

Abstract
Background:
Sleep/wake disturbances in patients with amyotrophic lateral sclerosis (ALS) are well-documented, however, no animal or mechanistic studies on these disturbances exist. Orexin is a crucial neurotransmitter in promoting wakefulness in sleep/wake regulation, and may play an important role in sleep disturbances in ALS. In this study, we used SOD1-G93A transgenic mice as an ALS mouse model to investigate the sleep/wake disturbances and their possible mechanisms in ALS.
Methods:
Electroencephalogram/electromyogram recordings were performed in SOD1-G93A transgenic mice and their littermate control mice at the ages of 90 and 120 days, and the samples obtained from these groups were subjected to quantitative reverse transcriptase-polymerase chain reaction, western blotting, and enzyme-linked immunosorbent assay.
Results:
For the first time in SOD1-G93A transgenic mice, we observed significantly increased wakefulness, reduced sleep time, and up-regulated orexins (prepro-orexin, orexin A and B) at both 90 and 120 days. Correlation analysis confirmed moderate to high correlations between sleep/wake time (total sleep time, wakefulness time, rapid eye movement [REM] sleep time, non-REM sleep time, and deep sleep time) and increase in orexins (prepro-orexin, orexin A and B).
Conclusion:
Sleep/wake disturbances occur before disease onset in this ALS mouse model. Increased orexins may promote wakefulness and result in these disturbances before and after disease onset, thus making them potential therapeutic targets for amelioration of sleep disturbances in ALS. Further studies are required to elucidate the underlying mechanisms in the future.
Keywords: Amyotrophic Lateral Sclerosis, Orexin, Sleep/Wake Disturbance
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a progressive and fatal neurodegenerative disease characterized by the loss of lower and upper motor neurons, leading to muscle atrophy, paralysis, and death.[1,2] Sleep disorders in patients with ALS are well-documented, and sleep-related complaints, such as insomnia, disturbed sleep, nightmare, and daytime sleepiness, have been frequently reported.[3,4] Moreover, several clinical studies on sleep disturbances in patients with ALS have been published, focusing on the frequency, characteristics, and severity of sleep problems.[4,5,6] However, there are no animal or mechanistic studies on sleep disturbances in ALS.
Animal and mechanistic studies on sleep disturbances in other neurodegenerative diseases, such as Alzheimer's disease (AD) and Parkinson disease (PD), might give us insights into sleep disturbances in ALS. Sleep problems in AD and PD involve disturbances in the neurotransmitter and hormone signaling, abnormal accumulations of neurotoxic proteins, and damage in the brain regions controlling the sleep/wake cycles,[7] which could exist in ALS as well. Orexin is a crucial neurotransmitter in the regulation of sleep and wakefulness. Precursor peptide prepro-orexin, which is produced in the hypothalamic neurons, matures into two peptides, orexin A and orexin B. These peptides promote wakefulness by activating wake-active neurons (WAN) in the hypothalamus and brain stem. The actions of orexins are mediated by two receptors, orexin-1 (OX1R) and orexin-2 (OX2R) receptors.[8,9]
We hypothesized that there are disturbances of sleep and wakefulness in the ALS mouse models and that orexin is an important molecule responsible for those disturbances. In the present study with SOD1-G93A transgenic mice, which are extensively used as animal model for mechanistic and therapeutic studies on ALS,[10] we used sleep/wake activity recordings and molecular techniques to test our hypothesis.
METHODS
Animals
Transgenic SOD1-G93A mice used in this study were bred from male hemizygous SOD1-G93 A mice (B6SJL-Tg [SOD1-G93A] 1 Gur/J) to female B6SJL/F1 hybrids. Their non-transgenic littermates served as controls. The genotyping of SOD1-G93A mice was performed by polymerase chain reaction (PCR), as previously reported.[11] Male hemizygous SOD1-G93A mice and female B6SJL/F1 hybrids were both purchased from the Jackson Laboratories (Bar Harbor, ME, USA). All mice were housed under a controlled temperature (22 ± 1°C) and 12 hours: 12 hours light-dark cycle.
All animal studies were approved by the Institutional Animal Care and Use Committee of Peking University Third Hospital, and conducted in accordance with the Guide for the Care and Use of Laboratory Animals of Peking University.
