cell membranes. It can be released from the membrane by activation of receptors that turn on phospholipase A2 which, in turn, hydrolyzes the sn-2 ester bond in the phospholipid, releasing AA as a free fatty acid.14 The release of AA initiates a cascade of events resulting in the generation of numerous lipid mediators that trigger inflammation, increased vascular permeability and platelet activation.15,16 These mediators can be generated either via nonenzymatic or enzymatic pathways. The nonenzymatic pathway involves free radicals generated when there is excess oxidative stress that causes the production of isoprostanes.17,18 Enzymatic breakdown of AA can occur either via the cytochrome P450s (P450s), lipoxygenase and/or the cyclooxygenase pathways that give rise to inflammatory mediators.19,20 P450s and LOX pathway can metabolize AA to give rise to hydroxyeicosatetranoic acids (HETEs) that are involved in increasing inflammation and play roles in platelet activation. Enzymatic breakdown of AA by lipoxygenases gives rise to leukotrienes that increase inflammation and vascular permeability. Finally, cyclooxygenases act on AA to give prostanoids such as the thromboxanes, prostacyclins, and prostaglandins that are crucial for skin physiology and hemostasis.21
Recently, we have shown that a mouse model in which the TNFSF14/LIGHT gene was deleted have impaired wound healing with characteristics of nonhealing ulcers similar to those observed in humans.22 We showed that the wounds of LIGHT−/− mice have high levels of proinflammatory chemokines and cytokines and consequently prolonged inflammation. The wounds have defective basement membrane, impaired dermal/epidermal interactions, leaky blood vessels and problems in granulation tissue formation. More recently we showed that very early after wounding, the LIGHT−/− wounds display elevated levels of oxidative stress due to increases in reactive oxygen species, reactive nitrogen species, and reduced levels of antioxidant enzymes.23 The increase in stress leads to lipid peroxidation, DNA damage and protein nitration suggesting broad-spectrum damage to the healing tissue. Furthermore, the increase in redox stress in the wound microenvironment and the presence of previously isolated biofilm-forming bacteria could lead to the development of chronic wounds.23 These studies suggest that increased redox stress shortly after injury coupled with presence of biofilm-forming bacteria can lead to wound chronicity.
In these studies, we have used the LIGHT−/− mice as a novel model to study impaired wound healing.22 Because we observed excessive inflammation and fibrin “cuffs” in the blood vessels of these mice and because it is known that many of the signaling lipids derived from AA metabolism can be involved in both processes, we hypothesized that AA metabolites may be major contributors to the underlying mechanism of impaired healing in these mice. We used a lipidomics approach in a single lipid platform, to investigate their levels during the course of impaired healing. Here we show that AA-derived metabolites, both via the enzymatic and nonenzymatic pathways, are significantly elevated during wound healing, but in particular in the first 48 hrs after injury. This is the first study deciphering the elevated levels of lipid metabolites in impaired healing. This information may help in furthering our knowledge on how to develop better therapeutic interventions, and perhaps manage possible side effects when treating impaired wounds.
METHODS
Dermal excisional wound model
Animals were housed at the University of California, Riverside (UCR) vivarium. All experimental protocols were approved by the UCR Institutional Animal Care and Use Committee. Experiments were performed using 4–5 month old mice. The procedure used was performed as previously described.22 Briefly, dorsal hair was removed using clippers and nair on control C57BL/6 and LIGHT−/− mice. Twenty four hours later, excisional wounds were performed on the dorsum of mice using a 7 mm biopsy punch (Acuderm, Inc, Fort Lauderdale, FL). Animals were anesthetized and wound tissues collected at various time points following injury using a 10-mm diameter biopsy punch. The tissues were flash frozen and stored in glass vials at −80°C until further analysis. The average tissue weight was 75 mg ± 25 mg with a n = 90 tissues.
Processing of tissues for lipid assay
1-mL of LCMS grade ethanol containing 0.05% BHT and 10 ng of each internal standard was added to frozen wound tissues. Samples were mixed using a bath sonicator incubated overnight at −20 °C for lipid extraction. The insoluble fraction was precipitated by centrifuging at 12,000×g for 20 minutes and the supernatant was transferred into a new glass tube.
Liquid chromatography mass spectrometry
For eicosanoid quantitation via UPLC ESI-MS/MS, the lipid extracts were then dried under vacuum and reconstituted in of LCMS grade 50:50 EtOH:H2O (100 μL). A 14 minutes reversed-phase LC method utilizing a Kinetex C18 column (100 × 2.1 mm, 1.7 μm) and a Shimadzu UPLC was used to separate the eicosanoids at a flow rate of 500 μL/minutes at 50°C. The column was first equilibrated with 100% Solvent A [acetonitrile:water:formic acid (20:80:0.02, v/v/v)] for 2 minutes and then 10 μL of sample was injected. 100% Solvent A was used for the first 2 minutes of elution. Solvent B [acetonitrile:isopropanol (20:80, v/v)] was increased in a linear gradient to 25% Solvent B to 3 minutes, to 30% by 6 minutes, to 55% by 6.1 minutes, to 70% by 10 minutes, and to 100% by 10.1 minutes. 100% Solvent B was held until 13 minutes, then decreased to 0% by 13.1 minutes and held at 0% until 14 minutes. The eluting eicosanoids were analyzed using a hybrid triple quadrupole linear ion trap mass analyzer (AB 6500 QTRAP) via multiple-reaction monitoring in negative-ion mode. Eicosanoids were monitored using species specific precursor → product MRM pairs. The mass spectrometer parameters were: curtain gas: 30; CAD: High; ion spray voltage: −3500 V; temperature: 300°C; Gas 1: 40; Gas 2: 60; declustering potential, collision energy, and cell exit potential were optimized per transition. Precursor-MRM transitions were obtained as previously described.12,24–28
Immunolabeling
The procedures used were previously reported by us.22 Briefly, frozen tissue sections were immunolabeled with antibodies to COX-2 (Abcam, Cambridge, United Kingdom), F4/80 (Abcam) and FITC or Alexa Fluor 594 conjugated secondary antibodies (Life Technologies, Grand Island, NY) and mounted with Vectashield. Images were viewed and recorded using Nikon Microphot-FXA fluorescence microscope with Nikon DS-Fi1 digital camera and Nikon NIS-Elements software (Nikon Instruments Inc., Melville, NY); for sections labeled with multiple dyes, images were merged in ImageJ software (National Institutes of Health, Bethesda, MD).
