

Abstract
Background:
Autophagy has been found to be involved in animal and cell models of atherosclerosis, but to date, it lacks general observation in human atherosclerotic plaques. Here, we investigated autophagy in smooth muscle cells (SMCs), endothelial cells (ECs), and macrophages in human atherosclerotic plaques via transmission electron microscopy (TEM), western blotting, and immunohistochemistry analysis.
Methods:
The histopathologic morphology of these plaques was observed via hematoxylin and eosin staining. The ultrastructural morphology of the SMCs, ECs, and macrophages in these plaques was observed via TEM. The localization of microtubule-associated protein 1 light chain 3 (MAP1-LC3), a relatively special maker of autophagy, in plaques was observed by double fluorescent immunochemistry and western blotting.
Results:
All of these human atherosclerotic plaques were considered advanced and unstable in histologically observation. By double fluorescent immunochemistry, the expression of LC3-II increased in the SMCs of the fibrous cap, the macrophages, and the microvascular ECs of the plaque shoulders. The protein level of LC3-II by western blotting significantly increased in plaques compared with normal controls. In addition, TEM observation of plaques revealed certain features of autophagy in SMCs, ECs, and macrophages including the formation of myelin figures, vacuolization, and the accumulation of inclusions in the cytosol. These results indicate that autophagy is activated in SMCs, ECs, and macrophages in human advanced atherosclerotic plaques.
Conclusions:
Our study is to demonstrate the existence of autophagy in human atherosclerotic plaques by different methods, which may contribute to the development of pharmacological approaches to stabilize vulnerable and rupture-prone lesions.
Keywords: Atherosclerosis, Autophagy, Endothelial Cells, Macrophages, Smooth Muscle Cells
INTRODUCTION
Atherosclerosis is a long-term inflammatory vascular disease in medium-large-sized arteries characterized by the formation of atherosclerotic plaques.[1] Many basic and clinical studies have focused on unstable atherosclerotic plaques because these unstable plaques are responsible for occlusion of the artery and more severe diseases such as acute coronary syndromes, stroke, or sudden death.[2,3] Therefore, the understanding the pathophysiology of unstable plaques has an important clinical significance for the prevention and treatment of atherosclerosis.
Autophagy plays an important role in the degradation of redundant or dysfunctional organelles and long-lived, mutant, or misfolded proteins in a lysosome-dependent manner, which involves in many diseases such as cancer, Alzheimer's disease, Parkinson's disease, Huntington's disease, and atherosclerosis.[4] There is evidence that autophagy is stimulated by oxidized lipids, inflammation, and metabolic stress conditions in advanced atherosclerotic plaques.[5] For example, it is well-known that macrophage infiltration contributes to the instability of atherosclerotic plaques while promoting macrophage autophagy contributed to the vulnerable atherosclerotic plaques stabilizing.[6] However, these studies were based on the cellular and animal model. The activation of autophagy in human atherosclerotic plaques was seldom studied due to the difficulty to collect human samples of atherosclerotic plaques. In this study, we observed the activation of autophagy in unstable atherosclerotic plaques from patients and analyzed the localization of autophagic cells in the plaque.
METHODS
Samples collection
We obtained atherosclerotic plaques from nine cases of carotid endarterectomy in patients of acute ischemic stroke with severe carotid artery stenosis in the Second Affiliated Hospital of Soochow University. These patients had the similar risk factors, such as old age, diabetes, hypertension, etc. All experiments were conducted in accordance with the Declaration of Helsinki, and the study protocol was approved by the Joint Ethical Committee of Soochow University. All subjects provided written informed consent before entering the study. In addition, three normal carotid artery samples from Neurosurgery Brain Tumor Research (Soochow University, China) were used as normal controls.
Samples were fixed by 4% paraformaldehyde and embedded in paraffin. 10 μm-thickness sections were sliced by a microtome (AO-820, American). Ten sections of each group were used for hematoxylin and eosin staining and screening under optical microscope.
Materials
Protease inhibitor cocktail was purchased from Sigma-Aldrich, St. Louis, MO, USA. Bovine serum albumin (BSA) protein assay kit was purchased from Pierce, Rockford, IL USA. In western blotting, antibodies against light chain 3 (LC3), β-actin were purchased from Abcam, Cambridge, UK. In immunofluorescence staining, mouse monoclonal microtubule-associated protein 1-LC3 (MAP1-LC3) antibody was purchased from Santa Cruz Biotechnology, Santa Cruz, CA, USA; Rabbit polyclonal alpha smooth muscle actin antibody was purchased from abcam, Cambridge, MA, USA; Rabbit polyclonal Von Willebrand factor (vWF) antibody and Rabbit polyclonal CD68 antibody were purchased from AbD Serotec, Oxford, UK. 40-6-diamidino-2-phenylindole (DAPI) was purchased from Serva, Heidelberg, Germany.
