

Abstract
Background. Encephalitis is parenchymal brain inflammation, commonly due to herpes simplex virus (HSV). Key host inflammatory mediators and their relationship to blood-brain barrier (BBB) permeability, neuroimaging changes, and disease outcome are poorly understood.
Methods. We measured levels of 38 mediators in serum (n = 78) and cerebrospinal fluid (n = 37) specimens from patients with encephalitis, including 17 with disease due to HSV infection. Outcome measures were Glasgow coma and outcome scores; CSF to serum albumin ratio, reflecting BBB permeability; and, in patients with HSV infection, magnetic resonance imaging–based temporal lobe volume.
Results. Serum interleukin 1 receptor antagonist (IL-1RA) levels were elevated in patients with a good outcome (P = .004). Among patients infected with HSV, the ratio of CSF IL-1β to IL-1RA was associated with a worse outcome (P = .009); a ratio of ≥0.55 pg/mL had high specificity and sensitivity for a poor outcome (100% and 83%; P = .015). Temporal lobe volume had a negative correlation with serum IL-1RA level (P = .012) and a positive correlation with serum IL-1α level (P = .0003) and CSF IL-1β level (P = .007). A normal coma score was associated with an elevated interleukin 10 (IL-10) level in serum specimens from HSV-infected patients (P = .007) and CSF specimens from all patients (P = .016); the IL-10 level correlated inversely with BBB permeability (P = .005).
Conclusions. A proinflammatory cytokine response is associated with greater clinical severity, BBB permeability, and neuroimaging damage during encephalitis. IL-1 antagonists should be investigated as adjunctive treatment in encephalitis.
Keywords: encephalitis, cytokine, chemokine, herpes simplex, blood-brain barrier
Encephalitis is a pathological inflammation of the brain parenchyma. The most common cause is herpes simplex virus (HSV). During HSV-associated encephalitis, HSV infection predominantly affects the temporal lobes and has a mortality rate of 10%–30% despite effective antiviral treatment, with neurological morbidity in at least 60% of survivors [1, 2]. Acyclovir effectively reduces viral load but does not inhibit immune-mediated pathogenesis [3]. There is mounting evidence that the host inflammatory response, particularly cytokines and associated mediators, may play a key role in pathogenesis [4, 5]. These mediators act directly on neuroglial cells, and they orchestrate permeability of the blood-brain barrier (BBB) and the influx of leucocytes, which is a key process in inflammation [6, 7].
Interleukin 1 (IL-1) is the prototypical proinflammatory cytokine and has elevated levels in murine and cell culture models of viral encephalitis, resulting in fever, increased BBB permeability, and the production of additional proinflammatory mediators [3, 8, 9]. Many of these actions are directly opposed by the IL-1 receptor antagonist (IL-1RA) and interleukin 10 (IL-10) [10–12]. However, these models only provide a limited representation of host responses, and the importance of investigation of clinical samples has been highlighted [13–16].
The key mediators underlying this inflammatory response remain unclear. Previous clinical studies have assessed a few mediators in a limited number of cases [4, 5, 14]. Moreover, the downstream effects on BBB permeability and parenchymal damage detected by neuroimaging have not been assessed [4, 5, 14]. In addition, HSV only accounts for 20% of cases, and it is not known whether these mediators are associated with disease severity or BBB permeability in encephalitis more broadly [17]. These mediators act in concert, and the importance of assessing their relative abundance has been demonstrated [18, 19]. Therefore, the ratio of proinflammatory cytokines to antiinflammatory cytokines has been evaluated: for example, the interleukin 6 (IL-6) to interleukin 4 (IL-4) ratio and the IL-6 to IL-10 ratio have strong correlations with outcome in Japanese encephalitis and cerebral malaria, respectively [14, 20].
Therefore, we assessed the relationship between cytokines and associated mediators in cerebrospinal fluid and serum with clinical disease severity and BBB permeability. Because HSV is the most common cause of encephalitis, and because there are stereotypic neuroimaging changes, we also assessed the relationship between these mediators and the volume of injury in this subgroup.
METHODS
Study Population and Measurement of Mediators
Patients were recruited prospectively from 24 centers over a 2-year period between 2005 and 2008 through the Health Protection Agency (HPA) Aetiological Study of Encephalitis in England; the study is described in detail elsewhere [17]. Of 203 patients recruited, serum and/or cerebrospinal fluid (CSF) specimens were used if >50 µL was available after diagnostic testing and aliquot archiving. Samples were collected at recruitment and stored at −80°C, and freeze-thaw cycles were minimized. Thirty-eight mediators were assessed in duplicate, using a commercial cytometric bead array (Procarta, Affymetrix, Milano, Italy), and were analyzed using the BioPlex Manager 4.1 (Bio-Rad Laboratories, Hemel Hempstead, United Kingdom). The balance between proinflammatory cytokines and antiinflammatory cytokines was assessed by measuring the IL-1 to IL-1RA and IL-1 to IL-10 ratios [12, 14, 20].
The HPA study was approved by the North and East Devon Multicentre Research Ethics Committee (reference 05/Q2102/22). This substudy was approved by the HPA Encephalitis Study Steering Group and the Pan-Manchester Research and Development Group for the University of Manchester.
Clinical Outcome Measures and BBB Permeability
The admission Glasgow coma scale (GCS) score was recorded; a score of 15 (out of 15) was defined as good, and scores of ≤14 were defined as poor [14]. The discharge Glasgow outcome scale (GOS) score was recorded, with good defined as a score of 5 (out of 5; indicating minor or no disability) and poor defined as scores of ≤4 (indicating moderate disability or death) [5]. CSF white blood cell count of <5 cells/µL was defined as normal. As a marker of BBB permeability, the ratio of CSF to serum albumin levels was determined on paired samples, using radial immunodiffusion (Binding Site, Birmingham, United Kingdom) [21].
Imaging Outcome
Magnetic resonance imaging (MRI) was performed when possible for patients with HSV encephalitis. Volumes were determined using stereology of temporal lobes on T1-weighted images (0.72–0.94-mm resolution), indicating swelling, and T2-weighted images (0.45–0.54 mm resolution), indicating tissue damage, with a 5-mm thickness, using a grid-size of 10 voxels and established anatomical boundaries (EasyMeasure) [22, 23]. Preliminary analysis compared the absolute volume on T1 with 18 healthy controls [24]. However, to account for individual differences, all analyses were undertaken with volumes as a percentage of the total intracranial volume. Measurements were made in duplicate and by another blinded person [25].
Statistical Analysis
The Mann–Whitney U test, the Kendall rank correlation coefficient, linear regression, and the Pearson correlation coefficient were used (SPSS 2011 and GraphPad 2014), and a P value of <.05 was defined as significant. Overall mediator data underwent a 1-way hierarchical cluster analysis; data from each outcome group underwent nearest neighbor analysis, using the Pearson correlation coefficient, to generate proximity matrices [19, 26]. A heat map was generated for those with a good outcome and those with a poor outcome. A separate heat map was generated by subtracting the values of the proximity matrix for those with a good outcome from values for those with a poor outcome, and a heat map was generated, as described previously [19, 26].
To avoid undetectable levels or missing data bias, only mediators detected in CSF or serum specimens from ≥80% of the samples were analyzed [27]. To minimize any potential for the influence of storage, concentrations of each mediator were median centered for each patient [19, 27]. Therefore, the concentration of each mediator was expressed and analyzed as a value relative to the median concentration of all of the mediators in that patient sample, as described previously [19, 27].