Elastase assay
Elastase activity of the wound samples at various time points after injury was done using a commercially available kit EnzChek Elastase Assay Kit (Life Technologies). A fluorescence-labeled elastin substrate (DQ elastin from bovine neck filament; 4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionic acid), was used as a substrate to be digested by the elastase in the samples to yield fluorescent products determined at Ex/Em of 480 nm/530 nm. Elastase from pig pancreas was used to create standard curve. Measurements were run in 96-well microplates (black, flat bottomed, Nunc, Denmark). To 50 μL reaction buffer and 50 μL of DQ elastin substrate (100 μg/mL), 100 μL of the equilibrated sample solution was added and incubated for 2hrs while the fluorescence was measured every 15 minutes at 22°C, using the Victor plate reader.
Tail bleeding time
Hemostasis was measured using the tail transection technique, following previously established protocols.29,30 Briefly, 5 months old mice were separated in two groups: LIGHT−/− (n = 5) and C57BL/6 (n = 5) were anesthetized by an intraperitoneal injection of ketamine (80 mg/kg) and xylazine (16 mg/kg). Mice were placed on a 37 °C heating blanket (Harvard Apparatus Limited, Edenbridge, KY) before the tail was transected using a sterile scalpel to make a clean cut at a distance of 5 mm from the tip. After transection, the tail was immediately immersed in warmed saline (37 °C, constant temperature). The bleeding time was followed visually and determined as the time from the tail transection to the moment the blood flow stopped and did not resume within the next 60 seconds. It is expected that less than 300 μL of blood is lost even if the bleeding does not stop within 15 minutes. When bleeding did not stop within 15 minutes, pressure was applied to the tail to seal the wound, thus avoiding excessive loss of blood. A bleeding time beyond 15 minutes was considered as the cutoff time for the purpose of statistical analysis.
Platelet Aggregation
These studies were performed as described previously.30,31 Groups of five to six mice from each group were anesthetized by an intraperitoneal injection of ketamine (80 mg/kg) and xylazine (16 mg/kg) before their blood was collected from the heart in 3.8% sodium citrate (1 part citrate to 9 parts blood) and pooled. Platelet rich plasma (PRP) was then isolated by differential centrifugation at 170 g for 10 minutes and platelet counts were adjusted to 3 × 108 with Tyrode’s buffer prior to each experiment. After establishing a baseline light transmission for 1 minute, platelets were stimulated with agonists. Aggregation of platelets was monitored using a Lumi-Aggregometer (Havertown, PA) at 37 °C under constant stirring at 1,200 rpm. To calculate the % aggregation increase with respect to control C57BL/6 mice, we used to following equation:
% increase in aggregation for LIGHT = [(B−A)/A]*100, where A% is the maximum aggregation for C57BL/6 mice and B% is the maximum aggregation for LIGHT−/− mice.
Statistical analysis
We used Graphpad Instat Software and Sigmaplot Software. Analysis of variance was used to test significance of group differences between two or more groups. Bleeding time analysis for day 3 and 7 were performed using GraphPad PRISM statistical software (San Diego, CA) and presented as mean ± SEM. The analysis was performed by unpaired t test using Mann–Whitney test to accommodate non-Gaussian data distribution. Significance was accepted at p<0.05 (two-tailed p value), unless stated otherwise. Mean of the data set is shown as + in the whisker box plots with all the points shown.
RESULTS
To further understand the effects of the redox stress present in the wounds of the LIGHT−/− mouse model of impaired healing,22,23 we took a lipidomics approach to evaluate the nonenzymatic and enzymatic breakdown of AA (Figure 1). The nonenzymatic breakdown of AA gives rise to isoprostanes whereas the enzymatic breakdown of AA can be accomplished via three major routes. P450s lead to the generation of HETEs and epoxyeicosatrienoic acids (EETs). Lipoxygenases that result in production of leukotrienes can also produce HETEs. Cyclooxygenases lead to the generation of prostanoids which consist of three key groups, thromboxane, prostacyclin, and prostaglandin. We will first describe our findings for the different eicosanoids during normal and impaired healing using our standard panel of eicosanoids that include transitions for greater than 150 metabolites.25 Individual LC-MS results obtained for each of the analytes were determined by calculating the ratio of the analyte peak area to that of the internal standard. We will then show the results for those involved in platelet aggregation and hemostasis.
Figure 1.

Schematic illustration of breakdown of AA through the nonenzymatic and enzymatic pathways. AA breaks down via the nonenzymatic pathways in the presence of elevated levels of oxidative stress to give rise to 8- and 5-isoprostanes. The enzymatic pathway of AA metabolism can occur via three categories. Cytochrome P450 (P450s) modify AA to result in epoxyeicosatrienoic acids (EETs) and hydroxyeicosatetranoic acids (16–20 series of HETEs). The same metabolite (HETE) can also be achieved via the lipoxygenase pathways of AA modification (5, 12, and 15 HETE’s). The lipoxygenase metabolizes AA to the leukotrienes, in particular cystenyl leukotrienes, cys-LTD4 and cys-LTE4. The cyclooxygenase (COX) pathway is subdivided to give AA breakdown products thromboxanes A2 (TXA2) and stable metabolite TXB2, prostacyclin (PGI2) and stable metabolite 6kPGF1α, prostaglandin E2 (PGE2) and PGF2α.
Proinflammatory AA-derived signaling lipids
The majority of the pro-inflammatory lipids derived from AA metabolism come from enzymatic reactions. We know that the impaired wounds in LIGHT−/− mice contain excess inflammation.22 Therefore, we looked for increases in pro-inflammatory lipids because it is known that they play an important role in the chemotaxis of inflammatory cells, in particular neutrophils. These leukocytes produce broad-spectrum serine proteases which cause extensive degradation of extracellular matrix and hence can cause poor development of the granulation tissue and lead to impaired healing.
Monooxygenation of AA can occur by lipoxygenase enzymes resulting in the production of hydroxyeicosatetranoic acids (HETEs). Furthermore CYP enzymes can give rise to HETEs and EETs via the ω-hydroxylases and epoxygenases respectively.32 15-HETE was significantly elevated in the LIGHT−/− mouse wounds throughout the first 7 days postwounding (Figure 2A). Moreover, two substantial peaks were detected at days 1 and 7 postwounding in the LIGHT−/− mice when compared to the control C57BL/6 mice. The initial peak in the control (C57BL/6) mouse wounds was observed at day 2. The levels in the control wounds increased subsequently but were not significantly different than those observed at day 2 postwounding. We also show that the potent leukocyte chemoattractant 11-HETE, was already significantly elevated at day 1 postwounding and remained elevated beyond day 7 (Figure 2B). However, in the control (C57BL/6) wounds, the level of 11-HETE peaked during the first three days postwounding and then returned to baseline levels by day 7. Significant increases during the first two days and an overall amplified presence of 11-HETE in LIGHT−/−wound may explain the prolonged and sustained influx of inflammatory cells in the wounds of these mice. Furthermore, 12-HETE, an important mediator of neutrophil chemotaxis,33 was found to be significantly elevated at days 1 and 3 postwounding in LIGHT−/− wounds (Figure 2C). By contrast, the level of 12-HETE in control (C57BL/6) wounds remained relatively lower and we saw no significant difference between control and LIGHT−/− wounds by day 7 and 12 (Figure 2C).