Experimental design
To monitor disease progression, all mice were tested using the Rota Rod test apparatus (Ugo Basile, Varese, Italy). The day on which a mouse first dropped off the Rota Rod within 600 seconds was designated as a day of disease onset for that mouse.[12] Using this testing criterion, we observed that all the SOD1-G93A transgenic mice in this study had disease onset between 90 and 120 days of age. Thus, we determined the two-time points in our study: 90-day representing the age before disease onset and 120-day representing the age after disease onset.
Twenty-eight mice were divided into four groups: SOD1-G93A group at 90-day (n = 8), control group at 90-day (n = 6), SOD1-G93A group at 120-day (n = 6), and control group at 120-day (n = 8). All mice received electroencephalogram/electromyogram (EEG/EMG) recordings at 90 or 120 days of age. After performing the recordings, the cerebrospinal fluid (CSF) and brain samples were collected. The hypothalamus and brain stem were isolated from the brain tissue for real-time reverse transcriptase (RT)-PCR, western blotting, and enzyme-linked immunosorbent assay (ELISA).
Sleep-wake activity recordings
Surgeries and EEG/EMG recordings were performed as previously described.[13] Briefly, after anesthesia, 28 mice were implanted with EEG and EMG electrodes. The EEG electrodes were placed epidurally on the cortex, and the EMG electrodes were placed in the dorsal neck muscles. Ten days after surgery, the electrodes were connected to recording cables attached to the MP150 system (Biopac, Goleta, CA, USA). EEG and EMG recordings were performed in this manner from the freely behaving mice for 24 hours. Data were analyzed using Sleep sign 2.0 software (Biopac, Goleta, CA, USA).
Quantitative reverse transcriptase-polymerase chain reaction
A description of quantitative RT-PCR has been previously published.[14] Total RNA from the hypothalamus and brainstem tissues was extracted using TRIzol reagent (Takara, Dalian, China) according to the manufacturer's instructions. First-strand cDNA was synthesized at 42°C with FastQuant RT Kit (Tiangen, Beijing, China) using 1 μg of total RNA. The amplification was performed using a SuperReal PreMix Plus Kit (Tiangen) and an ABI 7500 RT-PCR system (Applied Biosystems, Foster City, USA). The cDNA was amplified with an initial denaturation step (95°C, 15 minutes), and then with 40 PCR cycles consisting of a denaturation step (95°C, 10 seconds) and an annealing/extension step (58°C, 32 seconds). β-actin was used as internal control to calculate the relative abundance of each mRNA (n = 4–6/group). The specific sets of primers were as follows: Prepro-orexin: F: TGAACTTTCCTTCTACAAAGGTTC, R: CAACAGTTCGTAGAGACGGCA; Orexin1 receptor: F: CGCCAACCCTATCATCTACAA, R: GCTCTGCAAGGACAAGGACTT; Orexin2 receptor: F: GCTCACCAGCATAAGCACACT, R: TATCTCTTTGAGCAGACATGGG; β-actin: F: CCTAGCACCATGAAGATCAAGAT, R: ACTCATCGTACTCCTGCTTGCT.
Western blotting
Western blotting was performed as previously reported.[15] Total protein was extracted from the hypothalamus samples using the Total Protein Extraction Kit (Applygen, Beijing, China), consisting of radioimmunoprecipitation assay lysis buffer, phenylmethylsulfonyl fluoride, protease inhibitors, and phosphatase inhibitors, according to the manufacturer's instructions. Protein concentration was detected by the bicinchoninic acid protein assay using a protein Assay kit (Applygen). Samples containing equal amounts of total protein (20 μg) were separated by 12% SDS-PAGE and transferred to a polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA). The membrane was blocked with 3% bovine serum albumin (BSA) in tris buffered saline (TBS) containing 0.1% Tween 20 (TBS-T) for one hour, and then incubated overnight at 4°C with the primary orexin antibody (anti-orexin-prepro, 1:500, Millipore, Billerica, MA, USA) or primary β-actin antibody (anti-β-actin, 1:5000, EarthOx, San Francisco, CA, USA). β-actin was used as an internal loading control. The membrane was then incubated with secondary IRDye 800CW goat anti-rabbit (1:10,000, LI-COR, Lincoln, NE, USA) or goat anti-mouse (1:10,000, LI-COR, Lincoln, NE, USA) antibodies at 37°C for one hour. Quantitation of immunoreactive bands was performed using an Odyssey infrared imaging system (LI-COR, Lincoln).