Transmission electron microscopy
We used transmission electron microscopy (TEM) to detect ultrastructure of plaques. Samples were processed by 2.5% glutaraldehyde and 1% OsO4 double fixation. Dehydration in a gradient ethanol was repeated three times and flat embedding was performed in Araldite. Ultrathin sections (50 nm) were placed on grids (200 mesh) and double-stained with uranyl acetate and lead citrate. The grids containing the sections were observed on the electron microscope (H-600, Hitachi, Japan).
Western blotting for light chain 3
Protein samples were analyzed by western blotting to explore LC3 expression in plaque cells and normal arteries. Samples were lysed in lysis buffer (20 mmol/L Tris-HCl, 137 mmol/L NaCl, 2 mmol/L EDTA, 2% Nonidet P-40, 2% Triton-X100) and protease inhibitor cocktail. Extract protein amounts were quantified using the BSA protein assay kit. 10 μl of lysates with sodium dodecyl sulfate (SDS) sample buffer, boiled for 5 min, were ran by SDS–polyacrylamide gel electrophoresis (15%), transferred to polyvinylidene difluoride membrane, blocked with 5% milk in phosphate buffered saline (PBS) for 1 h at room temperature. The blots were incubated with primary antibodies including anti-LC3 antibody (1:1000) and anti-β-actin antibody (1:5000) at 4°C overnight. After washing and incubating with horse radish peroxidase-conjugated secondary antibodies for 1 h, the blots were treated with enhanced chemiluminescence (ECL) detection reagents. Relative intensity of each band was measured by Image J software.
Immunofluorescence double staining
We used immunofluorescence double staining for detection of LC3 expression in plaques and controls. After de-waxing and antigen retrieval, endogenous peroxidase was inactivated with H2O2, and sections were blocked with 5% BSA. The sections were incubated with primary antibodies including mouse anti-human MAP1-LC3 antibody (1:50), rabbit anti-human alpha smooth muscle actin antibody (1:100), rabbit anti-human vWF antibody (1:100), rabbit anti-human CD68 antibody (1:100) at 4°C overnight. Sections washed and incubated with corresponding secondary antibodies, and then incubated with 10 mg/ml DAPI for 5 minutes at room temperature. After several rinses, the sections were cover-slipped with Vectashield mounting medium for fluorescence (Vector Lab) and analyzed with a confocal microscope (Leica, Germany). In negative controls, the primary antibody was replaced with 1% PBS.
Statistical analysis
Data were expressed as mean ± standard deviation for at least three sets of independent experiments. Statistical analysis was performed by Statistical Package for the Social Sciences (SPSS) Version 6.1 software (SPSS, Chicago, IL, USA) using Student's t-test for each experimental group treated versus control. The difference was considered significant when the P < 0.05.
RESULTS
General and histological observation of unstable atherosclerotic plaques
General observation
The surfaces of all plaque samples were humped. Yellow atherosclerotic lipid deposition was found in subintim of all samples. Some samples showed composite secondary lesions, such as intima erosion, ulcerarions, or hemorrhage [Figure 1a]. In contrast, normal control showed smooth and flat intima with significant wall tension.
Figure 1.

Macroscopic and histological observation of unstable atherosclerotic plaque. (a) Plaque specimen taken by carotid endarterectomy (CEA) shows composite secondary lesions, such as intima erosion, ulcerasion, or hemorrhage. (b-d) Hematoxylin and eosin (H and E) staining: (b) Gross histological observation of plaque showed lipid-laden foam cells in intima (arrows). (c) Smooth muscle cells (SMCs) of the media adjacent to intima lesion were disarranged and proliferated (arrows). (d) Histological observation of normal carotid artery showed smooth intima, regularly arranged SMCs and integrity of adventitia.
Histological observation
All cases had atherosclerotic lesions characterized with fibrous plaques and lost endothelial cells (ECs). Intima of plaque showed lipid-laden foam cells [Figure 1b]. The fibrous cap on the surface of the plaque was thin and disrupted, mainly composed of a large number of collagen fibers, a small number of smooth muscle cells (SMCs) and proteoglycans. Many inflammatory cells and new capillary formation were shown in plaque shoulders.