Figure 2.

Cytochrome P450 derived AA metabolites are elevated in impaired healing. Lipoxygenase mediated conversion of AA-derived results in significant elevation of HETEs shortly after wounding of the LIGHT−/− mice. LIGHT−/− tissues collected at multiple time points after wounding were assayed for 15- HETE (A) which showed two peaks, one at day 1 and the other at day 7 postwounding and 11-HETE (B) which was overall elevated throughout the period of healing with respect to control C57BL/6 mice. 12-HETE (C) and 5,6 EET (D) were significantly elevated at days 1 and 3 postwounding. No other significant differences were noted at other time points. 11,12 EET (E) and 14,15 EET (F) were both significantly elevated at days 2 and 3 postwounding in the LIGHT−/− wounds. n = 4. All data are Mean ± SD. *p<0.05, **p<0.01, ***p<0.001.
The epoxygenase products of AA metabolism are a complicated group of eicosanoids with known roles in vascular tone, angiogenesis and inflammation.34–37 The epoxygenase attaches an oxygen atom to two of the carbons of a double bond on AA (5,6 or 11,12 or 14,15) and as the epoxide is formed the double bond reduces resulting in regioisomers (structural isomers) epoxygenase products. 5,6 EET levels in LIGHT−/− wounds was significantly elevated at days 1 and 3 postwounding when compared to control (C57BL/6) wounds (Figure 2D). Both 11,12 EET and 14,15 EET levels in the LIGHT−/− wounds had significant increase at days 2 and 3 when compared to the control (C57BL/6) wounds (Figure 2E,F). The level of 5,6 EET and 14,15 EET in the control wounds had a peak at day 2 and day 7 whereas 11,12 EET levels in control wounds were highest at day 2 postwounding and gradually decreased by day 12.
AA breakdown by lipoxygenases results in production of leukotrienes (LT) that are further metabolized by cysteinyl (cys) synthase to give rise to cys-LTD4. Inflammatory responses have been shown to be initiated in the presence of cystein leukotrienes.38 We show that the level of cys-LTD4 was significantly elevated during days 1 and 2 postwounding in the LIGHT−/− wounds whereas, no significant increases were observed in the control (C57BL/6) wounds (Figure 3A). Further cleaving of cys-LTD4 leads to cys-LTE4 production, which is highly elevated during the first three days of wounding in LIGHT−/− and remains higher than the control C57BL/6 wounds until day 7 (Figure 3B). The initial peak in cys-LTE4 was observed at day 1 in both LIGHT−/− and control (C57BL/6) mice. The increases in these lipids indicate an early and increased influx of inflammatory cells to the wound site.
Figure 3.

Enzymatically derived pro-inflammatory AA metabolites are elevated in impaired healing. (A) Lipoxygenase-mediated enzymatic breakdown product cystenyl leukotriene D4 (cys-LTD4) was significantly elevated during the first 2 days of healing in LIGHT−/− mice, suggesting a substantial influx of inflammatory cells. (B) Cystenyl leukotriene E4 (LTE4) is involved in increasing inflammation and was found to be elevated when compared to C57BL/6 mice throughout the course of healing. (C) Cyclooxygenase (COX)-mediated breakdown of AA gives rise to exacerbated levels of prostaglandin E2 (PGE2), which is involved in increased inflammation and neutrophil aggregation, was significantly elevated throughout the course of healing when compared to control C57BL/6 mice. (D) Prostaglandin F2α (PGF2α) levels had two significantly elevated peaks at days 1 and 3 postwounding. (E) Immunolabeling for COX-2, an enzyme responsible for conversion of AA to prostaglandins. (F) F4/80, a marker for macrophages, to illustrate the presence of inflammation. (G) Merger of (E) and (F) showing that macrophages produced COX-2. (H) COX-2 is shown to be expressed also by keratinocytes. The white line runs along the basement membrane. Propidium iodide staining identifies cell nuclei. (I) COX-2 is also expressed by endothelial cells lining the blood vessels. Scale bar. n = 4. All data are Mean ± SD. *p<0.05, **p<0.01, ***p<0.001. Scale bar 50 μm for (E)–(H) and 10 μm for (I).
Cyclooxygenase 2 (COX-2) gives rise to AA-derived prostaglandins (PGs). One of these PGs is PGE2 which has a variety of biological functions, and is one of the most abundant of the pro-inflammatory PGs.21 PGE2 was increased at days 1–7 postwounding in the LIGHT−/− mice compared to the control C57BL/6 wounds (Figure 3C). This prolonged presence of PGE2 most likely contributes to the presence of elevated inflammatory cells seen in the wounds of LIGHT−/− mice.22 Furthermore, the levels of PGE2 in the control (C57BL/6) wounds increased and peaked at day 2 postwounding, as opposed to the peak observed in LIGHT−/− wounds which occurred at day 1, supporting the early and prolonged increase in inflammation seen in LIGHT−/− mice wounds. Another pro-inflammatory PG is PGF2α which is synthesized from PGH2 via PGF synthase. PGF2α peaked at day 1 postwounding declining to baseline by 7 days (Figure 3D). This PG is a strong stimulator of neutrophil infiltration. The levels of PGF2α in the control (C57BL/6) wounds increased only during the first 2 days and returned to baseline levels by day 3 supporting the normal pattern of neutrophils chemotaxis during normal wound healing. Because we saw an increase in PGs and COX-2 expression that has been observed previously in inflammatory conditions,39 we performed double immunolabeling for COX-2 and for F4/80, an antigen present in macrophages. We show that some macrophages in the wound tissue expressed COX-2 suggesting them to be a source of PGs (Figure 3 E,F,G). Furthermore we found that the keratinocytes (Figure 3H) and endothelial cells of established blood vessels (Figure 3I) also produced COX-2.
Because we found so many pro-inflammatory lipids elevated after wounding and several of them increase chemotaxis of neutrophils, and we know that neutrophils secrete elastase, we investigated whether elastase activity was elevated in the LIGHT−/− mouse wounds during the course of healing. We show that the elastase activity is already elevated in the unwounded skin of the LIGHT−/− mice but after wounding it becomes further elevated reaching a peak at day 1 and then decreasing to normal levels until day 7 when it rises again and by day 12 postwounding it is just as high as at day 1 (Figure 4). These levels suggest a hostile proteolytic environment early after wounding which can put the healing process on an abnormal course and then again later when the wound tissue should begin to remodel. This course of events most likely will impact the development of the granulation tissue.