Enzyme-linked immunosorbent assay
Levels of orexin A and orexin B in mouse CSF and brain tissues were measured by orexin A/orexin B ELISA kits (BlueGene Biotech, Shanghai, China). Brain tissues were homogenized in 1 mL PBS (0.02 mol/L, pH 7.0–7.2) per 0.5 g tissue and centrifuged. Samples (CSF or brain homogenates supernatant) or standards were added to 96-well plates coated with anti-mouse orexin A/B antibody and ELISA was performed according to manufacturer's protocol.
Statistical analysis
Data are expressed as mean ± standard error. The independent-samples t-test and Pearson correlation analyses were performed using SPSS 19.0 (SPSS Inc., Chicago, IL, USA). A P < 0.05 was considered statistically significant.
RESULTS
Disturbances of sleep and wakefulness in SOD1-G93A transgenic mice
Sleep/wake recordings were performed in both SOD1-G93A transgenic mice and their control groups. In the 90-day SOD1-G93A transgenic mice, across a 24-hour recording period, the total sleep time (TST) was significantly decreased ([443.23 ± 40.42 minutes] vs. [569.97 ± 39.04 minutes], P < 0.05) and wakefulness was increased ([996.78 ± 40.42 minutes] vs. [870.03 ± 39.04 minutes], P < 0.05), compared to the littermate controls. Non-rapid eye movement (NREM) sleep ([411.29 ± 35.41 minutes] vs. [533.48 ± 37.01 minutes], P < 0.05) and deep sleep (DS) ([47.35 ± 9.55 minutes] vs. [107.35 ± 11.36 minutes], P < 0.01) were also reduced. No significant difference was found in REM sleep and light sleep between the two groups for the 24-hour period [Figure 1a].
Figure 1.

Disturbances in sleep and wakefulness in SOD1-G93A transgenic mice. EEG/EMG recordings for 24 hours from the SOD1-G93A transgenic mice and littermate control mice at 90 days and 120 days of age. (a) TST, NREM, and DS are significantly decreased in the 90-day SOD1-G93A transgenic mice. WAKE is significantly increased. (b) In the 120-day SOD1-G93A transgenic mice, WAKE is also significantly enhanced. Besides TST, NREM, and DS, REM is also significantly reduced. Data are expressed as mean ± standard error (n = 6–8/group). *P < 0.05 and †P < 0.01. TST: Total sleep time; WAKE: Wakefulness; REM: Rapid eye movement sleep; NREM: Non-rapid eye movement sleep; LS: Light sleep; DS: Deep sleep; EEG/EMG: Electroencephalogram/electromyogram.
In the 120-day SOD1-G93A transgenic mice, a remarkable increase in wakefulness ([1110.33 ± 38.16 minutes] vs. [804.29 ± 74.57 minutes], P < 0.01), and a decrease in TST ([329.67 ± 38.16 minutes] vs. [635.71 ± 74.57 minutes], P < 0.01), NREM ([306.03 ± 36.81 minutes] vs. [534.68 ± 82.68 minutes], P < 0.05) and DS ([22.88 ± 10.27 minutes] vs. [107.90 ± 25.21 minutes], P < 0.05) were observed. However, in contrast with the 90-day SOD1-G93A transgenic mice, REM sleep was decreased ([23.62 ± 9.96 minutes] vs. [101.08 ± 13.93 minutes], P < 0.01) in the 120-day SOD1-G93A transgenic mice compared to the 120-day littermate controls [Figure 1b].
Orexins increase in SOD1-G93A transgenic mice
Next, we evaluated the changes in the orexin system in the SOD1-G93A transgenic mice. First, we tested the level of mRNA and protein expression of prepro-orexin. Q-PCR showed increased levels of prepro-orexin mRNA in the hypothalamus of the 90-day (1.84 ± 0.24 vs. 1.00 ± 0.20, P < 0.05) and 120-day (2.06 ± 0.25 vs. 1.00 ± 0.14, P < 0.01) SOD1-G93A transgenic mice, compared to controls [Figure 2a]. The western blotting analysis also showed increased levels of prepro-orexin protein in the hypothalamus of both 90-day (2.33 ± 0.24 vs. 1.00 ± 0.18, P < 0.01) and 120-day (2.30 ± 0.14 vs. 1.00 ± 0.19, P < 0.01) SOD1-G93A transgenic mice as compared to littermate controls [Figure 2b].