RESULTS
Of 203 patients meeting the clinical case definition of encephalitis, 95 had sufficient serum and or CSF samples available for analysis. Serum samples were available for 78 patients; encephalitis had an infectious etiology in 38 (17 had HSV infection, 7 had varicella zoster virus [VZV] infection, 5 had tuberculosis, 3 had bacterial infection, 2 had dual infection [1 had tuberculosis and human immunodeficiency virus [HIV] infection and 1 had cryptococcal infection and VZV infection], 1 had influenza A virus infection, 1 had measles, 1 had HIV infection, and 1 had toxoplasmosis), was immune mediated in 20 (9 had antibody-mediated encephalitis, 8 had acute disseminated encephalomyelitis [ADEM], 1 had paraneoplastic encephalitis, 1 had vasculitis, and 1 had multiple sclerosis), and an unknown etiology in 20. For 37 patients, a CSF specimen was available; encephalitis had an infectious etiology in 20 (12 had HSV infection, 6 had VZV infection, 1 had JC virus infection, and 1 had toxoplasmosis), was immune mediated in 9 (5 had ADEM, 3 had antibody-mediated encephalitis, and 1 had paraneoplastic encephalitis), and had an unknown etiology in 8.
The following mediators were not identified in >80% of the cohort and were therefore removed from further analysis: tumor necrosis factor receptor 1 (TNFR1) and TNFR2, E-selectin, CXCL9, interleukin 17a, and vascular endothelial growth factor α. In addition, in CSF the following mediators were not detected at levels above the lower limits of quantification in >80% of the samples: granulocyte colony-stimulating factor, granulocyte macrophage colony-stimulating factor, interferon α2 (IFN-α2), IFN-β, leptin, and IL-6. In serum, the following mediators were also not detected at levels above the limits of quantification in >80%: IFN-α2 and IL-6.
Mediators Associated With Clinical Severity
For all patients with encephalitis, comparing those with a normal or reduced GCS score on admission, there was no significant difference in the IL-1 concentration in serum or CSF. However, the mediator most closely associated with GCS score on admission was IL-10 (Table 1). For all patients, the concentration of IL-10 in the CSF was significantly higher in those with a normal GCS score (P = .016) and IL-10 also had a positive correlation with GCS score (P = .017; Figure 1A). Assessing only the subset with HSV encephalitis, in the serum the mediator most closely associated with the GCS score was also IL-10, which was higher in those with a normal GCS score (P = .007), and serum IL-10 also had a positive correlation with the GCS score (P = .01) (Figure 1B).
Table 1.
Levels of Cytokines, Chemokines, and Associated Mediators in Relation to Glasgow Coma Scale (GCS) Score on Admission to the Hospital Among 89 Patients With Acute Encephalitis and a Subgroup of 26 Patients With Encephalitis Due to Herpes Simplex Virus (HSV)
| Specimen, Mediator | Level Among All Cases, pg/mL, Median (Range) |
Level Among Cases Due to HSV, pg/mL, Median (Range) |
||||
|---|---|---|---|---|---|---|
| GCS Score of 15 | GCS Score of <15 | P Value | GCS Score of 15 | GCS Score of <15 | P Value | |
| CSF | ||||||
| CCL2 | 0.39 (−0.10 to 0.37) | 0.65 (−1.19 to 1.04) | NS | 0.27 (0.09–0.96) | 0.62 (−0.24 to 1.93) | NS |
| CCL3 | −0.16 (−1.34 to −0.01) | −0.21 (−1.49 to −0.05) | NS | −0.35 (−0.45 to −0.08) | −0.15 (−1.67 to 0.10) | NS |
| CCL5 | −0.26 (−0.44 to 1.02) | −0.69 (−1.17 to 0.48) | .007 | −0.27 (−0.46 to 1.02) | −0.37 (−1.04 to 0.48) | NS |
| CXCL10 | 1.63 (−0.17 to 1.87) | 1.23 (−0.32 to 1.72) | NS | 1.69 (0.95–1.92) | 1.22 (0.37–2.17) | NS |
| ICAM | 1.58 (1.21–2.69) | 1.50 (1.24–2.64) | NS | 2.07 (1.21–2.17) | 1.458 (1.05–2.43) | NS |
| IFN-γ | −0.43 (−0.88 to −0.01) | −0.47 (−1.00 to 0.61) | NS | −0.47 (−0.88 to −0.11) | −0.47 (−0.99 to 0.13) | NS |
| IFN-ο | −0.38 (−0.88 to 0.46) | −0.37 (−0.78 to 0.39) | NS | −0.391 (−0.52 to −0.39) | −0.40 (−0.76 to −0.08) | NS |
| IL-1α | −0.18 (−0.46 to −0.03) | −0.16 (−0.88 to 0.04) | NS | −0.25 (−0.46 to −0.08) | −0.20 (−0.50 to 0.04) | NS |
| IL-1β | −0.48 (−1.10 to −0.11) | −0.56 (−0.95 to −0.24) | NS | −0.75 (−1.06 to −0.28) | −0.50 (−0.77 to −0.24) | NS |
| IL-1RA | 0.58 (0.31–1.84) | 0.69 (0.01–1.67) | NS | 1.5 (0.68–1.84) | 1.23 (0.30–1.67) | NS |
| IL-4 | −0.13 (−1.73 to 0.18) | −0.24 (−1.66 to 0.13) | NS | −0.49 (−0.80 to −0.03) | −0.18 (−0.35 to −0.01) | NS |
| IL-8 | 0.57 (−0.03 to 1.47) | 0.56 (−0.01 to 2.03) | NS | 0.67 (−0.03 to 1.47) | 0.58 (0.17–2.03) | NS |
| IL-10 | −0.48 (−0.58 to 0.10) | −0.69 (−1.70 to 0.01) | .016a | −0.31 (−0.48 to 0.36) | −0.49 (−0.66 to 0.01) | NS |
| MPO | 1.21 (0.39–1.62) | 1.0 (−0.87 to 2.00) | NS | 1.26 (1.03–1.62) | 1.04 (−0.87 to 1.42) | NS |
| TNF-α | −0.60 (−1.38 to −0.38) | −0.69 (−1.27 to −0.09) | NS | −0.69 (−1.11 to −0.38) | −0.67 (−1.00 to −0.37) | NS |
| TRAIL | 0.21 (−0.17 to 0.57) | 0.17 (−0.04 to 0.78) | NS | 0.19 (0.24–0.57) | 0.23 (−0.04 to 0.78) | NS |
| VCAM | 1.32 (−0.28 to 2.19) | 1.31 (−0.37 to 2.03) | NS | 1.67 (1.08–2.19) | 1.15 (0.88–2.03) | NS |
| IL-1α:IL-1RA | −0.22 (−0.84 to −0.04) | −0.30 (−29.57 to 0.03) | NS | −0.15 (−0.31 to −0.04) | −0.19 (−0.68 to 0.17) | NS |
| IL-1β:IL-1RA | −0.64 (−2.28 to −0.38) | −0.77 (−107.06 to −0.20) | NS | −0.61 (−0.88 to −0.38) | −0.53 (−0.37 to −1.59) | NS |
| IL-1α:IL-10 | 0.09 (−19.03 to 1.47) | 0.26 (−17.42 to 2.05) | NS | 0.007 (−19.03 to 1.47) | 0.26 (−17.43 to 2.05) | NS |
| IL-1β:IL-10 | 0.51 (−57.23 to 2.92) | 0.71 (−43.88 to 1.42) | NS | 0.59 (−57.23 to 2.92) | 0.69 (−43.88 to 3.65) | NS |