Figure 4.
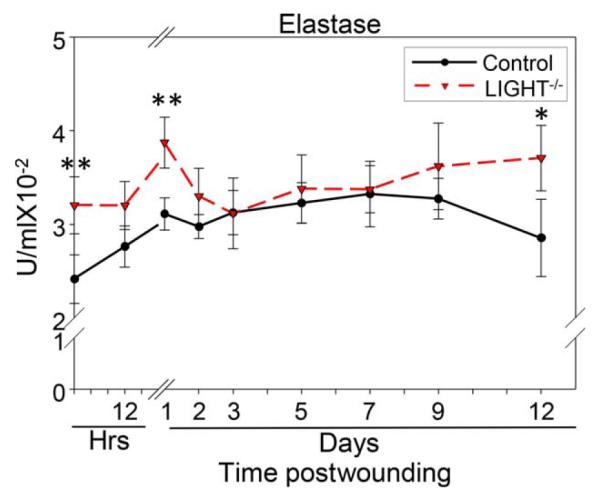
Elastase activity is elevated during the course of healing in impaired healing. Elastase activity was significantly elevated in the skin of LIGHT−/− mice when compared to C57BL/6 mice. This activity was exacerbated 1 day postwounding, potentially putting the wound on a course that leads to impaired healing. Moreover, the elastase activity was also significantly increased during the remodeling phase of healing contributing to impaired healing by interfering with the remodeling process. n = 4. All data are Mean ± SD. *p<0.05, **p<0.01, ***p<0.001.
AA-derived signaling lipids that are important in platelet aggregation and hemostasis
During our analysis of the pro-inflammatory lipids, we observed that many of them were also involved in activating or enhancing platelet aggregation. In particular, 8-isoprostane, PGE2, and 15-HETE are significantly involved in increasing platelet aggregation.40–42 This observation and the fact that the LIGHT−/− mouse wound tissues contain blood vessels with fibrin “cuffs” (essentially clots) led us to examine the presence of additional lipids involved in platelet function. Indeed, AA metabolites generated via cyclooxygenase 1 (COX-1) are mediators of platelet aggregation and vascular constriction. We found that thromboxane A2 (TXA2), one such metabolite that is rapidly transformed into its stable product TXB2, peaks at day 1 postwounding and is significantly increased at days 2 and 3 (Figure 5A). Furthermore, COX-2 is the leading source for generation of 6kPGF1α, the stable product of prostacyclin (PGI2) 43 We show that the levels of 6kPGF1α are significantly elevated after day 1, reach a peak at day 3 and then decline to normal levels by day 11 postwounding (Figure 5B). 6kPGF1α is a powerful inhibitor of platelet aggregation. The complementary elevation of TXB2 and 6kPGF1α suggests that upon elevation of platelet aggregation early after wounding by the various lipids described above, 6kPGF1α comes in to reestablish the equilibrium in platelet aggregation in the wounded tissue.
Figure 5.

Signaling lipids involved in platelet function are increased in impaired healing. (A) Cyclooxygenase (COX)-mediated AA metabolite thromboxane A2, measured by thromboxane B2 (TXB2) levels, are significant increased during the first 3 days of healing suggesting enhanced platelet activity and increases in platelet aggregation. (B) 6kPGF1α is stable metabolite of prostaglandin I2 (PGI2), was significantly increased from day three to day 7 postwounding as a response to the enhanced platelet aggregation. n = 4. All data are Mean ± SD. *p<0.05, **p<0.01, ***p<0.001.
Given these results, we performed a series of experiments to evaluate platelet aggregation and hemostasis in the LIGHT−/− mice. We show that platelet aggregation in response to the agonist ADP is significantly increased 3 days after wounding in the LIGHT−/− mice and that this enhancement is dose dependent when compared to the control C57BL/6 mice (Figure 6A; compare red with blue and green with black). To capture the long-term effects of elevated levels of these pro-aggregation lipids, we examined the intensity of aggregation at Day 7 postwounding. We show that at this time the LIGHT−/− mice still display enhanced level of platelet aggregation in a dose-dependent manner (Figure 6B). However, the intensity of aggregation is less pronounced than at day 3. Because we observed that platelet aggregation is enhanced in the LIGHT−/− mice, we examined bleeding time using tail bleeding and observed that indeed the bleeding time in wounded LIGHT−/− mice (days 3 and 7) is significantly reduced in comparison to that seen in the C57BL/6 mice (Figure 7A,B).
Figure 6.

Platelet aggregation is increased during impaired healing. Platelet aggregation was estimated using aggregometer and the agonist adenosine diphosphate (ADP). (A) Platelet aggregation 3 days postwounding was significantly enhanced in the LIGHT−/− mice and the aggregation occurred in an ADP dose-dependent manner. There was a 50% enhanced aggregation observed in LIGHT−/− mice when compared to control C57BL/6 mice for 0.25 μM ADP. (B) At day 7 postwounding there was a 40.7% enhanced aggregation observed in LIGHT−/− mice when compared to control C57BL/6 mice when 0.25 μM ADP was used. The enhanced aggregation seen in LIGHT−/− mice was reduced from day 3 to day 7. n = 5. There are no standard deviations because to measure % aggregation increase we needed to pool the blood of all 5 samples given that the amount of blood we obtain from each mouse is very small.
Figure 7.

Coagulation time is faster in impaired healing. Tail vein bleeding time in LIGHT−/− mice suggested faster clotting and reduced coagulation time when compared to C57BL/6 control C57BL/6 mice. (A) Bleeding time was significantly reduced in LIGHT−/− mice at 3 days postwounding. (B) Bleeding time is significantly faster in LIGHT−/− mice than in the control C56BL/6 mice. Although no significance difference was observed, the overall, bleeding time in LIGHT−/− wounds at day 7 increased from that seen in LIGHT−/− mice at day 3 postwounding. n = 5. All data are Mean ± SD. The data analysis was performed by unpaired t-test (Graphpad Instat software) using Mann–Whitney test to accommodate non-Gaussian data distribution. The graph was plotted as box and whisker plot with the scattergram and the mean is represented as +.
