

Abstract
Background:
Imaging mass spectrometry (IMS) generates molecular images directly from tissue sections to provide better diagnostic insights and expand the capabilities of clinical anatomic pathology. Although IMS technology has matured over recent years, the link between microscopy imaging currently used by pathologists and MS-based molecular imaging has not been established.
Methods:
We adapted the Vanderbilt University Tissue Core workflow for IMS into a web-based system that facilitates remote collaboration. The platform was designed to perform within acceptable web response times for viewing, annotating, and processing high resolution microscopy images.
Results:
We describe a microscopy-driven approach to tissue analysis by IMS.
Conclusion:
The Pathology Interface for Mass Spectrometry is designed to provide clinical access to IMS technology and deliver enhanced diagnostic value.
Keywords: Diagnostics, imaging mass spectrometry, molecular imaging
INTRODUCTION
Matrix-assisted laser desorption ionization (MALDI) imaging mass spectrometry (IMS) is a label-free molecular imaging technology that can be used to study a wide array of molecules (e.g., proteins, peptides, lipids, metabolites, and drugs) in their native tissue environment.[1,2] This technology is uniquely suited to the analysis of solid tissues because it provides targeted in situ molecular analysis while maintaining tissue morphological information upon which current anatomical pathology standards are constructed. Histology-directed IMS (Histo-IMS) is one modality of IMS that focuses on characterizing analyte distributions in discrete tissue locations, such as tumor versus nontumor regions or cell-type specific areas within a tissue section.[3,4] In contrast to whole specimen imaging, implementation of Histo-IMS improves the specificity and efficiency of the analysis making it possible to measure 100-1,000s of specimens per day as would be required for clinical diagnostic applications.[5]
The molecular specificity and sensitivity of modern MS provide unique capabilities to accurately measure disease markers that have clinical application. In recent years, liquid chromatography ([LC]-MS) has become a routine technology in the clinical laboratory[6,7] and is now responsible for returning millions of tests annually in the US alone.[8] Notwithstanding these enormous strides in the development of clinical MS, few examples of the application of these technologies to the diagnosis of disease in solid tissues have become standard clinical practice. This translational gap is the result of several factors including (1) increased difficulty and complexity of preparing solid tissues for mass spectrometric analysis and (2) the lack of accessibility to the technology for those individuals with training in pathology and access to specimens. Further, the image manipulation process required to go from marked serial histology sections to pixel coordinates for image acquisition requires considerable time and expertise[9] and is a limiting factor for high-throughput analysis. Current approaches to the design and control of MS-based experiments are insufficient to overcome these problems. Thus, the opportunity exists to develop a user interface for MS suitable for the clinical environment to deliver quality, clinically actionable results to the physician.
RESULTS AND DISCUSSION
We have developed a novel workflow automation web-based software tool – the pathology interface for MS (PIMS) – that overcomes these challenges and facilitates collaboration between anatomical pathologists and analytical scientists through seamless integration of microscopy, sample preparation, and MS analysis [Figure 1]. Importantly, the interface allows biologists and pathologists who are untrained in IMS technology to control the acquisition of MS data. Image annotation performed by the pathologist drives histology-directed sample preparation and IMS analysis,[10,11,12] making this technology easy to use and ultimately accessible to physicians worldwide.
Figure 1.
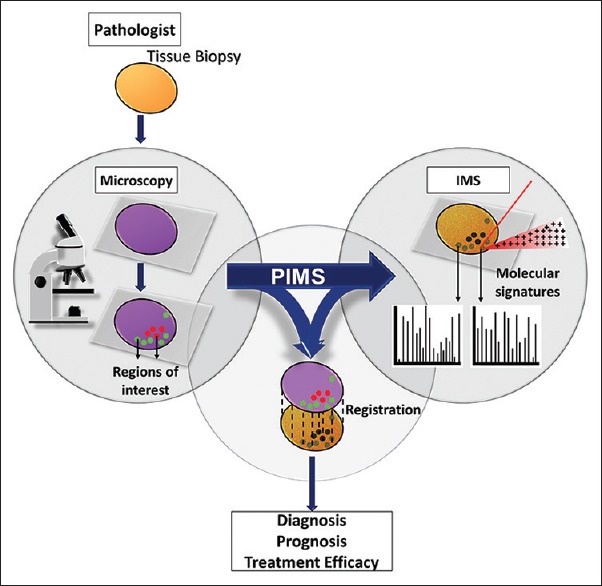
Pathology interface for mass spectrometry transforms the current IMS approach into one practical for the clinical setting, integrating microscopy, and IMS analyses to deliver molecularly specific, clinically valuable results
The core of the software is an image server that permits remote viewing and annotation of high-resolution micrographs by pathologists and biologists anywhere in the world. Other software features include automation tools to permit control of instruments based on annotations and a project management tool to facilitate the transfer of relevant sample information. PIMS incorporates a histological examination of the tissue, sample preparation and analysis, and reporting while maintaining the important link between the histology and the molecular measurements that can further guide diagnosis. PIMS facilitates rapid collaboration, reduces experimental error, and increases productivity, saving time and money for the laboratory.