Inside of the plaque, there were mainly proliferation of connective tissue, homogeneous glass-like materials, and a large amount of hemosiderin, cholesterol crystals, and lipid droplets. Some plaques also showed hemorrhage or thrombus, and lipofuscin deposition. SMCs of the media adjacent to intima lesion were disarranged and proliferated, indicating SMCs migration [Figure 1c]. Shrinkage of adventitia showed accumulation of a few of inflammatory cells. Intima of control was smooth and flat, SMCs were regularly arranged in tunica media, and adventitia is integrity [Figure 1d].
According to the American Heart Association histological classifications of atherosclerosis,[7] the nine specimens taken in this study were advanced plaques and classified as type IV or above, containing 1 type IV, 1 type Vb, 1 type Vc, 4 type V, and 2 type VI.
Observation of autophagy in unstable atherosclerotic plaque via TEM
We observed the typical ultrastructural features of autophagy via TEM, such as vacuolization and formation of myelin figures in the nuclei of SMCs and macrophages [Figures 2a–e]. However, no abnormal chromatin aggregation was found in the nuclei of SMCs [Figures 2a–c] and macrophages [Figure 2d and e]. In ECs [Figure 2f], autophagolysosome was also found.
Figure 2.
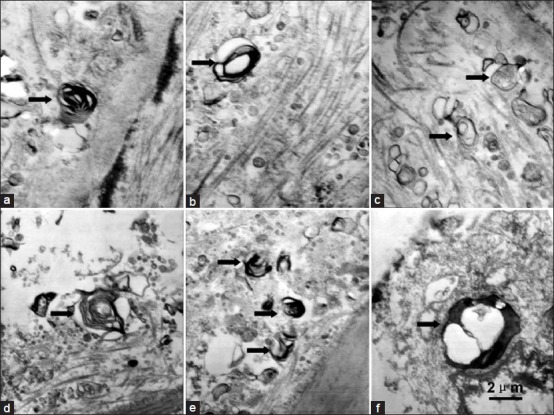
Observation of autophagy in unstable atherosclerotic plaque via transmission electron microscopy (TEM). Autophagic vacuoles with myelin structures (arrows) (a-b) and autophagic vacuoles with organelle inclusions (arrows) (c) were detectable in the cytoplasm of SMCs in the media adjacent to intima. Myelin structures were detectable in macrophages in fibrous cap and shoulder of plaque (d-e). The formation of autophagolysosomes was detected in endothelial cells (ECs) (f). Scale bars=2 μm.
Detection of autophagy in unstable atherosclerotic plaques by western blotting
LC3-II is a widely used marker for autophagy. In atherosclerotic plaques, the protein level of LC3-II was obviously increased compared with normal controls [Figure 3]. This finding was consistent with the following results of MAP1-LC3 staining in the plaques.
Figure 3.
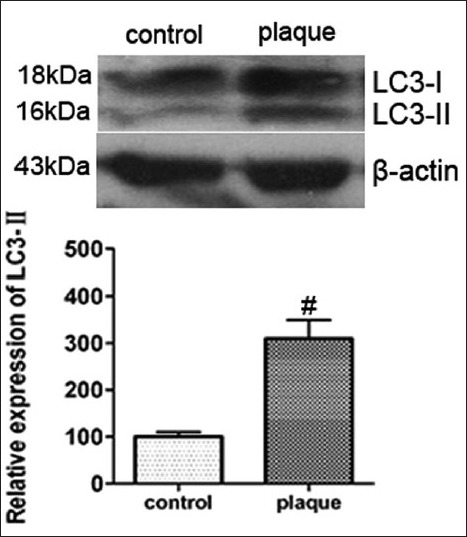
Expression of LC3-I/-II in unstable atherosclerotic plaque in patients with atherosclerosis was measured by western blotting. β-actin was used as a loading control. Upper panels show examples of protein levels from western blots; lower panels show statistics of optical density measurements. Values represent mean ± SD (n = 3). *represents P < 0.05 versus control group.
Expression of MAP1-LC3 in the different cells of unstable atherosclerotic plaques by immunofluorescence double staining
In macrophages, we found a large number of macrophages expressing MAP1-LC3 were significantly accumulated in the fibrous caps and shoulders of plaques [Figure 4]. In normal vessels, there was no macrophage infiltration [Figure 4].
Figure 4.