| Serum | ||||||
| CCL2 | 0.09 (−0.1 to 0.49) | 0.05 (−0.65 to 1.04) | NS | 0.15 (0.09–0.27) | 0.15 (−0.65 to 0.41) | NS |
| CCL3 | −0.53 (−1.34 to −0.01) | −0.45 (−1.19 to −0.05) | NS | −0.46 (−0.53 to −0.25) | −0.29 (−0.91 to −0.05) | NS |
| CXCL10 | 0.54 (−0.17 to 1.69) | 0.53 (−0.32 to 1.72) | NS | 1.63 (0.30–1.69) | 0.58 (−0.04 to 1.72) | NS |
| G-CSF | −0.11 (−1.0 to 0.45) | −0.57 (−1.15 to 0.14) | NS | −0.06 (−0.11 to 0.45) | −0.28 (−1.15 to 0.12) | NS |
| GM-CSF | 0.11 (−1.14 to 0.54) | −0.41 (−1.36 to 0.33) | NS | 0.10 (−0.13 to 0.54) | −0.05 (−1.36 to 0.33 ) | NS |
| Granzyme B | −0.25 (−1.30 to 0.86) | −0.40 (−1.82 to 0.31) | NS | 0.32 (−0.25 to 0.86) | −0.34 (−1.15 to −0.13) | NS |
| ICAM | 2.04 (−0.30 to 2.78) | 2.05 (0.14–2.92) | NS | 2.07 (0.40–2.12) | 2.04 (0.78–2.53) | NS |
| IFN-β | −0.39 (−2.42 to 0.05) | −1.17 (−2.01 to 0.28) | NS | −0.39 (−0.36 to −0.18) | −1.17 (−1.89 to 0.0) | NS |
| IFN-γ | −0.67 (−1.04 to −0.29) | −0.84 (−1.60 to −0.06) | .02 | −0.71 (−0.88 to −0.59) | −0.81 (−1.56 to −0.61) | NS |
| IFN-ο | −0.05 (−0.77 to 0.60) | −0.28 (−0.67 to 0.54) | NS | 0.25 (0.10–0.41) | −0.23 (−0.70 to 0.26) | .04 |
| IL-1α | −0.91 (−1.55 to −0.08) | −0.85 (−1.69 to 0.11) | NS | −0.08 (−0.25 to −0.03) | −0.55 (−1.34 to 0.10) | NS |
| IL-1β | −0.88 (−1.77 to −0.07) | −1.14 (−2.14 to −0.27) | NS | −0.6 (−0.75 to −0.07) | −0.62 (−2.02 to 0.27) | NS |
| IL-1RA | 0.68 (−0.22 to 1.61) | 0.24 (−0.26 to 1.02) | .049 | 0.68 (0.15–1.61) | 0.22 (−0.11 to 1.02) | NS |
| IL-4 | −0.49 (−1.25 to 0.19) | −0.53 (−1.39 to 0.04) | NS | −0.29 (−0.49 to 0.19) | −0.30 (−1.32 to −0.01) | NS |
| IL-8 | −0.34 (−1.60 to 2.21) | −0.27 (−2.27 to 0.48) | NS | 0.07 (−0.03 to 0.67) | −0.12 (−1.91 to 1.19) | NS |
| IL-10 | −1.12 (−2.33 to 0.01) | −0.83 (−2.45 to 0.16) | NS | −0.33 (−0.43 to 0.01) | −0.81 (−2.45 to −0.44) | .007a |
| Leptin | 0.98 (−0.01 to 2.62) | 1.08 (0.04–2.41) | NS | 0.38 (−0.01 to 1.01) | 1.37 (0.34–1.99) | NS |
| MPO | 1.14 (−0.30 to 2.24) | 1.15 (−0.43 to 2.29) | NS | 1.03 (0.57–1.63) | 1.44 (0.30–2.22) | NS |
| TNF-α | −0.69 (−1.61 to −0.10) | −0.98 (−1.67 to −0.26) | NS | −0.50 (−0.69 to −0.10) | −0.88 (−1.67 to −0.54) | NS |
| TRAIL | 0.26 (0.01–0.64) | 0.24 (−0.11 to 1.30) | NS | 0.24 (0.17–0.49) | 0.33 (0.08–0.73) | NS |
| VCAM | 1.75 (−0.20 to 2.40) | 1.75 (−0.48 to 2.55) | NS | 1.67 (−0.20 to 2.19) | 1.56 (−0.39 to 2.13) | NS |
| IL-1α:IL-1RA | −1.10 (−29.16 to 2.50) | −1.45 (−56.12 to 24.51) | NS | −0.15 (−0.51 to −0.04) | −0.80 (−56.12 to 24.51) | NS |
| IL-1β:IL-1RA | −1.40 (−31.10 to 3.45) | −2.01 (−84.69 to 20.63) | NS | −0.47 (−0.88 to −0.46) | −1.11 (−84.69 to 59.58) | NS |
| IL-1α:IL-10 | 0.81 (−37.20 to 1.30) | 0.71 (−3.17 to 8.25) | NS | 0.07 (−37.2 to 0.24) | 0.55 (−0.23 to 1.03) | .04 |
| IL-1β:IL-10 | 1.09 (−112.50 to 1.36) | 1.01 (−6.84 to 2.59) | NS | 0.22 (−112.50 to 1.39) | 0.88 (0.41–1.23) | .44 |
GCS score was available for 59 serum samples (76%) and 30 CSF samples (81%). Of those infected with HSV, the GCS score was available for 14 serum specimens (82%) and 12 CSF samples (100%). A GCS score of 15 was considered good, and a GCS score of <15 was considered poor.
Abbreviations: CCL2, monocyte chemotactic protein 1; CCL3, monocyte inflammatory protein 1α; CCL5, regulated on activation normal T-cell expressed and secreted; CXCL10, inducible protein 10; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte macrophage colony-stimulating factor; ICAM, intracellular adhesion molecule; IFN-γ, interferon γ; IFN-ο, interferon ο; IL-1RA, interleukin 1 receptor antagonist; IL-1α, interleukin 1α; IL-1β, interleukin 1β; IL-4, interleukin 4; IL-8, interleukin 8; IL-10, interleukin 10; MPO, myeloperoxidase; NS, not significant; TNF-α, tumor necrosis factor α; TRAIL, tumor necrosis factor α–related apoptosis-inducing ligand; VCAM, vascular cell adhesion molecule.
a Also correlated with GCS score.
Figure 1.

A and B, Relationship between Glasgow coma scale (GCS) score on admission and interleukin 10 (IL-10) concentration in cerebrospinal fluid in patients with encephalitis (A) and serum IL-10 level in patients with encephalitis due to herpes simplex virus (HSV) infection (B). C and D, Relationship between Glasgow outcome scale (GOS) score at discharge and serum interleukin 1 receptor antagonist (IL-1RA) in patients with encephalitis (C) and the ratio of IL-1β to IL-1RA levels in cerebrospinal fluid in patients with HSV encephalitis (D). Abbreviation: CI, confidence interval.
For the cohort overall comparing those with a good and poor GOS score, there was no significant difference in IL-1 concentration in the CSF or serum. However, the serum mediator associated most significantly with outcome was IL-1RA (Table 2). Higher serum concentrations of IL-1RA were seen in those with a good as opposed to poor outcome (P = .004). Also there was a significant positive correlation between IL-1RA and the GOS score (P = .01) (Figure 1C). Assessing only the subset with HSV encephalitis, IL1RA was also the mediator most significantly associated with the GOS score. The CSF IL-1β:IL-1RA ratio was significantly higher in those with a worse outcome score (P = .009). There was also a significant inverse correlation with GOS score (P = .003; Figure 1D). Applying a CSF IL-1β:IL-1RA cut-off of >−0.55 pg/mL had a high specificity and sensitivity for distinguishing patients with a good from a poor outcome (100% and 83% respectively; P = .015).
Table 2.