In a normal pathology workflow, tissue samples are sent to the laboratory for analysis. Thin serial sections (approximately 5–10 μm) are prepared for analysis by sectioning on a microtome. One of the serial sections is stained using normal procedures for histological analysis and subsequently imaged using a high-resolution slide scanner, typically at ×20–40 magnification. One of the issues encountered is that high-resolution optical micrographs suitable for interpretation by pathologists are often larger than 30 GB without data compression. Workflows requiring the transfer of these images to a remote collaborator and return of the image after annotation are slow and inefficient, making this process impractical for clinical work that requires rapid testing.
In contrast, the PIMS interface does not necessitate the transfer of a compressed image to capture the annotations needed to execute the required experiment. After tissue sections are imaged, users upload images to the cloud from which they are automatically imported into the PIMS database, eliminating the need to reduce image size and resolution for E-mail transfer of images. Collaborators can open the PIMS interface through any web browser (Chrome, Firefox, Internet Explorer, Safari, etc.,) and therefore, it can be used on PC or Mac desktops, laptops, or tablets with no additional installs required. Images can be composed – viewed, cropped, rotated, and annotated – using the Image Markup Viewer [Figure 2a-c]. The image server supports layered tiling to optimize performance in viewing large, high-resolution (gigapixel) images so that users can efficiently view and annotate tissue areas. Opening images, this size can take hours on a common laptop/desktop setup, but PIMS accomplishes this task in seconds.
Figure 2.
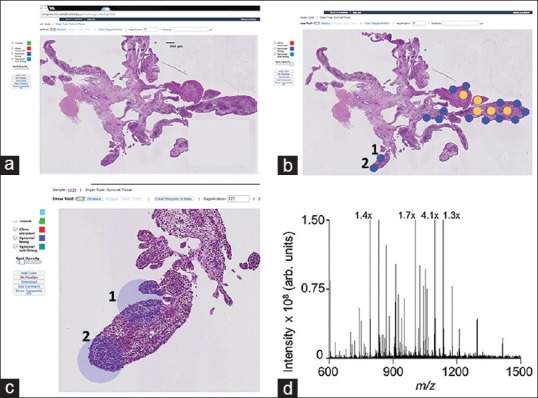
Screenshots from the pathology interface for mass spectrometry interface highlight the software features and workflow. (a) A scanned H and E stained tissue section can be viewed through the image markup screen and examined remotely by a pathologist. (b) The pathologist can then annotate the tissue via a user-defined color palette to distinguish regions of interest, for example, tissue classes (e.g., normal versus disease) or cell types. Annotations can be made as discrete spots (as shown) or regions. (c) Pathology interface for mass spectrometry can zoom to regions of interest. Scale bars and the magnification level are automatically rescaled according to the zoom level. (d) A mass spectrometry spectrum acquired from the spot labeled “1” in panels b and c
Once the coordinates for each annotated region of interest are recorded, the annotated image is spatially registered with an optical image of the serial unstained tissue section. Currently, registration of the annotated image to the optical image of an unstained tissue section is performed manually. Pixel coordinates of the annotated regions are transferred to the coordinate system of the spotting instrument (e.g., Labcyte Portrait 630) for matrix application to the unstained tissue section and subsequent MS data acquisition [Figure 2d]. There are no internal, software automated controls to verify the accuracy of the overlaid annotations or the position of the resulting matrix spots. However, the relative alignment between the matrix spots and the annotations are checked using overlaid optical images by both the analyst and pathologist to ensure accuracy and importantly that the cell population underlying the matrix spot is homogeneous. If the sample preparation is not accurate, the specimen is prepared again for analysis. Development of the registration feature is ongoing, and automated registration of images is being developed for future versions of PIMS.