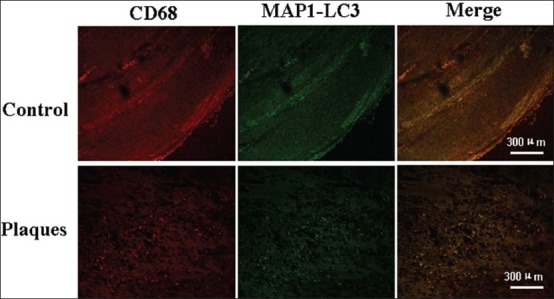
Photomicrographs of the vascular wall cells stained with MAP1-LC3 (green) and CD68 (marker of macrophages, red) in control (lower panels) and plaque shoulder (upper panels), Scale bars=300 μm.
SMCs expressing MAP1-LC3 were found in the fibrous cap of the plaque [Figure 5]. In normal vessels, there were no migrated SMCs to the intima and SMCs did not express MAP1-LC3 [Figure 5].
Figure 5.
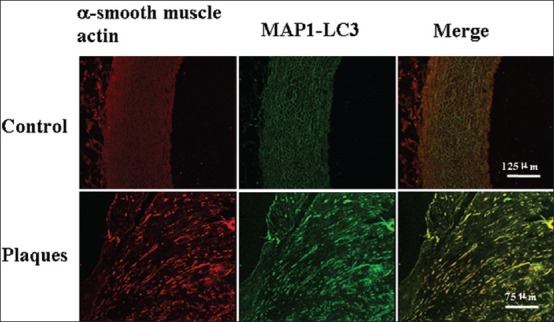
Photomicrographs of the media adjacent to intima stained with MAP1-LC3 (green) and a-smooth muscle actin (red) in control (upper panels, scale bars= 125 μm) and plaque shoulder (lower panels, scale bars=75 μm).
As to ECs, the expression of MAP1-LC3 was found in new capillaries of the shoulders [Figure 6]. In normal vascular endothelial cells (VECs), there was no expression of MAP1-LC3 [Figure 6].
Figure 6.
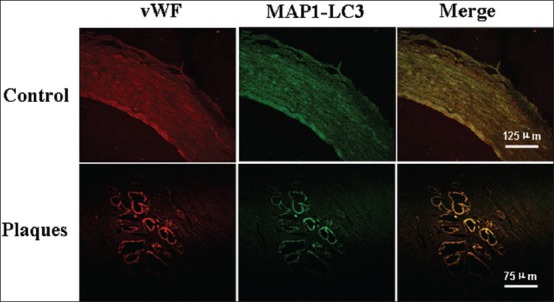
Photomicrographs of the vascular wall cells stained with MAP1-LC3 (green) and Von Willebrand factor (vWF) (marker of vascular endothelia cells, red) in control (upper panels, scale bars=125 μm) and plaque shoulder (lower panels, scale bars=75 μm).
DISCUSSION
Plaque stability has a close relationship with SMCs in the fibrous cap and the survival of VECs.[5] In previous studies, more attention was paid to investigate on these cells and apoptosis, or type I programmed cell death.[8] However, recent studies have found that autophagy or type II programmed cell death might also be involved in the progression of atherosclerosis in the regulation of cell death and survival.[5,9] Therefore, this study confirmed the existence and localization of autophagy in human plaques.
At present, TEM was recognized as the “gold standard” for the detection of autophagy. LC3 is a marker of the autophagic-lysosomal pathway, in which its lipidated (phosphatidylethanolamine-conjugated) forms (LC3-II in mammalian cells) appear to be predominantly associated with autophagic organelles.[10]
Results of TEM showed that some myelin structures were arranged in concentric rings in the cytoplasma of both SMCs adjacent to the ECs layer and macrophages infiltrated in plaques, which were autophagosome and composed of double membrane wrapped non-lysosomal membranous cellular components (e.g. endoplasmic reticulum).[10] In peri-nucleus of ECs, a high nonhomogeneous density of cellular components was detected and regarded as autophagolysosome. By immunofluorescence double staining, we found the increased expression of MAP1-LC3 in ECs, macrophages, and SMCs in plaques, but no MAP1-LC3 expressed in normal carotid artery. LC3-II is a membrane-bound form of LC3 and formed by LC3-I interacting with phosphatidylethanolamine, an abundant membrane phospholipid. The conformational change of LC3 is critical in autophagosome formation. Therefore, the conversion from LC3-I to LC3-II indicates autophagy activity.[5] The protein levels of LC3-II in these plaques were up-regulated compared with normal arteries. This further confirmed that autophagy is activated in atherosclerosis.