Levels of Cytokines, Chemokines, and Associated Mediators in Relation to Glasgow Outcome Scale (GOS) Score at Discharge Among 95 Patients With Acute Encephalitis and a Subgroup of 29 Patients With Encephalitis Due to Herpes Simplex Virus (HSV)
| Specimen, Mediator | Level Among All Cases, pg/mL, Median (Range) |
Level Among Cases Due to HSV, pg/mL, Median (Range) |
||||
|---|---|---|---|---|---|---|
| GOS Score of 5 | GOS Score of <5 | P Value | GOS Score of 5 | GOS Score of <5 | P Value | |
| CSF | ||||||
| CCL2 | 0.36 (0.01–0.88) | 0.7 (−0.24 to 1.93) | .02 | 0.49 (0.09–0.72) | 0.78 (−0.24 to 1.93) | NS |
| CCL3 | −0.15 (−0.88 to −0.01) | −0.19 (−1.67 to 0.16) | NS | −0.27 (−0.45 to −0.11) | −0.13 (−1.67 to −0.08) | NS |
| CCL5 | −0.40 (−1.17 to 0.71) | −0.57 (−1.04 to 1.02) | NS | −0.44 (−0.71 to 0.48) | −0.22 (−1.04 to 1.02) | NS |
| CXCL10 | 1.35 (0–2.28) | 1.56 (−0.07 to 2.35) | NS | 1.37 (0.37–1.92) | 1.18 (0.41–1.96) | NS |
| ICAM | 1.48 (1.05–2.64) | 1.49 (0.79–2.69) | NS | 1.46 (1.05–2.07) | 1.44 (1.21–2.43) | NS |
| IFN-γ | −0.47 (−0.88 to 0) | −0.47 (−0.99 to 0.61) | NS | −0.53 (−0.88 to −0.11) | −0.02 (−0.99 to 0.13) | NS |
| IFN-ο | −0.39 (−0.94 to 0.41) | −0.34 (−0.76 to 0.39) | NS | −0.33 (−0.52 to 0.41) | −0.46 (−0.56 to 0.08) | NS |
| IL-1α | −0.16 (−0.88 to 0.12) | −0.16 (−0.50 to 0.06) | NS | −0.04 (−0.43 to 0.04) | −0.25 (−0.50 to 0) | NS |
| IL-1β | −0.41 (−1.10 to −0.16) | −0.51 (−0.95 to −0.11) | NS | −0.43 (−1.06 to −0.24) | −0.65 (−0.92 to −0.28) | NS |
| IL-1RA | 0.54 (0.13–1.83) | 0.63 (−0.01 to 1.67) | NS | 0.54 (0.24–1.84) | 1.43 (0.73–1.67) | NS |
| IL-4 | −0.16 (−1.73 to 0.13) | −0.22 (−1.66 to 0.18) | NS | −0.2 (−0.80 to −0.01) | −0.27 (−0.57 to 0) | NS |
| IL-8 | 0.58 (−0.03 to 1.47) | 0.55 (−0.42 to 2.03) | NS | 0.58 (−0.03 to 1.47) | 0.73 (0.17–2.03) | NS |
| IL-10 | −0.48 (−1.70 to 0.10) | −0.53 (−0.75 to 0.01) | NS | −0.44 (−0.66 to 0.36) | −0.31 (−0.63 to 0) | NS |
| MPO | 1.01 (0–1.49) | 1.02 (−0.87 to 2.00) | NS | 1.03 (0.49–1.42) | 1.08 (−0.87 to 1.62) | NS |
| TNF-α | −0.58 (−1.52 to −0.37) | −0.68 (−1.26 to −0.09) | NS | −0.55 (−1.11 to −0.37) | −0.81 (−1.00 to −0.38) | NS |
| TRAIL | 0.14 (−0.17 to 0.27) | 0.24 (−0.04 to 0.69) | NS | 0.17 (0.09–0.24) | 0.3 (−0.04 to 0.78) | NS |
| VCAM | 1.2 (−0.37 to 2.19) | 1.44 (−0.29 to 2.03) | NS | 1.43 (0.98–2.19) | 1.31 (0.88–2.03) | NS |
| IL-1α/IL1-RA | −0.3 (−1.18 to 0.25) | −0.19 (−29.57 to 14.83) | NS | −0.07 (−0.68 to 0.17) | −0.24 (−0.31 to −0.10) | NS |
| IL-1β/IL-1RA | −0.74 (−1.59 to −0.43) | −0.6 (−107.06 to 25.02) | NS | −0.82 (−1.59 to −0.57) | −0.46 (−0.61 to −0.37) | .009a |
| IL-1α/IL-10 | 0.07 (−19.03 to 1.45) | 0.27 (−17.42 to 2.05) | NS | 0.05 (−19.03 to 0.46) | 0.8 (−17.42 to 2.05) | NS |
| IL-1β/IL-10 | 0.52 (−57.23 to 1.45) | 0.91 (−43.88 to 3.65) | .039 | 0.66 (−57.23 to 1.39) | 1.42 (−43.88 to 3.65) | NS |
| Serum | ||||||
| CCL2 | 0.08 (−0.45 to 0.56) | 0.10 (−0.79 to 1.04) | NS | 0.21 (−0.28 to 0.41) | 0.14 (−0.04 to 1.30) | NS |
| CCL3 | −0.33 (−1.49 to −0.01) | −0.48 (−1.34 to −0.05) | NS | −0.35 (−0.53 to −0.23) | −0.27 (−0.91 to −0.05) | NS |
| CXCL10 | 0.48 (−0.32 to 1.72) | 0.56 (−0.17 to 1.87) | NS | 1.37 (0.13–1.72) | 0.58 (−0.04 to 1.30) | NS |
| G-CSF | −0.36 (−1.11 to 0.05) | −0.49 (−1.29 to 0.77) | NS | −0.15 (−0.45 to −0.06) | −0.42 (1.2–0.45) | NS |
| GM-CSF | −0.07 (−1.26 to 0.40) | −0.37 (−1.39 to 0.54) | NS | 0.03 (−0.70 to 0.33) | −0.06 (−1.36 to 0.54) | NS |
| Granzyme B | −0.33 (−1.30 to 0.86) | −0.37 (−1.182 to 0.32) | NS | −0.24 (−0.34 to 0.86) | −0.81 (−1.15 to 0.32) | NS |
| ICAM | 2.06 (0.78–2.85) | 2.03 (−0.30 to 4.40) | NS | 2.06 (0.78–2.39) | 1.90 (0.40–2.80) | NS |
| IFN-β | −0.38 (−2.43 to 0.28) | −1.3 (−2.55 to 0.09) | NS | −0.21 (−0.91 to 0) | −0.18 (−1.89 to −0.09) | NS |
| IFN-γ | −0.78 (−1.16 to −0.43) | −0.82 (−1.66 to 0.18) | NS | −0.70 (−1.00 to −0.59) | −0.83 (−1.56 to −0.29) | NS |
| IFN-ο | −0.28 (−0.77 to 0.88) | −0.19 (−0.71 to 0.60) | NS | 0.17 (−0.56 to 0.41) | −0.31 (−0.70 to 0.26) | NS |
| IL-1α | −0.86 (−1.69 to 0.11) | −0.86 (−1.93 to 0.06) | NS | −0.25 (−0.95 to 0.10) | −0.55 (−1.60 to −0.08) | NS |
| IL-1β | −0.95 (−1.85 to −0.36) | −1.22 (−2.34 to 0.07) | NS | −0.61 (−1.37 to −0.36) | −0.73 (−2.14 to −0.07) | NS |
| IL-1RA | 0.62 (−0.15 to 1.61) | 0.23 (−0.26 to 1.04) | .004a | 0.48 (−0.01 to 1.61) | 0.04 (−0.11 to 1.02) | NS |
| IL-4 | −0.46 (−1.27 to −0.03) | −0.56 (−1.39 to 0.19) | NS | −0.34 (−0.67 to −0.10) | −0.25 (−1.11 to 0.19) | NS |
| IL-8 | −0.13 (−1.51 to 2.21) | −0.59 (−2.47 to 1.19) | .02 | −0.07 (−0.83 to 0.67) | −0.04 (−2.12 to 1.19) | NS |
| IL-10 | −0.89 (−1.62 to 0.16) | −0.88 (−2.45 to 0.05) | NS | −0.52 (−1.26 to 0.01) | −0.91 (−2.10 to −0.33) | NS |
| Leptin | 1.00 (−0.01–2.62) | 1.08 (0.01–2.41) | NS | 0.38 (−0.01–1.01) | 1.37 (0.34–2.0) | NS |
| MPO | 1.33 (−0.04 to 2.28) | 1.01 (−0.43 to 2.89) | .02 | 1.54 (0.30–1.82) | 0.92 (0.57–2.22) | NS |
| TNF-α | −0.8 (−0.91 to −0.37) | −0.89 (−1.67 to −0.10) | NS | −0.61 (−0.91 to −0.26) | −1.02 (−1.67 to −0.10) | NS |
| TRAIL | 0.27 (−0.06 to 1.13) | 0.21 (−0.11 to 1.30) | NS | 0.4 (0.16–0.49) | 0.28 (0.02–0.73) | NS |
| VCAM | 1.75 (0.68–2.30) | 1.77 (−0.48 to 2.55) | NS | 1.9 (1.03–2.30) | 1.31 (−0.43 to 1.99) | NS |
| IL-1α/IL-1RA | −0.95 (−5.06 to 10.75) | −1.22 (−243.56 to 34.72) | NS | −0.47 (−2.18 to 1.97) | −0.62 (−56.12 to 24.51) | NS |
| IL-1β/IL-1RA | −1.39 (−9.66 to 59.58) | −2.02 (−239.23 to 59.24) | NS | −0.99 (−6.99 to 59.58) | −0.79 (−84.69 to 32.78) | NS |
| IL-1α/IL-10 | 0.78 (−37.20 to 8.61) | 0.72 (−13.43 to 8.25) | NS | 0.03 (−37.20 to 0.76) | 0.55 (0.24–1.34) | .03 |
| IL-1β/IL-10 | 0.99 (–112.50 to 7.70) | 1.02 (−25.61 to 9.29) | NS | 0.93 (−112.50 to 1.39) | 0.85 (0.22–1.25) | NS |
GOS score at discharge was available for 95 patients (100%). A GOS score of 5 denotes minimal disability, and a GOS score of <5 denotes moderate disability or death.