DISCUSSION
Understanding the impairment in overlapping factors during the course of healing can aid in finding potential targeted therapeutics.2 Although lipid biology has been widely studied in artherosclerosis, cancer, platelet biology, oxidative stress and neurological settings,17,38,44,45 use of modern lipidomics approaches to study AA-derived signaling lipids in cutaneous injury has only caught momentum in recent years.46–49 Here we present for the first time data on the lipid metabolite profile in a mouse model of impaired healing, the LIGHT−/− mouse. We show that the pro-inflammatory lipid metabolites in LIGHT−/− wounds, as a result of enzymatic breakdown of AA, are increased very early during the course of healing when compared to the control and could potentially be involved in derailing proper healing. Also, we show that as a result of chemo-taxis of inflammatory cells, especially neutrophils, there was an increased activity of elastase that can degrade the extracellular proteins and disrupt the formation and remodeling of the healing tissue. We also show significant increases in lipid metabolites that were involved in increased platelet aggregation; LIGHT−/− mice have significantly enhanced platelet aggregation and a shortened bleeding time.
The HETEs are products of AA metabolism that are derived from P450s via the ω-oxidation of the carbon chain and/or from the lipoxygenase pathways.50,51 Particu larly present in platelets and neutrophils, 11-HETE has been shown to be chemotactic for human neutrophils and hence its prolonged presence can lead to persistent inflammation.52 In addition to the increases in 11-HETE, we also show the elevated presence of 15-HETE in the LIGHT−/− wounds. 15-HETE is known to increase platelet aggregation and thrombin generation,41 two major players in clot formation. Furthermore, levels of 15-HETE have been shown to increase with increases in monocytes and macrophages.53 Also 12-HETE is a potent initiator of neutrophil chemotaxis, and we found it to be elevated during the early days postwounding in LIGHT−/− wounds.33 We have previously reported increases in macrophages and neutrophils in the LIGHT−/− wounds,22 and show here that not only macrophages but also keratinocytes and endothelial cells of developed blood vessels were sources of COX-2 production. Although the majority of HETE production is attributed to P450 and LOX catalysis, nonenzymatic processes might contribute to the elevated levels we see.
EETs are the epoxygenase metabolites of AA metabolism under the P450 enzymatic pathway and are primarily produced by endothelial cells.36,54,55 EETs have been shown to have an anti-inflammatory function.34 Although it was reported that 11,12 EET inhibits the expression of surface adhesion molecules (CAM) including vascular cell adhesion molecule 1 (VCAM-1), E-selectin and intercellular adhesion molecule 1 (ICAM-1), 14,15 EET increase the adherence of monocytes to endothelial cells.34 The significant increases in both 11,12 EET and 14,15 EET at both days 2 and 3 postwounding in LIGHT−/− wounds suggests differences in inflammatory responses in the impaired wounds. 5,6 EETs have been shown to increase endothelial cell migration and capillary tube formation56 which could explain why we see more blood vessel sprouts in the LIGHT−/− mice than in the control
Application of cys-LTD4 in human skin has been shown to cause infiltration of neutrophils.57 This LT can be secreted by macrophages, which are increased in LIGHT−/− wounds,16 and prompt β-catenin translocation to the nucleus and activation of the target genes c-myc and cyclin D1.58 Significant increase of expression of the latter two genes has been shown to be present in the nonhealing wound edge of chronic ulcers and to inhibit epithelialization.59 Moreover, although the levels of cys-LTD4 peak in the first 2 days postwounding and then decline to normal levels for the remaining of the healing period, the levels of LTE4, the stable metabolite of LTD4, remain elevated during the first 7 days postwounding. Furthermore, LTE4 can act as an agonist for mast cells that are prominently present in chronic ulcers and cause chronic inflammation and fibrosis.60,61 Both LTD4 and LTE4 can also elicit changes in microvasculature and reduce blood pressure. LTE4 does so by stimulating endothelial cell contraction which opens gaps in the endothelium, leading to vascular leakage.62 The cumulative data of the leukotrienes suggest that they may be responsible for the increases in permeability and leaky vasculature seen in the LIGHT−/− wounds.57,62
Elastase is a broad spectrum serine protease that degrades most extracellular and membrane proteins in addition to degrading elastin, and therefore, plays an important role in the damage to the connective tissue during inflammatory processes.63 Elastase has been shown to degrade fibronectin in chronic wounds in humans.64 Furthermore, the elevated levels of elastase that increase with age in human chronic wounds have been shown to break down peptide growth factors used to heal chronic wounds.65,66 The presence of higher than normal levels of elastase later in the healing process may also be responsible for the impaired remodeling observed in the LIGHT−/− mice wounds.
Polyunsaturated fatty acid derived prostanoids are crucial for skin physiology and hemostasis. The two classes of compounds, PGs and TXA2 are collectively termed prostanoids and are produced as a result of the cyclooxygenase enzymatic pathway.21 PGE2, the most abundant of the prostaglandins, is produced by a diverse number of cells, including epidermal keratinocytes, dermal fibroblasts, and macrophages (in particular M1 macrophages).67,68 Increased levels of PGE2 in the LIGHT−/− wounds has strong implications in generating a pro-inflammatory wound microenvironment with increased neutrophil infiltration, increased microvascular permeability and platelet aggregation.21 In addition, increased levels of prostaglandin PGF2α also increased neutrophil infiltration.4 TXA2, a strong direct activator of platelet function leads to irreversible aggregation18,44 and has a very short half-life and hence converts to TXB2. The significantly high levels of TXA2 in the wounds of LIGHT−/− mice indicate complications in platelet behavior and hence improper healing outcomes. It is to note that along with TXA2, many of the previously discussed inflammatory lipids, including 8 isoprostane, 15-HETE, and PGE2, all increase platelet activation and aggregation.
Aggregation of platelets was significantly enhanced in LIGHT−/− than C57BL/6 mice. This was observed regardless of the dose used of the agonist ADP both at days 3 and 7. Enhanced aggregation was more prominent in mice at 3 days postwounding when compared to 7 days post-wounding. This could be attributed to the fact that PGI2, measured via the secondary stable metabolite 6kPGF1α, inhibits platelet aggregation.69 This PG becomes elevated when the levels of the other lipids that induce platelet aggregation decrease; in fact, its levels remain significantly elevated even at 7 days postwounding. This observation may also derive from the finding that levels of isoprostanes are more elevated at day 3 compared to day 7 as previously reported,23 and the fact that they are known to enhance platelet function (e.g., in response to the agonist ADP).40 Platelet activation is required for aggregation that is critical for clot formation during an episode of bleeding that may follow injury. Increases in lipids causing platelet aggregation reduce the time of bleeding due to a faster hemostasis response. The significant reduction in bleeding time in the LIGHT−/− mice, especially at day 3 postwounding, confirms the hyperactive platelet behavior.