Within the Image Markup Viewer, many custom features have been specifically designed for the IMS workflow such as a custom color palette to distinguish the classification of annotated regions and cell types, a measurement tool to calculate the distance between sample features, a polygon tool to designate regions of interest, and a text annotation tool for comments. These features are highlighted in a screenshot of the Image Markup Viewer [Figure 2a-c]. The classification of specific features (e.g., cell type and disease state) into a specific color group has been designed to prevent errors in annotation and classification for projects with multiple experiments. For example, if a specific feature is designated by a certain color in the first experiment that color can only represent that feature in subsequent related experiments. In addition, the software guides the proximity of two annotated areas based on achievable spatial resolutions for the project. Annotations are saved as a separate data layer to allow for easy editing, and custom annotated images are generated when a user chooses to save a snapshot of the viewport in the Image Markup Viewer. Once the analysis has been completed, the annotations can be color coded to represent the classification of the cells of interest in accordance with the MS results.
PIMS also streamlines data management throughout the entire project. For example, in the Vanderbilt University Tissue Core, project information is included such as grant and billing information, project PI, lead project scientist, affiliated scientists, and resource information. Notifications of the project status can be sent through PIMS, thereby expediting the sample preparation process. For example, the project scientist can notify the clinical collaborator that samples were received and enter the planned schedule for sample preparation and analysis into PIMS; the collaborator will be notified and can view the schedule. The collaborator is also notified when samples are ready for viewing and annotation. As soon as the images have been annotated, the project scientist is notified. Furthermore, information managed through PIMS is secured using a single sign-on protocol Central Authentication Server that supports multiple authentication methods and contains five access levels: System administrator, core manager, core user, user, and billing.
PIMS is built on a distributed processing architecture to handle concurrent requests for composing annotated images while maintaining acceptable response times. Supplementary Figure 1 (871.2KB, tif) and the accompanying text illustrate the processing architecture and provide detailed descriptions of the technology stack and development environment. Development and improvement of PIMS are ongoing. To provide the latest version, source code will be shared with interested collaborators by request to the corresponding author. In its current implementation, no patient data is stored in the system. A planned further development will involve security measures that meet HIPAA security standards and integration with existing digital pathology platforms so that the molecular classification provided by MS can be integrated into a single clinical report with the histopathological results.
Architecture for pathology interface for mass spectrometry software. Pathology interface for mass spectrometry is built on a distributed processing architecture to handle concurrent requests for composing annotated images while maintaining performance and throughput. The system incorporates open-source technologies and is modularized to facilitate future development and optimization
PIMS is currently the central touchstone for collaborators to direct analytical efforts within our laboratory. Currently, there are thirty projects managed through PIMS, and feedback from these collaborators permits continual refinement of the software to ensure a relevant and user-friendly product. Examples of published work that used PIMS include the analysis of racial disparities in Wilms Tumor by IMS[10,11] and tissue profiling and diagnosis of melanocytic skin tumors by IMS.[12] In the latter case, IMS was able to detect spatial, diagnostically useful differences in vimentin and actin expression in spitz nevi and spitzoid malignant melanomas, but neither conventional immunohistochemistry nor automated quantitative analysis of tissue sections could distinguish these differences.[13] These results highlight the power and utility of IMS for diagnostic medicine.
CONCLUSIONS
This report describes a first generation software system for the control of histology-directed MS that was designed and built specifically with pathologists’ needs in mind. This control paradigm places the tissue and the visualization of the cell morphology at the heart of the experiment. This approach to the design and execution of the experiment allows for biologists and pathologists, whose expertise is not necessarily this instrumentation, to guide the collection of molecular data from cells of interest in tissues with minimal training. Likewise, collaboration among groups of chemists and pathologists is made easier even across great distances. While the PIMS software described above was implemented for MALDI-IMS, this software platform could be used to guide similar experiments using a number of surface sampling/ionization technologies in MS, such as desorption electrospray ionization. Further, the PIMS technology could be adapted for the remote operation of laser-capture microscopy to facilitate other molecular analysis techniques including genomics, transcript analysis, and LC-based MS. As highly specialized analytical platforms continue to transition to diagnostic applications, software solutions that facilitate access to these technologies will be necessary to connect patients and physicians to important and potentially life-saving results.