Abbreviations: CCL2, monocyte chemotactic protein 1; CCL3, monocyte inflammatory protein 1α; CCL5, regulated on activation normal T-cell expressed and secreted; CXCL10, inducible protein 10; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte macrophage colony-stimulating factor; ICAM, intracellular adhesion molecule; IFN-γ, interferon γ; IFN-ο, interferon ο; IL-1RA, interleukin 1 receptor antagonist; IL-1α, interleukin 1α; IL-1β, interleukin 1β; IL-4, interleukin 4; IL-8, interleukin 8; IL-10, interleukin 10; MPO, myeloperoxidase; NS, not significant; TNF-α, tumor necrosis factor α; TRAIL, tumor necrosis factor α–related apoptosis-inducing ligand; VCAM, vascular cell adhesion molecule.
a Also correlated with GOS score.
Hierarchical analysis of the data showed that the mediators clustered into three broad groups; cluster one contained IL-10 and IL-1RA, amongst other mediators, cluster two contained the chemokines CCL2, CCL3 and CXCL10, and cluster three contained IL-1α and IL-1β in addition to other mediators including the adhesion molecules VCAM and ICAM (Figure 2). There was a negative correlation between group one and group three mediators in those with a good outcome which was less apparent in those with a poor outcome, this was particularly highlighted in the extraction heat-map. This suggests that in those with a good outcome greater concentrations of the antiinflammatory mediators IL-1RA and IL-10 were associated with lower concentrations of the proinflammatory mediators, IL-1α and IL-1β, and that this relationship was less evident in those with a poor outcome.
Figure 2.
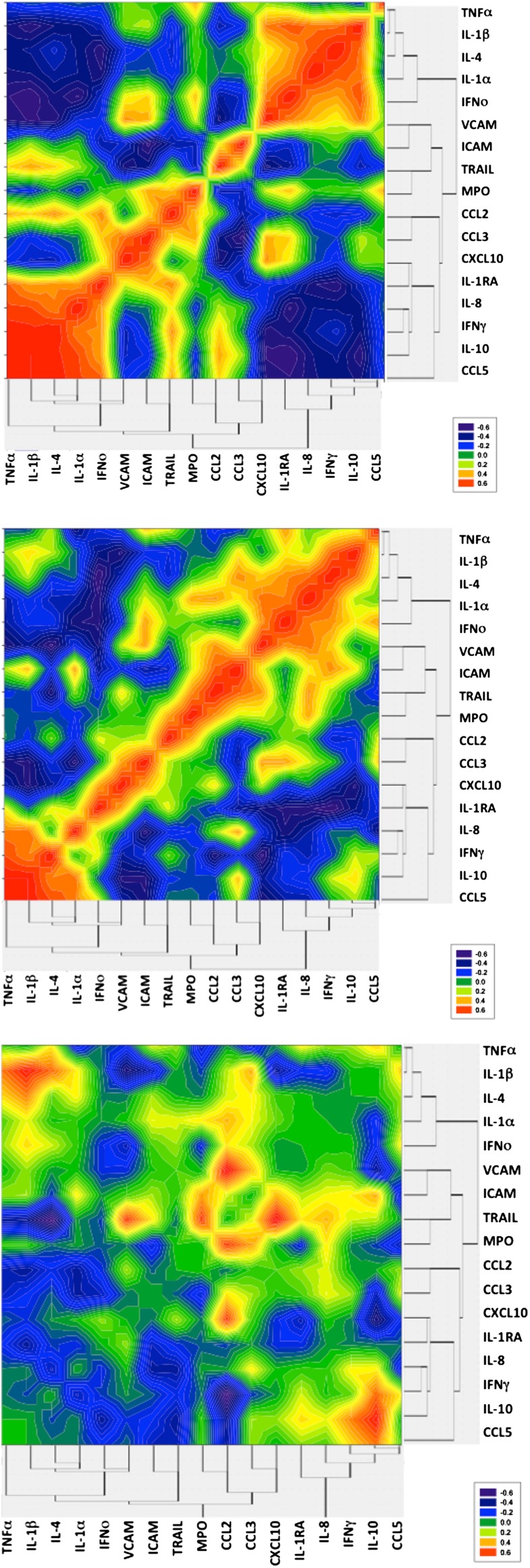
Heat map representation of mediator interaction in the cerebrospinal fluid of patients with encephalitis with a good (A) or poor (B) outcome score, as well as the subtraction heat map (C). Abbreviations: CCL2, monocyte chemotactic protein 1; CCL3, monocyte inflammatory protein 1α; CCL5, regulated on activation normal T-cell expressed and secreted; CXCL10, inducible protein 10; ICAM, intracellular adhesion molecule; IFN-γ, interferon γ; IFN-ο, interferon ο; IL-1RA, interleukin 1 receptor antagonist; IL-1α, interleukin 1α; IL-1β, interleukin 1β; IL-4, interleukin 4; IL-8, interleukin 8; IL-10, interleukin 10; MPO, myeloperoxidase; TNF-α, tumor necrosis factor α; TRAIL, tumor necrosis factor α–related apoptosis-inducing ligand; VCAM, vascular cell adhesion molecule.
Mediators Associated With BBB Permeability
Paired CSF and serum samples from 20 patients (8 with infective encephalitis, 6 with immune-mediated encephalitis, and 6 with encephalitis of unknown etiology) were available for determining the albumin ratio. For all mediators in serum and for most mediators in CSF, there was not a significant association with the ratio of CSF to serum albumin levels (Supplementary Table 3). However, the CSF mediator with the strongest negative correlation with the ratio of CSF to serum albumin levels was IL-10 (tau-b statistic, −0.49 [95% confidence interval {CI}, −.79 to −.20]; P = .0045) and that with the strongest positive correlation was vascular cell adhesion protein (VCAM; tau-b statistic, 0.50 [95% CI, 0.20–0.81]; P = .0035; Figure 3). There was no association between the ratio of CSF to serum albumin levels and the GCS or GOS scores. There was a trend toward a higher median CSF albumin concentration in those with an elevated CSF white blood cell count (250 mg/L [range, 67.5–905 mg/L] vs 149 mg/L [range 50.65–348.5 mg/L]; P = .049).