Figure 2.
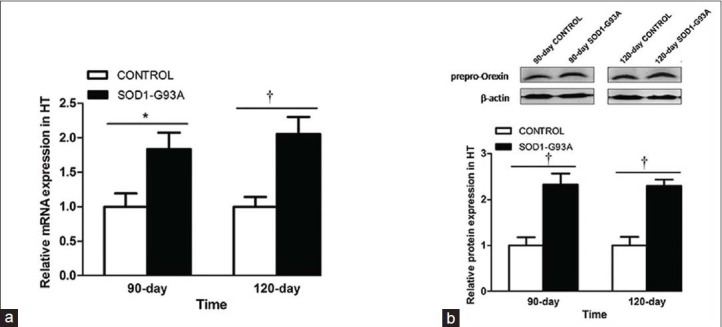
Prepro-orexin increases in SOD1-G93A transgenic mice. (a) Q-PCR was performed in the hypothalamic tissues from the SOD1-G93A transgenic mice and control groups. Prepro-orexin mRNA is significantly elevated in the hypothalamus of the 90 and 120 days SOD1-G93A transgenic mice, compared to control. (b) Western blotting was performed with antibody anti-orexin-prepro in the SOD1-G93A transgenic mice and control groups. Prepro-orexin expression is increased in the hypothalamus of the 90 and 120 days SOD1-G93A transgenic mice as compared to control. Data are expressed as mean ± standard error of mean (n = 4–6/group). *P < 0.05 and †P < 0.01. HT: hypothalamus; PCR: Polymerase chain reaction.
We then tested the protein levels of orexin A and B, using ELISA. In the hypothalamus, the level of orexin A was significantly enhanced in the 90-day ([2126.47 ± 70.65 pg/mg] vs. [1591.52 ± 61.25 pg/mg] protein, P < 0.01) and 120-day ([2166.32 ± 115.98 pg/mg] vs. [1446.18 ± 41.31 pg/mg] protein, P < 0.01) SOD1-G93A transgenic mice, compared to their respective littermate control mice [Figure 3a]. The level of orexin A was also elevated significantly in the brain stem of the 90-day ([1540.93 ± 34.87 pg/mg] vs. [1170.04 ± 47.73 pg/mg] protein, P < 0.01) and 120-day ([1583.96 ± 21.64 pg/mg] vs. [1224.87 ± 51.62 pg/mg] protein, P < 0.01) transgenic groups [Figure 3b]. There was no significant difference in orexin A levels between the SOD1-G93A transgenic mice and control groups in the CSF, at either time points [Figure 3c]. Similarly, orexin B levels were also significantly increased in the hypothalamus of the 90-day ([3485.35 ± 74.62 pg/mg] vs. [2352.45 ± 142.56 pg/mg] protein, P < 0.01) and 120-day ([3656.34 ± 139.17 pg/mg] vs. [2411.57 ± 117.70 pg/mg] protein, P < 0.01) SOD1-G93A transgenic mice [Figure 3d]. In the brain stem, orexin B was also significantly enhanced in both transgenic groups (90-day: [2726.79 ± 34.06 pg/mg] vs. [2411.70 ± 64.18 pg/mg] protein, P < 0.01 and 120-day: [2736.45 ± 57.32 pg/mg] vs. [2369.33 ± 83.57 pg/mg] protein, P < 0.05) as compared to littermate controls [Figure 3e]. In the CSF, no significant difference was found in orexin B levels between the respective transgenic and control groups [Figure 3f].
Figure 3.

Orexin A and B increase in SOD1-G93A transgenic mice. (a-c) ELISA reveals that orexin A levels are enhanced in the hypothalamus and brain stem of the 90 and 120 days SOD1-G93A transgenic mice than controls, but there is no significant difference for orexin A in the CSF. (d-f) ELISA shows increased the level of orexin B in the hypothalamus and brain stem of the 90 and 120 days SOD1-G93A transgenic mice compared with control, but no significant difference is seen in the CSF. Data are expressed as mean ± standard error (n = 4–6/group). *P < 0.05 and †P < 0.01. HT: Hypothalamus; BS: Brain stem; CSF: Cerebrospinal fluid; ELISA: Enzyme-linked immunosorbent assay.