In conclusion, the studies presented here in the LIGHT−/− mouse model of impaired healing are the first to use lipidomics approaches to elucidate the lipid profile and behavior during the course of impaired healing. In particular, platelet activating and pro-inflammatory lipids were elevated early after wounding resulting in increased platelet activation/aggregation, reduced time for bleeding and increased inflammation in the LIGHT−/− mice. Our previous work on the impaired characteristics of LIGHT−/− wounds and its similarities to human chronic wounds make this mouse model of importance for wound healing studies. Furthermore, the results presented here have major implication for our understanding of impaired healing because they put into prospective a class of molecules that has so far not been taken into consideration when trying to understand how impaired healing develops. We anticipate that further investigation of both proinflammatory and anti-inflammatory lipid markers during impaired/chronic wounds in mouse models of wound healing and extending these studies to human wounds will contribute to our understanding of the etiology of how these wounds develop which could lead to the use of existing therapeutics such as nonsteroidal anti-inflammatory agents or to the development of new therapies to treat problematic wounds.
Acknowledgments
The contents of this manuscript do not represent the views of the Department of Veterans Affairs or the United States Government.
Source of funding: This work was supported in part by research grants from the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development (Career Development Award CDA1 to DSW, VA Merit Award BX001792 to C.E.C. and a Research Career Scientist Award to C.E.C.); from the National Institutes of Health via HL072925 (C.E.C.); NH1C06-RR17393 (to Virginia Commonwealth University for renovation). Services and products in support of the research project were generated by the VCU Massey Cancer Center Lipidomics Shared Resource (Developing Core), supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059 as well as a shared resource grant (S10RR031535 to C.E.C.) from the National Institutes of Health.
Footnotes
Conflict of interest: None.
References
- 1.Lazarus GS, Cooper DM, Knighton DR, Margolis DJ, Pecoraro RE, Rodeheaver G, Robson MC. Definitions and guidelines for assessment of wounds and evaluation of healing. Wound Repair Regen. 1994;2:165–70. doi: 10.1046/j.1524-475X.1994.20305.x. [DOI] [PubMed] [Google Scholar]
- 2.Mustoe TA, O’Shaughnessy K, Kloeters O. Chronic wound pathogenesis and current treatment strategies: a unifying hypothesis. Plast Reconstr Surg. 2006;117:35S–41S. doi: 10.1097/01.prs.0000225431.63010.1b. [DOI] [PubMed] [Google Scholar]
- 3.Demidova-Rice T, Hamblin MR, Herman IM. Acute and impaired wound healing: pathophysiology and current methods for drug delivery, part 2: role of growth factors in normal and pathological wound. Adv Skin Wound Care. 2012;25:349–70. doi: 10.1097/01.ASW.0000418541.31366.a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicolaou A. Eicosanoids in skin inflammation. Prostaglandins Leukot Essent Fatty Acids. 2013;88:131–8. doi: 10.1016/j.plefa.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 5.Arturson G. Prostaglandins in human burn-wound secretion. Burns. 1977;3:112–8. [Google Scholar]
- 6.Turner SR. Polymorphonulcear leukocyte chemotaxis toward oxidized lipid components of cell membranes. J Exp Med. 1975;141:1437–41. doi: 10.1084/jem.141.6.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goetzl EJ, Woods JM, Gorman RR. Stimulation of human eosinophil and neutrophil polymorphonuclear leukocyte chemotaxis and random migration by 12-L-hydroxy-5,8,10,14-eicosatetraenoic acid. J Clin Invest. 1977;59:179–83. doi: 10.1172/JCI108617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moncada S, Gryglewski RJ, Bunting S, Vane JR. A lipid peroxide inhibits the enzyme in blood vessel microsomes that generates from prostaglandin endoperoxides the substance (prostaglandin X) which prevents platelet aggregation. Prostaglandins. 1976;12:715–37. doi: 10.1016/0090-6980(76)90048-4. [DOI] [PubMed] [Google Scholar]
- 9.Moncada S. Human arterial and venous tissues generate prostacyclin (prostaglandin X), a potent inhibitor of platelet aggregation. Lancet. 1977;309:18–21. doi: 10.1016/s0140-6736(77)91655-5. [DOI] [PubMed] [Google Scholar]
- 10.Saynor R, Verel D, Gillott T. The long-term effect of dietary supplementation with fish lipid concentrate on serum lipids, bleeding time, platelets and angina. Atherosclerosis. 1984;50:3–10. doi: 10.1016/0021-9150(84)90003-0. [DOI] [PubMed] [Google Scholar]
- 11.Wenk MR. The emerging field of lipidomics. Nat Rev Drug Discov. 2005;4:594–610. doi: 10.1038/nrd1776. [DOI] [PubMed] [Google Scholar]
- 12.Wijesinghe DS, Chalfant CE. Systems-level lipid analysis methodologies for qualitative and quantitative investigation of lipid signaling events during wound healing. Adv Wound Care. 2013;2:538–48. doi: 10.1089/wound.2012.0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meikle PJ, Wong G, Barlow CK, Kingwell BA. Lipidomics: potential role in risk prediction and therapeutic monitoring for diabetes and cardiovascular disease. Pharmacol Ther. 2014;143:12–23. doi: 10.1016/j.pharmthera.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 14.Balsinde J, Winstead MV, Dennis EA. Phospholipase A2 regulation of arachidonic acid mobilization. FEBS Lett. 2002;531:2–6. doi: 10.1016/s0014-5793(02)03413-0. [DOI] [PubMed] [Google Scholar]
- 15.Kuehl F, Egan R. Prostaglandins, arachidonic acid, and inflammation. Science. 1980;210:978–84. doi: 10.1126/science.6254151. [DOI] [PubMed] [Google Scholar]
- 16.Mruwat R, Cohen Y, Yedgar S. Phospholipase A 2 inhibition as potential therapy for inflammatory skin diseases. Immunotherapy. 2013;5:315–7. doi: 10.2217/imt.13.18. [DOI] [PubMed] [Google Scholar]
- 17.Montuschi P, Barnes PJ, Roberts LJ. Isoprostanes: markers and mediators of oxidative stress. FASEB J. 2004;18:1791–800. doi: 10.1096/fj.04-2330rev. [DOI] [PubMed] [Google Scholar]