Figure 3.

The relationships between the ratio of cerebrospinal fluid (CSF) to serum albumin levels and the concentration of vascular cell adhesion molecule (VCAM; A) and interleukin 10 (IL-10) in the CSF (B) among patients with encephalitis.
Mediators Associated With Temporal Lobe Volume in Patients With HSV Encephalitis
MRI findings were available for 8 patients with HSV encephalitis; 6 underwent MRI on the first day of admission, 1 underwent MRI on day 13, and 1 underwent MRI on day 33. All images showed enlarged temporal lobes and tissue damage. Temporal lobe volume ranged between 9.5% and 13.9% of the total intracranial volume, and global tissue damage ranged from 0.4% to 11.0%. This represents a mean increase (±SD) of 8.5% ± 0.9% in temporal lobe volume in comparison to normal values in healthy controls [24]. There was good intraobserver agreement (95% limit of agreement, 5%), but interobserver agreement was less consistent (95% limit of agreement, 11%). Larger studies, which are suitably powered to address the question of the optimal number of investigators reviewing MRI findings for patients with encephalitis, are needed. There was no significant correlation between temporal lobe volume and the volume of damaged tissue. Serum mediator data were available for all cases, and CSF data were available for 2 cases.
In serum, there was a strong positive correlation between temporal lobe volume and the IL-1α level (P = .0003) and a negative correlation with the IL-1RA level (P = .012; Supplementary Table 4 and Figure 4). No other mediators in serum or CSF correlated with temporal lobe volume or damaged tissue volume. There was no correlation between either the temporal lobe or damaged tissue volumes and the GCS or GOS scores.
Figure 4.

The relationship between temporal lobe damage as a percentage of total brain volume revealed by magnetic resonance imaging and the concentration of interleukin 1α (IL-1α) and IL-1 receptor antagonist (IL-1RA) in the serum of patients with encephalitis due to herpes simplex virus.
DISCUSSION
This study found that, in a cohort of patients with encephalitis, including a subset with encephalitis due to HSV, the balance between the IL-1 level and levels of its antagonists, IL-1RA and IL-10, was associated with clinical severity, BBB permeability, and, in HSV-infected patients, the volume of temporal lobes. Specifically, the level of IL-1RA was associated with a better outcome in the cohort overall and among those with encephalitis due to HSV, as well as with reduced temporal lobe swelling in those with encephalitis due to HSV. The IL-10 level was associated with a better coma score on admission in the cohort overall and also in those with encephalitis due to HSV. Elevated levels of IL-10 were also associated with a lesser degree of BBB permeability. These may be proxy markers of clinical severity and outcome, or they may represent potential avenues for adjunctive therapy in HSV encephalitis, which may have implications for encephalitis more broadly.
Encephalitis is a devastating condition of brain parenchymal inflammation [28]. The majority of cases are due to infection, most commonly with HSV, although other cases are antibody mediated or of unknown etiology [2, 17]. The pathophysiology of the host inflammatory response is poorly understood, and current treatments are limited to antiviral therapy for HSV-associated cases and nonspecific immune suppression for antibody-mediated cases [2, 29].
Despite antiviral therapy, the mortality from HSV encephalitis is 10%–30%, and neurological sequelae are common, with <20% returning to work [1, 30]. There is mounting evidence from both animal models and clinical studies that the cytokine-mediated inflammatory response may play an important role in pathogenesis [3, 5, 14]. These mediators modulate the innate and adaptive inflammatory responses and facilitate BBB permeability, which is vital for leukocyte infiltration and edema [15, 16]. However, the key mediators in HSV encephalitis and in encephalitis in general remain unclear. The numbers of patients and mediators assessed have limited previous studies, and markers of BBB permeability and parenchymal inflammation have not been assessed. The significance of many mediators, such as IL-1, is dependent on the relative concentration of natural antagonists and has also not been assessed [4, 5, 7, 12, 31]. An improved understanding of the pathophysiology of this inflammatory response could pave the way for utilization of novel or existing adjunctive immunomodulatory therapies [32, 33].
Therefore, we analyzed CSF and serum samples from patients prospectively recruited in a multicenter study in England to determine whether mediator profiles correlated with clinical severity, with downstream markers of BBB permeability, and, in the subset with HSV encephalitis, with neuroimaging changes. This study identified a potential role for the antiinflammatory IL-1 antagonists, IL-1RA and IL-10. The serum concentration of IL-1RA was elevated in those with a better outcome score at discharge in the cohort overall, and, among patients with HSV encephalitis, the ratio of IL-1β to IL-1RA levels in CSF was raised in those with a worse outcome. Moreover, in patients with HSV encephalitis, the serum IL-1RA concentration was inversely associated with the volume of the temporal lobes on MRI, and the serum IL-1α level was associated with a greater volume. The CSF IL-10 level was elevated in patients with a higher admission GCS score and, in the subset with HSV encephalitis, a higher concentration of serum IL-10 was found in those with a higher GCS score. In addition, the IL-10 level was associated with less BBB permeability. That the IL-1RA level correlated with temporal lobe volume but not damaged tissue indicates that there may be different mechanisms underlying these changes in MRI findings. For example, because IL-10 was associated with both GCS score and BBB permeability, this may occur through a common mechanism of raised intracranial pressure, because both are reported to have a reciprocal relationship [14], whereas the mechanisms underlying the action of IL-1 may be broader.
Members of the IL-1 family, particularly IL-1β are the prototypical proinflammatory cytokines. IL-1α and IL-1β act through the IL-1 receptor 1, which is blocked by IL-1RA [10, 12]. In vitro studies of HSV infection have found marked upregulation of IL-1β predominantly by human microglia [3, 12, 31, 34]. Also, IL-1α, IL-1β, and IL-1RA have been identified in murine models of HSV encephalitis in association with cerebral edema [35]. IL-1 has an important role in upregulating many proinflammatory mediators, including adhesion molecules and chemokines, which further mediate BBB permeability [10, 12, 34]. Two previous studies have attempted to assess IL-1 in the CSF of patients with HSV encephalitis, and neither identified concentrations above the limit of detection in the majority of the 20 and 9 adults in each study [5, 36]. Although many patients in both studies received steroids, the timing of treatment was not always clear. Because steroid use and later sampling are both associated with lower levels of IL-1, this may account for these findings. Elevated concentrations of IL-10 were identified in the majority, and this did not differ between those with moderate sequelae/death and those with mild/no disability, although it was not possible to assess this relative to the IL-1 concentration or BBB permeability [5, 36].
In our study, it is not surprising that the IL-1 antagonists were found at higher concentrations and with more consistent associations than IL-1, as it is well recognized that upregulation of IL-1 in the central nervous system is early and transient and that the action is predominantly autocrine and paracrine, with a lesser spillover into the peripheral circulation [12]. As the early upregulation of IL-1 production may be pivotal, future studies require assessment of critical time frames within which any therapy may be efficacious, as has been found in murine models of glucocorticoid therapy [30, 37]. A degree of IL-1β production may be protective in HSV encephalitis in the absence of acyclovir treatment, as IL-1β knockout mice die of infection [37]. However, IL-1β has not been demonstrated to suppress infection of human astrocytes with HSV and associated viruses [3]. Exogenous IL-1RA (Anakinra) is currently contraindicated in active infection, but the risk of promoting replication among Herpesviridae may be low [38]. Interestingly, HSV has been used as a gene-therapy vector to induce IL-1RA production, ameliorating experimental autoimmune encephalomyelitis [39]. In addition, a phase 2 trial of IL-1RA demonstrated improved outcomes in patients with inflammation due to cortical infarcts [32]. Moreover, IL-1RA expression and corresponding IL-1β inhibition due to glatiramer acetate has been found to reduce central nervous system inflammation in multiple sclerosis [40]. Inhibition of IL-1 production has also been demonstrated with corticosteroids [34]. Indeed, steroids were routinely used to treat HSV encephalitis before acyclovir was established, and in one study of 45 acyclovir-treated patients, not receiving corticosteroids was associated with a poor outcome [41, 42]. Moreover, administration of adjunctive steroids in a murine HSV model reduced the severity of neuroimaging findings without increasing viral load [43].