Q-PCR analysis revealed no significant difference in the mRNA levels of OX1R [Figure 4a and 4b] and OX2R [Figure 4c and 4d] in the hypothalamus or the brain stem between the SOD1-G93A transgenic mice and controls.
Figure 4.
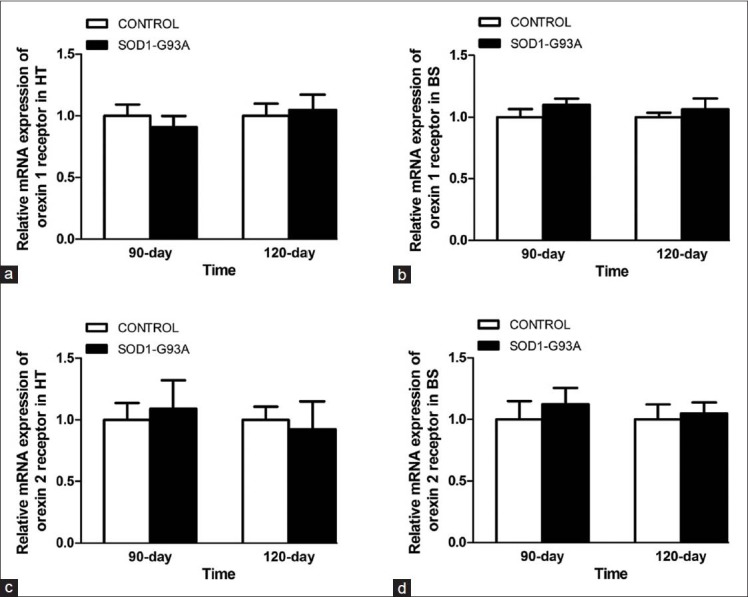
mRNA levels of orexin receptors. Q-PCR of orexin-1 receptor mRNA show no significant difference in the hypothalamus (a) and brain stem (b) of the 90 and 120 days SOD1-G93A transgenic mice than controls. No significant difference is found in orexin-2 receptor mRNA in the hypothalamus (c) and brain stem (d) of the 90 and 120 days SOD1-G93A transgenic mice than controls. HT: Hypothalamus; BS: Brain stem; PCR: Polymerase chain reaction.
Correlation analyses
Pearson correlation analyses were performed between sleep/wake time (TST, WAKE, REM, NREM, and DS) and expression levels of orexins (prepro-orexin, orexin A and B). Absolute values of the correlation coefficient confirmed high correlation between the sleep/wake time (TST, WAKE, and NREM) and expression levels of orexins. Meanwhile, there was a moderate correlation between REM/DS and orexins [Table 1].
Table 1.
Pearson correlation coefficient of sleep/wake time and orexins
| Variables | TST | WAKE | REM | NREM | DS |
|---|---|---|---|---|---|
| Prepro-orexin mRNA | −0.866 | 0.866 | −0.384 | −0.848 | −0.524 |
| Prepro-orexin protein | −0.904 | 0.904 | −0.432 | −0.879 | −0.585 |
| Orexin A HT | −0.879 | 0.879 | −0.643 | −0.827 | −0.691 |
| Orexin A BS | −0.859 | 0.859 | −0.469 | −0.844 | −0.632 |
| Orexin B HT | −0.887 | 0.887 | −0.538 | −0.859 | −0.674 |
| Orexin B BS | −0.890 | 0.890 | −0.521 | −0.867 | −0.652 |
TST: Total sleep time; WAKE: Wakefulness; REM: Rapid eyes movement sleep; NREM: Nonrapid eyes movement sleep; DS: Deep sleep; HT: Hypothalamus; BS: Brain stem.
DISCUSSION
In summary, we observed marked sleep disturbances in the SOD1-G93A mouse model of ALS, and demonstrated that the increase in expression of orexins correlated with these sleep disturbances in these mice.