Therefore, based on the results of TEM, western blotting and immunofluorescence staining, we concluded that autophagy is activated in human atherosclerotic plaques. Some studies showed that both starvation and mild oxidative stress induce autophagy to remove the damaged organelles in SMCs.[11] In animal models, morphological feature of autophagy was detected by TEM in SMCs of the fibrous cap of atherosclerotic plaque.[12] In cell models, 7-ketocholesterol, one of the major oxysterols presented in oxidized low-density lipoprotein (ox-LDL), triggered not only oxidative damage but also extensive vacuolization and MAP1-LC3 formation in SMCs,[13] indicating that oxidative stress induces autophagy in SMC. Therefore, up-regulation of autophagy is a cellular protective approach to attenuate 7-KC-induced cell death in human aortic SMCs.[14]
Meanwhile, we found myelin structures in concentric rings and the expression of MAP1-LC3 in macrophages of the plaque shoulders, which indicated that autophagy was induced in macrophage of unstable plaques. Macrophage produces matrix metalloproteinases that degrade the extracellular matrix and induce SMCs death, which indicates that they play a major role in plaque destabilization and rupture. Macrophage could induce cell death in an autophagy-dependent manner by phagocytosis of plant sterols.[15] A recent study showed that imiquimod-inducing autophagy activation in macrophage stimulated plaque progression and enhanced inflammatory cell infiltration.[16] These demonstrated that in response to either intrinsic or extrinsic stimulations, macrophagy plays a role in the progress of atherosclerosis by autophagy activation.
In VECs, we also found the presence of autophagy. Ox-LDL aggregated in human umbilical vein ECs (HUVECs) and involved in the initiation and progression of atherosclerosis. Our previous study had shown that ox-LDL significantly induces autophagy in HUVECs.[17]
Recently, more attention has been paid on the role of autophagy in atherosclerosis. Some researchers believed that autophagy is a protective mechanism to promote cell survival rather than cell death, suggesting that autophagy induced in SMCs of advanced plaque is potentially an important mechanism to maintaining plaque stability. Autophagy protects vascular cells in plaques against oxidative stress and apoptosis by degrading damaged organelles.[11] Conversely, if autophagy is not engaged as part of the oxidative stress response in plaques, or when oxidative injury overcomes the cellular defenses, cells probably die via apoptosis. A recent study has proved that apolipoprotein L6, a pro-apoptotic BH3-only member of the B-cell lymphoma-2 family, regulated both autophagy and apoptosis in SMCs.[18] In a pharmacological study, it illustrated the protective effects of autophagy in atherosclerosis by using statins and 7-ketocholesterol. Low concentrations of statins induced cell death of SMC, which was attenuated by the autophagy inducer 7-ketocholesterol,[19] suggesting that stimulation of autophagy could avoid SMCs from death. The mechanisms of this suppression might be that the engulfment of defective mitochondria by autophagosomes limited the release of proapoptotic proteins such as cytochrome c and apoptosis-inducing factors into the cytosol.[20]
However, excessive activation of autophagy may cause autophagic SMCs death, which conversely results in plaque destabilization. Moreover, autophagic death of VECs may be detrimental for the structure of the plaque as endothelial injury or death represents a primary mechanism for acute clinical events by promoting lesional thrombosis.[21] Therefore, proper autophagy activation may be beneficial to stabilize atherosclerotic plaques.
In this study, we demonstrated that autophagy occurred in human unstable atherosclerotic plaques. However, the role of autophagy in atherosclerosis needs to be further studied, which will help to develop the pharmacological approaches for stabilizing vulnerable and rupture-prone lesions.
Footnotes
Edited by: De Wang
Source of Support: This work is supported by grants from the National Natural Science Fund of China (No. 81200894, No. 81471195); the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions; the Province Natural Science Fund of Jiangsu of China (No. BK2012172); Suzhou Science and Technology Development Program (No.SZS201205); Suzhou Foundation of Science and Technology Development Plan (No.SYSD2012083).
Conflict of Interest: None declared.