Financial Support and Sponsorship
This work was funded through NIH, NIGMS Grant number P41 GM103391.
Conflicts of Interest
There are no conflicts of interest.
Footnotes
Available FREE in open access from: http://www.jpathinformatics.org/text.asp?2016/7/1/13/179903
REFERENCES
- 1.Caprioli RM, Farmer TB, Gile J. Molecular imaging of biological samples: Localization of peptides and proteins using MALDI-TOF MS. Anal Chem. 1997;69:4751–60. doi: 10.1021/ac970888i. [DOI] [PubMed] [Google Scholar]
- 2.Norris JL, Caprioli RM. Analysis of tissue specimens by matrix-assisted laser desorption/ionization imaging mass spectrometry in biological and clinical research. Chem Rev. 2013;113:2309–42. doi: 10.1021/cr3004295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cornett DS, Reyzer ML, Chaurand P, Caprioli RM. MALDI imaging mass spectrometry: Molecular snapshots of biochemical systems. Nat Methods. 2007;4:828–33. doi: 10.1038/nmeth1094. [DOI] [PubMed] [Google Scholar]
- 4.Taverna D, Boraldi F, De Santis G, Caprioli RM, Quaglino D. Histology-directed and imaging mass spectrometry: An emerging technology in ectopic calcification. Bone. 2015;74:83–94. doi: 10.1016/j.bone.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cornett DS, Mobley JA, Dias EC, Andersson M, Arteaga CL, Sanders ME, et al. A novel histology-directed strategy for MALDI-MS tissue profiling that improves throughput and cellular specificity in human breast cancer. Mol Cell Proteomics. 2006;5:1975–83. doi: 10.1074/mcp.M600119-MCP200. [DOI] [PubMed] [Google Scholar]
- 6.van den Ouweland JM, Kema IP. The role of liquid chromatography-tandem mass spectrometry in the clinical laboratory. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;883-884:18–32. doi: 10.1016/j.jchromb.2011.11.044. [DOI] [PubMed] [Google Scholar]
- 7.Vogeser M, Seger C. A decade of HPLC-MS/MS in the routine clinical laboratory – Goals for further developments. Clin Biochem. 2008;41:649–62. doi: 10.1016/j.clinbiochem.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 8.Grebe SK, Singh RJ. LC-MS/MS in the clinical laboratory – Where to from here? Clin Biochem Rev. 2011;32:5–31. [PMC free article] [PubMed] [Google Scholar]
- 9.Samsi SS, Krishnamurthy AK, Groseclose M, Caprioli RM, Lozanski G, Gurcan MN. Minneapolis, MN: IEEE; 2009. Imaging Mass Spectrometry Analysis for Follicular Lymphoma Grading. Proceedings of the 31st Annual International Conference of the IEEE Engineering in Medicine and Biology Society, 2009 September 2-6. [DOI] [PubMed] [Google Scholar]
- 10.Axt J, Murphy AJ, Seeley EH, Martin CA, Taylor C, Pierce J, et al. Race disparities in Wilms tumor incidence and biology. J Surg Res. 2011;170:112–9. doi: 10.1016/j.jss.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Libes JM, Seeley EH, Li M, Axt JR, Pierce J, Correa H, et al. Race disparities in peptide profiles of North American and Kenyan Wilms tumor specimens. J Am Coll Surg. 2014;218:707–20. doi: 10.1016/j.jamcollsurg.2013.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lazova R, Seeley EH, Keenan M, Gueorguieva R, Caprioli RM. Imaging mass spectrometry – A new and promising method to differentiate spitz nevi from spitzoid malignant melanomas. Am J Dermatopathol. 2012;34:82–90. doi: 10.1097/DAD.0b013e31823df1e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alomari AK, Klump V, Neumeister V, Ariyan S, Narayan D, Lazova R. Comparison of the expression of vimentin and actin in spitz nevi and spitzoid malignant melanomas. Am J Dermatopathol. 2015;37:46–51. doi: 10.1097/DAD.0000000000000147. [DOI] [PubMed] [Google Scholar]