- 18.Ting HJ, Khasawneh FT. Platelet function and isoprostane biology. Should isoprostanes be the newest member of the orphan-ligand family? J Biomed Sci. 2010;17:24. doi: 10.1186/1423-0127-17-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spite M, Clària J, Serhan CN. Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell Metab. 2014;19:21–36. doi: 10.1016/j.cmet.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwab JM, Serhan CN. Lipoxins and new lipid mediators in the resolution of inflammation. Curr Opin Pharmacol. 2006;6:414–20. doi: 10.1016/j.coph.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petreaca ML, Do D, Dhall S, McLelland D, Serafino A, Lyubovitsky J, Schiller N, Martins-Green MM. Deletion of a tumor necrosis superfamily gene in mice leads to impaired healing that mimics chronic wounds in humans. Wound Repair Regen. 2012;20:353–66. doi: 10.1111/j.1524-475X.2012.00785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dhall S, Do D, Garcia M, Wijesinghe DS, Brandon A, Kim J. A novel model of chronic wounds: importance of redox imbalance and biofilm-forming bacteria for establishment of chronicity. PLoS One. 2014;9:e109848. doi: 10.1371/journal.pone.0109848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mietla JA, Wijesinghe DS, Hoeferlin LA, Shultz MD, Natarajan R, Fowler AA., 3rd Characterization of eicosanoid synthesis in a genetic ablation model of ceramide kinase. J Lipid Res. 2013;54:1834–47. doi: 10.1194/jlr.M035683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simanshu DK, Kamlekar RK, Wijesinghe DS, Zou X, Zhai X, Mishra SK. Non-vesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoids. Nature. 2013;500:463–7. doi: 10.1038/nature12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Norton SK, Wijesinghe DS, Dellinger A, Sturgill J, Zhou Z, Barbour S. Epoxyeicosatrienoic acids are involved in the C70 fullerene derivative–induced control of allergic asthma. J Allergy Clin Immunol. 2012;130:761–9.e2. doi: 10.1016/j.jaci.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wijesinghe DS, Brentnall M, Mietla JA, Hoeferlin LA, Diegelmann RF, Boise LH. Ceramide kinase is required for a normal eicosanoid response and the subsequent orderly migration of fibroblasts. J Lipid Res. 2014;55:1298–309. doi: 10.1194/jlr.M048207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoeferlin LA, Huynh QK, Mietla JA, Sell SA, Tucker J, Chalfant CE. The lipid portion of activated platelet-rich plasma significantly contributes to its wound healing properties. Adv Wound Care. 2015;4:100–9. doi: 10.1089/wound.2014.0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paez Espinosa EV, Murad JP, Ting HJ, Khasawneh FT. Mouse transient receptor potential channel 6: role in hemostasis and thrombogenesis. Biochem Biophys Res Commun. 2012;417:853–6. doi: 10.1016/j.bbrc.2011.12.058. [DOI] [PubMed] [Google Scholar]
- 30.Lin OA, Karim ZA, Vemana HP, Espinosa EVP, Khasawneh FT. The antidepressant 5-HT2A receptor antagonists pizotifen and cyproheptadine inhibit serotonin-enhanced platelet function. PLoS One. 2014;9:e87026. doi: 10.1371/journal.pone.0087026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murad JP, Espinosa EVP, Ting HJ, Khasawneh FT. The C-terminal segment of the second extracellular loop of the thromboxane A2 receptor plays an important role in platelet aggregation. Biochem Pharmacol. 2012;83:88–96. doi: 10.1016/j.bcp.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 32.Panigrahy D, Greene ER, Pozzi A, Wang DW, Zeldin DC. EET signaling in cancer. Cancer Metastasis Rev. 2011;30:525–40. doi: 10.1007/s10555-011-9315-y. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Turner SR, Tainer JA, Lynn WS. Biogenesis of chemotactic molecules by the arachidonate lipoxygenase system of platelets. Nature. 1975;257:680–1. doi: 10.1038/257680a0. [DOI] [PubMed] [Google Scholar]
- 34.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K. Anti-inflammatory Properties of Derived Eicosanoids. Science. 1999;285:1276–9. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Medhora M, Daniels J, Mundey K, Fisslthaler B, Busse R, Jacobs ER. Epoxygenase-driven angiogenesis in human lung microvascular endothelial cells. Am J Physiol Heart Circ Physiol. 2003;284:H215–24. doi: 10.1152/ajpheart.01118.2001. [DOI] [PubMed] [Google Scholar]
- 36.Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. AJP Cell Physiol. 2006;292:C996–1012. doi: 10.1152/ajpcell.00402.2006. [DOI] [PubMed] [Google Scholar]
- 37.Greene ER, Huang S, Serhan CN, Panigrahy D. Regulation of inflammation in cancer by eicosanoids. Prostaglandins Other Lipid Mediat. 2011;96:27–36. doi: 10.1016/j.prostaglandins.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Samuelsson B, Dahlén SE, Lindgren JA, Rouzer CA, Serhan CN. Leukiotrienes and lipoxins: structures, biosynthesis, and biological effects. Science. 1987;237:1171–1176. doi: 10.1126/science.2820055. [DOI] [PubMed] [Google Scholar]
- 39.Hull MA, Booth JK, Tisbury A, Scott N, Bonifer C, Markham AF, Coletta PL. Cyclooxygenase 2 is up-regulated and localized to macrophages in the intestine of Min mice. Br J Cancer. 1999;79:1399–405. doi: 10.1038/sj.bjc.6690224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khasawneh FT, Huang JS, Mir F, Srinivasan S, Tiruppathi C, Le Breton GC. Characterization of isoprostane signaling: evidence for a unique coordination profile of 8-iso-PGF(2alpha) with the thromboxane A(2) receptor, and activation of a separate cAMP-dependent inhibitory pathway in human platelets. Biochem Pharmacol. 2008;75:2301–15. doi: 10.1016/j.bcp.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vijil C, Hermansson C, Jeppsson A, Bergström G, Hultén LM. Arachidonate 15-lipoxygenase enzyme products increase platelet aggregation and thrombin generation. PLoS One. 2014;9:e88546. doi: 10.1371/journal.pone.0088546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vezza R, Roberti R, Nenci GG, Gresele P. Prostaglandin E2 potentiates platelet aggregation by priming protein kinase C. Blood. 1993;82:2704–13. [PubMed] [Google Scholar]