Sleep/wake disturbances are often reported by patients with ALS,[2,3,4,16,17] but no related animal studies have been published. The exact mechanism behind the sleep/wake disturbances in ALS remains to be determined. In the present study, for the first time, we observed enhanced wakefulness and reduced sleep time (including TST, NREM, and DS) in both 90-day and 120-day SOD1-G93A transgenic mice. These results indicate that sleep/wake disturbances are early symptoms in these SOD1-G93A transgenic mice, and these disturbances may be correlated with the onset of ALS. In addition, REM sleep time was significantly decreased in the 120-day SOD1-G93A transgenic mice as compared to the 90-day SOD1-G93A transgenic mice, suggesting an aggravation and the possible link of sleep disturbances with disease progression. Therefore, studies on sleep disturbances in ALS are of significance to elucidate the etiology and pathogenesis of ALS. Moreover, considering the early occurrence of sleep disorders in AD and PD,[7] we speculate that the dysregulation of sleep might be common in the early stages of neurodegenerative diseases.
To further investigate the mechanism of sleep/wake disturbances in ALS, we detected the level of the sleep regulator, orexin. Sleep is a complicated behavior regulated by wakefulness promoting systems, sleep promoting systems, and circadian rhythms.[9,18] Wakefulness is promoted by multiple groups of WAN, which are mainly located in the brain stem and hypothalamus, including cholinergic neurons, orexinergic neurons, and dopaminergic neurons. Neurotransmitters in these WAN, including acetylcholine, dopamine, glutamate, histamine, norepinephrine, orexin, and serotonin, promote wakefulness.[19,20] Inhibition of these systems promotes sleep. The orexin system is an important component of wakefulness-promoting systems. Prepro-orexin, which is produced in hypothalamic neurons, matures into two neuropeptides, orexin A and orexin B. These two peptides activate monoaminergic and cholinergic neurons in the hypothalamus and brain stem to maintain a long, consolidated awake period.[8]
In our study, prepro-orexin was increased in the hypothalamus in the 90 and 120 days SOD1-G93A transgenic mice. Orexin A and B were significantly enhanced in the hypothalamus and brain stem of the 90 days and 120 days SOD1-G93A transgenic mice compared with littermate controls. These results suggest a strong association between increased expression of orexins and the extension of wakefulness in ALS mice. Pearson correlation analysis, which showed moderate to high correlations between sleep/wake time and expression of orexins, further confirmed this association. These results indicate that the extension of wakefulness and sleep/wake disturbances in the ALS mice may be caused, at least partly, by increase in expression of orexins. According to the absolute value of the correlation coefficient in Table 1, NREM had the highest correlation with expression of orexins among all stages of sleep. However, no differences were detected in the levels of orexin receptors in the hypothalamus and brain stem between the ALS mice and control groups, suggesting that enhanced wakefulness and other sleep disturbances in these transgenic mice were not promoted by upregulation of orexin receptors. In addition, the levels of orexin A and B were only elevated in the brain tissue other than the CSF in the SOD1-G93A transgenic mice. This is consistent with the clinical study by Van Rooij et al.,[21] which demonstrated that CSF orexin levels were normal in patients with ALS.
Sleep disturbances occur before disease onset in the ALS SOD1-G93A transgenic mice. Increased expression of orexins may promote wakefulness and result in sleep/wake disturbances in these mice. Orexins might be potential targets for ameliorating the sleep disturbances in ALS. Further experiments need to be carried out in the future to investigate the underlying mechanisms.
ACKNOWLEDGMENTS
We are grateful to Institute for Experimental Animals of Peking University Health Science Center.
Footnotes
Edited by: Huan Liu
Source of Support: This work was supported by a grant from the National Natural Science Foundation of China (No. 81030019).
Conflict of Interest: None declared.