This study also identified a potentially important role for IL-10. One previous study identified higher CSF concentrations of IL-10 in patients with HSV encephalitis, compared with noninfectious controls, although this study did not assess disease severity [4]. This potent antiinflammatory cytokine produced by glial cells and lymphocytes reduces production of proinflammatory cytokines, particularly IL-1, and promotes survival signaling, including in in vitro studies of HSV-infected human microglia [13, 44]. Also, the IL-10 level was elevated in a model of murine encephalitis due to Japanese encephalitis virus and correlated inversely with the IL-1β level and the severity of histopathological findings [8, 9]. Moreover, exogenous IL-10 also decreases cyclooxygenase-2 production, which is important for BBB permeability and for reducing neuronal death in murine models of viral encephalitis [44, 45]. In a BBB model, exogenous IL-1β has been associated with increased expression of adhesion molecules and permeability, as determined by the endothelial electrical resistance [46]. This supports our findings that the mediator with the strongest negative correlation with BBB permeability was IL-10 and that the mediator with the strongest positive correlation was VCAM. However, IL-10 did not correlate with outcome, suggesting that BBB permeability is only part of the determinant of neurological injury, which is also due to cytotoxic edema. The combined volume of vasogenic and cytotoxic edema seen on MRI was most closely associated with IL-1α and IL-1RA.
High levels of IL-10 potentially increase susceptibility to intracellular pathogens [47]. However, IL-10 treatment may reduce levels of proinflammatory cytokines and infiltrate in murine HSV keratitis without impairing viral clearance [48]. Interestingly, the protection from otherwise fatal murine HSV encephalitis demonstrated with intravenous immunoglobulin is not achieved in IL-10 knockout mice [49]. Our study did not identify any association with IL-6 or the ratio of IL-6 to IL-4 levels, and the level of IL-6 was not identified above the limits of detection in >20% of the cohort. Whilst this has been identified in previous studies of severe infection, these have been with viremia, with Japanese encephalitis virus, or a parasitemia, with malaria [14, 20]. This is pathophysiologically different from HSV encephalitis, in which brain parenchymal dysfunction follows neurotropic migration [2].
This study adds to our understanding of the complex interplay of cytokines and associated mediators in encephalitis in relation to disease severity, and it identified associations between these same mediators and BBB permeability and the degree of parenchymal swelling revealed by MRI. These may represent modifiable mediators with the potential to improve outcomes in HSV encephalitis and encephalitis more broadly and warrant further investigation in animal models, which may open the door to clinical studies.
Supplementary Data
Supplementary materials are available at http://jid.oxfordjournals.org. Consisting of data provided by the author to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the author, so questions or comments should be addressed to the author.
Notes
Acknowledgments. We thank Prof Dame Nancy Rothwell for expert advice on the research and manuscript.
Disclaimer. The views expressed are those of the authors and not necessarily those of the National Health Service, the National Institute of Health Research (NIHR), the Department of Health, or Public Health England.
Financial support. This work was supported by the laboratory team at the Vaccine Evaluation Unit, Public Health England, Manchester, United Kingdom; the NIHR, United Kingdom (doctoral research fellowship to B. D. M.); and the NIHR Health Protection Research Unit in Emerging and Zoonotic Infections, Liverpool, United Kingdom (to T. S.); and the MRC (to T. S.).
Potential conflicts of interest. All authors: No reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Whitley RJ, Alford CA, Hirsch MS et al. Vidarabine versus acyclovir therapy in herpes simplex encephalitis. N Engl J Med 1986; 314:144–9. [DOI] [PubMed] [Google Scholar]
- 2.Solomon T, Michel BD, Smith PE et al. Management of suspected viral encephalitis in adults: Association of British Neurologists and British Infection Association National Guideline. J Infect 2012; 64:347–73. [DOI] [PubMed] [Google Scholar]
- 3.Lokensgard JR, Hu S, Sheng W et al. Robust expression of TNF-alpha, IL-1beta, RANTES, and IP-10 by human microglial cells during nonproductive infection with herpes simplex virus. J Neurovirol 2001; 7:208–19. [DOI] [PubMed] [Google Scholar]
- 4.Ichiyama T, Shoki H, Takahashi Y et al. Cerebrospinal fluid levels of cytokines in non-herpetic acute limbic encephalitis: comparison with herpes simplex encephalitis. Cytokine 2008; 44:149–53. [DOI] [PubMed] [Google Scholar]
- 5.Kamei S, Taira N, Ishihara M et al. Prognostic value of cerebrospinal fluid cytokine changes in herpes simplex virus encephalitis. Cytokine 2009; 46:187–93. [DOI] [PubMed] [Google Scholar]
- 6.Wright JL, Merchant RE. Blood-brain barrier changes following intracerebral injection of human recombinant tumor necrosis factor-alpha in the rat. J Neurooncol 1994; 20:17–25. [DOI] [PubMed] [Google Scholar]
- 7.Szelenyi J. Cytokines and the central nervous system. Brain Res Bull 2001; 54:329–38. [DOI] [PubMed] [Google Scholar]
- 8.Saxena V, Mathur A, Krishnani N et al. An insufficient anti-inflammatory cytokine response in mouse brain is associated with increased tissue pathology and viral load during Japanese encephalitis virus infection. Arch Virol 2008; 153:283–92. [DOI] [PubMed] [Google Scholar]
- 9.Saxena V, Mathur A, Krishnani N et al. Kinetics of cytokine profile during intraperitoneal inoculation of Japanese encephalitis virus in BALB/c mice model. Microbes Infect 2008; 10:1210–7. [DOI] [PubMed] [Google Scholar]
- 10.Alheim K, Bartfai T. The interleukin-1 system: receptors, ligands, and ICE in the brain and their involvement in the fever response. Ann N Y Acad Sci 1998; 840:51–8. [DOI] [PubMed] [Google Scholar]
- 11.Dietrich WD, Busto R, Bethea JR et al. Postischemic hypothermia and IL-10 treatment provide long-lasting neuroprotection of CA1 hippocampus following transient global ischemia in rats. Exp Neurol 1999; 158:444–50. [DOI] [PubMed] [Google Scholar]
- 12.Boutin H, Kimber I, Rothwell NJ, Pinteaux E. The expanding interleukin-1 family and its receptors: do alternative IL-1 receptor/signaling pathways exist in the brain? Mol Neurobiol 2003; 27:239–48. [DOI] [PubMed] [Google Scholar]
- 13.Vitkovic L, Maeda S, Sternberg E et al. Anti-inflammatory cytokines: expression and action in the brain. Neuroimmunomodulation 2001; 9:295–312. [DOI] [PubMed] [Google Scholar]
- 14.Winter PM, Dung N, Loan HT et al. Proinflammatory cytokines and chemokines in humans with Japanese encephalitis. J Infect Dis 2004; 190:1618–26. [DOI] [PubMed] [Google Scholar]
- 15.Ellermann-Eriksen S. Macrophages and cytokines in the early defence against herpes simplex virus. Virol J 2005; 2:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conrady CD, Drevets DA, Carr DJ. Herpes simplex type I (HSV-1) infection of the nervous system: is an immune response a good thing? J Neuroimmunol 2010; 220:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Granerod J, Ambrose H, Davies NWS et al. Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis 2010; 10:835–44. [DOI] [PubMed] [Google Scholar]