- 43.Libby P, Warner S, Friedman G. Interleukin 1: a mitogen for human vascular smooth muscle cells that induces the release of growth-inhibitory prostanoids. J Clin Invest. 1988;81:487–98. doi: 10.1172/JCI113346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hamberg M, Svensson J, Samuelsson B. Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. Proc Natl Acad Sci USA. 1975;72:2994–8. doi: 10.1073/pnas.72.8.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stevens MK, Yaksh TL. Time course of release in vivo of PGE2, PGF2a, 6-Keto ¬ PGF1a, and TxB2 into the brain extracellular space after 15 min of complete global ischemia in the presence and absence of cyclooxygenase inhibition. J Cereb Blood Flow Metab. 1988;8:790–8. doi: 10.1038/jcbfm.1988.134. [DOI] [PubMed] [Google Scholar]
- 46.Wijesinghe DS, Brentnall M, Boise LH, Chalfant CE. A lipidomics approach towards understanding eicosanoid signaling events and the role of ceramide kinase during the wound healing process. FASEB J. 2011;25:551–15. [Google Scholar]
- 47.Lu Y, Tian H, Hong S. Novel 14,21-dihydroxy-docosahexaenoic acids: structures, formation pathways, and enhancement of wound healing. J Lipid Res. 2010;51:923–32. doi: 10.1194/jlr.M000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patil K, Bellner L, Cullaro G, Gotlinger KH, Dunn MW, Schwartzman ML. Heme oxygenase-1 induction attenuates corneal inflammation and accelerates wound healing after epithelial injury. Invest Ophthalmol Vis Sci. 2008;49:3379–86. doi: 10.1167/iovs.07-1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Norling LV, Spite M, Yang R, Flower RJ, Perretti M, Serhan CN. Cutting edge: humanized nano-proresolving medicines mimic inflammation-resolution and enhance wound healing. J Immunol. 2011;186:5543–7. doi: 10.4049/jimmunol.1003865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spector AA, Gordon JA, Moore SA. Hydroxyeicosatetraenoic acids (HETEs) Prog Lipid Res. 1988;27:271–323. doi: 10.1016/0163-7827(88)90009-4. [DOI] [PubMed] [Google Scholar]
- 51.Capdevila J, Marnett LJ, Chacos N, Prough RA, Estabrook RW. Cytochrome P-450-dependent oxygenation of arachidonic acid to hydroxyicosatetraenoic acids. Proc Natl Acad Sci USA. 1982;79:767–70. doi: 10.1073/pnas.79.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goetzl EJ, Brash AR, Tauber AI, Oates JA, Howard WCHT. Modulation of human neutrophil function by monohydroxy-eicosatetraenoic acids. Immunology. 1980;30:491–7. [PMC free article] [PubMed] [Google Scholar]
- 53.Conrad DJ, Kuhn H, Mulkins M, Highland E, Sigal E. Specific inflammatory cytokines regulate the expression of human monocyte 15-lipoxygenase. Proc Natl Acad Sci USA. 1992;89:217–21. doi: 10.1073/pnas.89.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosolowsky M, Campbell WB. Role of PGI2 and epoxyeicosatrienoic acids in relaxation of bovine coronary arteries to arachidonic acid. Am J Physiol. 1993;264:H327–35. doi: 10.1152/ajpheart.1993.264.2.H327. [DOI] [PubMed] [Google Scholar]
- 55.Panigrahy D, Kalish BT, Huang S, Bielenberg DR, Le HD, Yang J. Epoxyeicosanoids promote organ and tissue regeneration. Proc Natl Acad Sci USA. 2013;110:13528–33. doi: 10.1073/pnas.1311565110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pozzi A, Macias-Perez I, Abair T, Wei S, Su Y, Zent R. Characterization of 5,6- and 8,9-Epoxyeicosatrienoic Acids (5,6- and 8,9-EET) as Potent in Vivo Angiogenic Lipids. J Biol Chem. 2005;280:27138–46. doi: 10.1074/jbc.M501730200. [DOI] [PubMed] [Google Scholar]
- 57.Soter NA, Lewis RA, Corey EJ, Austen KF. Local effects of synthetic leukotrienes (LTC4,LTD4, and LTB4) in human skin. J Invest Dermatol. 1983;80:115–9. doi: 10.1111/1523-1747.ep12531738. [DOI] [PubMed] [Google Scholar]
- 58.Salim T, Sand-Dejmek J, Sjölander A. The inflammatory mediator leukotriene D4 induces subcellular β-catenin translocation and migration of colon cancer cells. Exp Cell Res. 2014;321:255–66. doi: 10.1016/j.yexcr.2013.10.021. [DOI] [PubMed] [Google Scholar]
- 59.Stojadinovic O, Brem H, Vouthounis C, Lee B, Fallon J, Stallcup M. Molecular pathogenesis of chronic wounds: the role of beta-catenin and c-myc in the inhibition of epithelialization and wound healing. Am J Pathol. 2005;167:59–69. doi: 10.1016/s0002-9440(10)62953-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paruchuri S, Tashimo H, Feng C, Maekawa A, Xing W, Jiang Y. Leukotriene E4-induced pulmonary inflammation is mediated by the P2Y12 receptor. J Exp Med. 2009;206:2543–55. doi: 10.1084/jem.20091240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abd-El-Aleem SA, Morgan C, Mccollum CN, Ireland GW. Spatial distribution of mast cells in chronic venous leg ulcers. Eur J Histochem. 2005;49:265–72. [PubMed] [Google Scholar]
- 62.Joris I, Majno G, Corey EJ, Lewis RA. The mechanism of vascular leakage induced by leukotriene E4. Endothelial contraction. Am J Pathol. 1987;126:19–24. [PMC free article] [PubMed] [Google Scholar]
- 63.Söder B. Neutrophil elastase activity, levels of prostaglandin E 2, and matrix metalloproteinase-8 in refractory periodontitis sites in smokers and non-smokers. Acta Odontol Scand. 1999;57:77–82. doi: 10.1080/000163599428940. [DOI] [PubMed] [Google Scholar]
- 64.Grinnell F, Zhu M. Fibronectin degradation in chronic wounds depends on the relative levels of elastase, 1-proteinase inhibitor, and 2-macroglobulin. J Invest Dermatol. 1996;106:335–41. doi: 10.1111/1523-1747.ep12342990. [DOI] [PubMed] [Google Scholar]
- 65.Chen SM, Ward SI, Olutoye OO, Diegelmann RF, Kelman Cohen I. Ability of chronic wound fluids to degrade peptide growth factors is associated with increased levels of elastase activity and diminished levels of proteinase inhibitors. Wound Repair Regen. 1997;5:23–32. doi: 10.1046/j.1524-475X.1997.50108.x. [DOI] [PubMed] [Google Scholar]
- 66.Herrick S, Ashcroft G, Ireland G, Horan M, McCollum C, Ferguson M. Up-regulation of elastase in acute wounds of healthy aged humans and chronic venous leg ulcers are associated with matrix degradation. Lab Invest. 1997;77:281–8. [PubMed] [Google Scholar]
- 67.Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends Immunol. 2002;23:144–50. doi: 10.1016/s1471-4906(01)02154-8. [DOI] [PubMed] [Google Scholar]
- 68.Norris PC, Dennis EA. A lipidomic perspective on inflammatory macrophage eicosanoid signaling. Adv Biol Regul. 2014;54:99–110. doi: 10.1016/j.jbior.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Needleman P, Wyche A, Raz A. Platelet and blood vessel arachidonate metabolism and interactions. J Clin Invest. 1979;63:345–9. doi: 10.1172/JCI109309. [DOI] [PMC free article] [PubMed] [Google Scholar]