REFERENCES
- 1.Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 2.Feng XH, Yuan W, Peng Y, Liu MS, Cui LY. Therapeutic effects of dl-3-n-butylphthalide in a transgenic mouse model of amyotrophic lateral sclerosis. Chin Med J. 2012;125:1760–6. [PubMed] [Google Scholar]
- 3.Hetta J, Jansson I. Sleep in patients with amyotrophic lateral sclerosis. J Neurol. 1997;244:S7–9. doi: 10.1007/BF03160565. [DOI] [PubMed] [Google Scholar]
- 4.Lo Coco D, Mattaliano P, Spataro R, Mattaliano A, La Bella V. Sleep-wake disturbances in patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2011;82:839–42. doi: 10.1136/jnnp.2010.228007. [DOI] [PubMed] [Google Scholar]
- 5.Atalaia A, De Carvalho M, Evangelista T, Pinto A. Sleep characteristics of amyotrophic lateral sclerosis in patients with preserved diaphragmatic function. Amyotroph Lateral Scler. 2007;8:101–5. doi: 10.1080/17482960601029883. [DOI] [PubMed] [Google Scholar]
- 6.Ebben MR, Shahbazi M, Lange DJ, Krieger AC. REM behavior disorder associated with familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2012;13:473–4. doi: 10.3109/17482968.2012.673172. [DOI] [PubMed] [Google Scholar]
- 7.Rothman SM, Mattson MP. Sleep disturbances in Alzheimer's and Parkinson's diseases. Neuromolecular Med. 2012;14:194–204. doi: 10.1007/s12017-012-8181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakurai T. The neural circuit of orexin (hypocretin): maintaining sleep and wakefulness. Nat Rev Neurosci. 2007;8:171–81. doi: 10.1038/nrn2092. [DOI] [PubMed] [Google Scholar]
- 9.Wright KP, Lowry CA, Lebourgeois MK. Circadian and wakefulness-sleep modulation of cognition in humans. Front Mol Neurosci. 2012;5:50. doi: 10.3389/fnmol.2012.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mancuso R, Oliván S, Mancera P, Pastén-Zamorano A, Manzano R, Casas C, et al. Effect of genetic background on onset and disease progression in the SOD1-G93A model of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2012;13:302–10. doi: 10.3109/17482968.2012.662688. [DOI] [PubMed] [Google Scholar]
- 11.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–5. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 12.Takata M, Tanaka H, Kimura M, Nagahara Y, Tanaka K, Kawasaki K, et al. Fasudil, a rho kinase inhibitor, limits motor neuron loss in experimental models of amyotrophic lateral sclerosis. Br J Pharmacol. 2013;170:341–51. doi: 10.1111/bph.12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szentirmai E. Central but not systemic administration of ghrelin induces wakefulness in mice. PLoS One. 2012;7:e41172. doi: 10.1371/journal.pone.0041172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu R, Zhang Z, Lu Z, Borlongan C, Pan J, Chen J, et al. Human umbilical cord stem cells ameliorate experimental autoimmune encephalomyelitis by regulating immunoinflammation and remyelination. Stem Cells Dev. 2013;22:1053–62. doi: 10.1089/scd.2012.0463. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Li L, Wang T, Zang H, An Y, Li L, et al. Analysis of epidermal growth factor signaling in nasal mucosa epithelial cell proliferation involved in chronic rhinosinusitis. Chin Med J. 2014;127:3449–53. [PubMed] [Google Scholar]
- 16.Culebras A. Sleep and neuromuscular disorders. Neurol Clin. 2005;23:1209–23. doi: 10.1016/j.ncl.2005.08.004. ix. [DOI] [PubMed] [Google Scholar]
- 17.Jenkins TM, Hollinger H, McDermott CJ. The evidence for symptomatic treatments in amyotrophic lateral sclerosis. Curr Opin Neurol. 2014;27:524–31. doi: 10.1097/WCO.0000000000000135. [DOI] [PubMed] [Google Scholar]
- 18.Abbott SM, Arnold JM, Chang Q, Miao H, Ota N, Cecala C, et al. Signals from the brainstem sleep/wake centers regulate behavioral timing via the circadian clock. PLoS One. 2013;8:e70481. doi: 10.1371/journal.pone.0070481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, Panossian LA, Zhang J, Zhu Y, Zhan G, Chou YT, et al. Effects of chronic sleep fragmentation on wake-active neurons and the hypercapnic arousal response. Sleep. 2014;37:51–64. doi: 10.5665/sleep.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuller PM, Gooley JJ, Saper CB. Neurobiology of the sleep-wake cycle: Sleep architecture, circadian regulation, and regulatory feedback. J Biol Rhythms. 2006;21:482–93. doi: 10.1177/0748730406294627. [DOI] [PubMed] [Google Scholar]
- 21.Van Rooij FG, Schelhaas HJ, Lammers GJ, Verbeek MM, Overeem S. CSF hypocretin-1 levels are normal in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10:487–9. doi: 10.3109/17482960802315024. [DOI] [PubMed] [Google Scholar]