- 18.Marcotte TD, Deutsch R, Michael BD et al. A concise panel of biomarkers identifies neurocognitive functioning changes in HIV-infected individuals. J Neuroimmune Pharmacol 2013; 8:1123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Michael BD, Elsone L, Griffiths MJ et al. Post-acute serum eosinophil and neutrophil-associated cytokine/chemokine profile can distinguish between patients with neuromyelitis optica and multiple sclerosis; and identifies potential pathophysiological mechanisms - a pilot study. Cytokine 2013; 64:90–6. [DOI] [PubMed] [Google Scholar]
- 20.Day NP, Hein TT, Schollaardt T et al. The prognostic and pathophysiologic role of pro- and antiinflammatory cytokines in severe malaria. J Infect Dis 1999; 180:1288–97. [DOI] [PubMed] [Google Scholar]
- 21.Reiber H. External quality assessment in clinical neurochemistry: survey of analysis for cerebrospinal fluid (CSF) proteins based on CSF/serum quotients. Clin Chem 1995; 41:256–63. [PubMed] [Google Scholar]
- 22.Demaerel P, Wilms G, Roberecht W et al. MRI of herpes simplex virus. Neuroradiol 1992; 34:490–3. [DOI] [PubMed] [Google Scholar]
- 23.Mackay CE, Webb JA, Eldridge PR et al. Quantitative magnetic resonance imaging in consecutive patients evaluated for surgical treatment of temporal lobe epilepsy. Magn Reson Imaging 2000; 18:1187–99. [DOI] [PubMed] [Google Scholar]
- 24.Hallan BP, Craig MC, Toal F et al. In vivo brain anatomy of adult males with Fragile X syndrome: an MRI study. Neuroimage 2011; 54:16–24. [DOI] [PubMed] [Google Scholar]
- 25.Bland JM, Altman DG. Measuring agreement in method comparison studies. Stat Methods Med Res 1999; 8:135–60. [DOI] [PubMed] [Google Scholar]
- 26.Szodoray P, Alex P, Chappell-Woodward CM et al. Circulating cytokines in Norwegian patients with psoriatic arthritis determined by a multiplex cytokine array system. Rheumatology 2007; 46:417–25. [DOI] [PubMed] [Google Scholar]
- 27.Griffiths MJ, Ooi M, Wong SC et al. In enterovirus 71 encephalitis with cardio-respiratory compromise, elevated interleukin 1beta, interleukin 1 receptor antagonist, and granulocyte colony-stimulating factor levels are markers of poor prognosis. J Infect Dis 2012; 206:881–92. [DOI] [PubMed] [Google Scholar]
- 28.Kennedy PG. Viral encephalitis. J Neurol 2005; 252:268–72. [DOI] [PubMed] [Google Scholar]
- 29.Tunkel AR, Glase CA, Bloch KC et al. The management of encephalitis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis 2008; 47:303–27. [DOI] [PubMed] [Google Scholar]
- 30.Raschilas F, Wolff M, Delatour F et al. Outcome of and prognostic factors for herpes simplex encephalitis in adult patients: results of a multicenter study. Clin Infect Dis 2002; 35:254–60. [DOI] [PubMed] [Google Scholar]
- 31.Rosler A, Pohl M, Braune HJ et al. Time course of chemokines in the cerebrospinal fluid and serum during herpes simplex type 1 encephalitis. J Neurol Sci 1998; 157:82–9. [DOI] [PubMed] [Google Scholar]
- 32.Emsley HC, Smith C, Georgiou RF et al. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry 2005; 76:1366–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burton JM, O'Connor PW, Hohol M et al. Oral versus intravenous steroids for treatment of relapses in multiple sclerosis. Cochrane Database Syst Rev 2012; 12:CD006921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gibson RM, Rothwell N, Le Feuvre RA. CNS injury: the role of the cytokine IL-1. Vet J 2004; 168:230–7. [DOI] [PubMed] [Google Scholar]
- 35.Tse MC, Lane C, Mot K et al. ICAM-5 modulates cytokine/chemokine production in the CNS during the course of herpes simplex virus type 1 infection. J Neuroimmunol 2009; 213:12–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aurelius E, Ansersson B, Forsgren M et al. Cytokines and other markers of intrathecal immune response in patients with herpes simplex encephalitis. J Infect Dis 1994; 170:678–81. [DOI] [PubMed] [Google Scholar]
- 37.Sergerie Y, Rivest S, Boivin G et al. Tumor necrosis factor-alpha and interleukin-1 beta play a critical role in the resistance against lethal herpes simplex virus encephalitis. J Infect Dis 2007; 196:853–60. [DOI] [PubMed] [Google Scholar]
- 38.Salliot C, Dougados M, Gossec L et al. Risk of serious infections during rituximab, abatacept and anakinra treatments for rheumatoid arthritis: meta-analyses of randomised placebo-controlled trials. Ann Rheum Dis 2009; 68:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Furlan R, Bergami A, Brambilla E et al. HSV-1-mediated IL-1 receptor antagonist gene therapy ameliorates MOG(35–55)-induced experimental autoimmune encephalomyelitis in C57BL/6 mice. Gene Ther 2007; 14:93–8. [DOI] [PubMed] [Google Scholar]
- 40.Burger D, Molnarfi N, Weber MS et al. Glatiramer acetate increases IL-1 receptor antagonist but decreases T cell-induced IL-1beta in human monocytes and multiple sclerosis. Proc Natl Acad Sci U S A 2009; 106:4355–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Upton AR, Foster JB, Barwick DD. Dexamethasone treatment in herpes-simplex encephalitis. Lancet 1971; 1:861. [DOI] [PubMed] [Google Scholar]
- 42.Kamei S, Sekizawa T, Shiota H et al. Evaluation of combination therapy using aci- clovir and corticosteroid in adult patients with herpes simplex virus encephalitis. J Neurol Neurosurg Psychiatry 2005; 76:1544–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyding-Lamade UK, Oberlinner C, Rau PR et al. Experimental herpes simplex virus encephalitis: a combination therapy of acyclovir and glucocorticoids reduces long-term magnetic resonance imaging abnormalities. J Neurovirol 2003; 9:118–25. [DOI] [PubMed] [Google Scholar]
- 44.Marques CP, Hu S, Sheng W, Cheeran MC et al. Interleukin-10 attenuates production of HSV-induced inflammatory mediators by human microglia. Glia 2004; 47:358–66. [DOI] [PubMed] [Google Scholar]
- 45.Molina-Holgado E, Arevalo-Martin A, Ortiz S et al. Theiler's virus infection induces the expression of cyclooxygenase-2 in murine astrocytes: inhibition by the anti-inflammatory cytokines interleukin-4 and interleukin-10. Neurosci Lett 2002; 324:237–41. [DOI] [PubMed] [Google Scholar]
- 46.Labus J, Hackel S, Lucka L et al. Interleukin-1beta induces an inflammatory response and the breakdown of the endothelial cell layer in an improved human THBMEC-based in vitro blood-brain model. J Neurosci Methods 2014; 228:35–45. [DOI] [PubMed] [Google Scholar]
- 47.Wilson EH, Wille-Reece U, Dzierszinski F et al. A critical role for IL-10 in limiting inflammation during toxoplasmic encephalitis. J Neuroimmunol 2005; 165:63–74. [DOI] [PubMed] [Google Scholar]
- 48.Tumpey TM, Elner VM, Chen SH et al. Interleukin-10 treatment can suppress stromal keratitis induced by herpes simplex virus type 1. J Immunol 1994; 153:2258–65. [PubMed] [Google Scholar]
- 49.Ramakrishna C, Newo A, Shen YW et al. Passively administered pooled human immunoglobulins exert IL-10 dependent anti-inflammatory effects that protect against fatal HSV encephalitis. PLoS Pathog 2011; 7:1002071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.