REFERENCES
- 1.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–41. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. 2010;30:1282–92. doi: 10.1161/ATVBAHA.108.179739. [DOI] [PubMed] [Google Scholar]
- 3.Heo SH, Cho CH, Kim HO, Jo YH, Yoon KS, Lee JH, et al. Plaque rupture is a determinant of vascular events in carotid artery atherosclerotic disease: Involvement of matrix metalloproteinases 2 and 9. J Clin Neurol. 2011;7:69–76. doi: 10.3988/jcn.2011.7.2.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baehrecke EH. Autophagy: Dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–10. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- 5.Martinet W, De Meyer GR. Autophagy in atherosclerosis: A cell survival and death phenomenon with therapeutic potential. Circ Res. 2009;104:304–17. doi: 10.1161/CIRCRESAHA.108.188318. [DOI] [PubMed] [Google Scholar]
- 6.Zhai C, Cheng J, Mujahid H, Wang H, Kong J, Yin Y, et al. Selective inhibition of PI3K/Akt/mTOR signaling pathway regulates autophagy of macrophage and vulnerability of atherosclerotic plaque. PLoS One. 2014;9:e90563. doi: 10.1371/journal.pone.0090563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995;92:1355–74. doi: 10.1161/01.cir.92.5.1355. [DOI] [PubMed] [Google Scholar]
- 8.Mallat Z, Tedgui A. Apoptosis in the vasculature: Mechanisms and functional importance. Br J Pharmacol. 2000;130:947–62. doi: 10.1038/sj.bjp.0703407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Meyer GR, Martinet W. Autophagy in the cardiovascular system. Biochim Biophys Acta. 2009;1793:1485–95. doi: 10.1016/j.bbamcr.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 10.Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3:181–206. doi: 10.4161/auto.3678. [DOI] [PubMed] [Google Scholar]
- 11.Kiffin R, Bandyopadhyay U, Cuervo AM. Oxidative stress and autophagy. Antioxid Redox Signal. 2006;8:152–62. doi: 10.1089/ars.2006.8.152. [DOI] [PubMed] [Google Scholar]
- 12.Kockx MM, De Meyer GR, Buyssens N, Knaapen MW, Bult H, Herman AG. Cell composition, replication, and apoptosis in atherosclerotic plaques after 6 months of cholesterol withdrawal. Circ Res. 1998;83:378–87. doi: 10.1161/01.res.83.4.378. [DOI] [PubMed] [Google Scholar]
- 13.Martinet W, De Bie M, Schrijvers DM, De Meyer GR, Herman AG, Kockx MM. 7-ketocholesterol induces protein ubiquitination, myelin figure formation, and light chain 3 processing in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:2296–301. doi: 10.1161/01.ATV.0000146266.65820.a1. [DOI] [PubMed] [Google Scholar]
- 14.He C, Zhu H, Zhang W, Okon I, Wang Q, Li H, et al. 7-Ketocholesterol induces autophagy in vascular smooth muscle cells through No×4 and Atg4B. Am J Pathol. 2013;183:626–37. doi: 10.1016/j.ajpath.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bao L, Li Y, Deng SX, Landry D, Tabas I. Sitosterol-containing lipoproteins trigger free sterol-induced caspase-independent death in ACAT-competent macrophages. J Biol Chem. 2006;281:33635–49. doi: 10.1074/jbc.M606339200. [DOI] [PubMed] [Google Scholar]
- 16.De Meyer I, Martinet W, Schrijvers DM, Timmermans JP, Bult H, De Meyer GR. Toll-like receptor 7 stimulation by imiquimod induces macrophage autophagy and inflammation in atherosclerotic plaques. Basic Res Cardiol. 2012;107:269. doi: 10.1007/s00395-012-0269-1. [DOI] [PubMed] [Google Scholar]
- 17.Zhang YL, Cao YJ, Zhang X, Liu HH, Tong T, Xiao GD, et al. The autophagy-lysosome pathway: A novel mechanism involved in the processing of oxidized LDL in human vascular endothelial cells. Biochem Biophys Res Commun. 2010;394:377–82. doi: 10.1016/j.bbrc.2010.03.026. [DOI] [PubMed] [Google Scholar]
- 18.Zhaorigetu S, Yang Z, Toma I, McCaffrey TA, Hu CA. Apolipoprotein L6, induced in atherosclerotic lesions, promotes apoptosis and blocks Beclin 1-dependent autophagy in atherosclerotic cells. J Biol Chem. 2011;286:27389–98. doi: 10.1074/jbc.M110.210245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinet W, Schrijvers DM, Timmermans JP, Bult H. Interactions between cell death induced by statins and 7-ketocholesterol in rabbit aorta smooth muscle cells. Br J Pharmacol. 2008;154:1236–46. doi: 10.1038/bjp.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 21.Debnath J, Baehrecke EH, Kroemer G. Does autophagy contribute to cell death? Autophagy. 2005;1:66–74. doi: 10.4161/auto.1.2.1738. [DOI] [PubMed] [Google Scholar]