

Abstract
Background
Jumping translocations (JT) are uncommon cytogenetic abnormalities involving non-reciprocal translocations of a single donor chromosome onto two or more chromosomes. The clinical characteristics and prognosis of JTs in patients with myeloid malignancies are not well described.
Materials and Methods
We searched our cytogenetic database from 2003 to 2014 to identify cases of myeloid malignancies associated with a JT. These cases were cross-referenced with our clinical databases to determine patient characteristics, response to treatment and overall survival.
Results
We identified 10 patients with myeloid malignancies and a JT: 4 cases of acute myeloid leukemia (AML) with MDS-related changes, 4 cases of myelodysplastic syndrome (MDS) and 2 cases of post-polycythemia myelofibrosis. The donor segment was derived from chromosome 1 in every case. The acquisition of a JT was a late ocurrence, with a median time to JT development of 24.9 months (range 0-248 months) from diagnosis. The overall response to treatment was poor, with no patients achieving a response to conventional chemotherapy or hypomethylating agents. The median overall survival for the group was 9 months (95% CI 2.5-15.5) after identification of a JT.
Conclusion
The acquisition of a JT in patients with myeloid malignancies appears to be a late event and is associated with myelodysplasia. In our series, this was associated with a poor prognosis with patients having a poor response to treatment, disease transformation to AML and short overall survival.
Keywords: Jumping translocations, myeloid neoplasms, leukemia, myelodysplastic syndromes, prognosis
Introduction
Jumping translocations (JT) are non-reciprocal chromosomal translocations of a single donor chromosome or chromosome fragment onto two or more recipient chromosomes 1. This was first identified in Prader-Willi Syndrome in the 1970s but has subsequently been described as an acquired cytogenetic change in several human cancers 2. Jumping translocations are uncommon but have been recurrently observed in solid and hematologic cancers, including several cases involving myeloid malignancies 1, 3-6. Jumping translocations involving chromosome 1 as the donor segment are the most frequently described, although the reason for this predilection is unknown. Several other chromosomes including 3, 11, 15, and 21 have also been reported to act as a donor chromosome 1, 3, 5, 6. Case reports and small series of JTs in myeloid malignancies report poor outcomes; however the implications of this unusual cytogenetic event are poorly understood. We report on a series of patients with myeloid neoplasms and JT with the goal of determining the clinicopathologic characteristics and response to treatment.
Methods
We searched our cytogenetic database for “jumping translocation” from the 2003 to 2014 to identify cases of JTs within our institution. This database was cross-referenced with a clinical database containing patient characteristics, treatments and outcomes. Disease subtypes were classified according to 2008 WHO classification 7. Response to treatment for acute myeloid leukemia (AML) was determined according to IWG 2003 criteria, response for myelodysplastic syndrome (MDS) was determined according to IWG 2006 criteria and response for polycythemia vera (PV) and myelofibrosis (MF) according to IWG 2013 criteria 8-11. Overall survival was calculated from the time of the first acquisition of a JT using the Kaplan-Meier method and data was censored at death or the last follow-up. Conventional chromosomal analysis was performed on G-banded metaphase cells from unstimulated 24-hour and 48-hour bone marrow aspirate cultures using standard techniques. Twenty metaphases were analyzed when possible and the results were reported using the international System for Human Cytogenetic Nomenclature (ISCN 2013)12.
Results
Baseline clinical characteristics and prior therapy
We identified 10 patients with myeloid neoplasms with a JT in our database from 2003 to 2014 (tables 1-3). The database contained approximately 20,000 individual patient cases with myeloid neoplasms during this time period. At the time of JT identification, 4 patients had acute myeloid leukemia (AML), 4 patients had myelodysplastic syndrome (MDS) and 2 patients had post-polycythemic myelofibrosis (post PV-MF). Of the 4 patients with AML all were classified as AML with myelodysplasia-related changes (AML-MRC), and 2 had a preceding diagnosis of MDS. Both patients with post PV-MF had the JAK2 V617F mutation. Only one patient had a JT identified on cytogenetic analysis at the time of their initial diagnostic bone marrow exam. The median time from initial diagnosis to identification of a JT was 24.9 months (range 0-248 months).
Table 1.
Clinical features and characteristics of jumping translocations.
| Pt. | Disease | Age/Sex | Jumping translocation | ||
|---|---|---|---|---|---|
|
| |||||
| Interval* (months) | Donor chromosome | Recipient chromosome | |||
| 1 | AML-MRC | 63/F | 1.4 | 1p22 | 3p21, 5q13, 11p15 |
|
| |||||
| 2 | AML-MRC | 62/F | 18.2 | 1q25 | 5q35, 6p25 17q36 |
|
| |||||
| 3 | AML-MRC | 82/F | 48.7 | 1q10 | 13q10, 14q10, 15q10 |
| 49.6 | 1q10 | 15q10, 22q10 | |||
| 52.4 | 1q10 | 13q10, 15q10, 22q10 | |||
| 53.8 | 1q10 | 15q10, 22q10 | |||
| 54.7 | 1q10 | 13q10, 15q10, 22q10 | |||
|
| |||||
| 4 | AML-MRC | 44/M | 12.7 | 1q21 | Yp11.2, 6p25, 7q22, 10q22, 12q15 |
|
| |||||
| 5 | MDS-RCMD | 75/M | 23.0 | 1q11 | Yq11.23, 12p11.2, 13p11.2, 16q11.1 |
| 26.1 | 1q11 | Yq11.23, 13p11.2, 16q11.2, 18q23 | |||
|
| |||||
| 6 | Therapy-related MDS | 65/F | 27.4 | 1q21 | 3p25, 13q34 |
| 29.4 | 1q21 | 3p25, 6q26, 7q26, 13q34, 14p11.2, 18q22 | |||
| 30.9 | 1q21 | 3p25, 13q34, 14p11.2 | |||
| 32.0 | 1q21 | 3q25, 13q34, 14p11.2 | |||
|
| |||||
| 7 | MDS-RCMD | 58/F | 26.8 | 1p11 | 5p11, 13q11, 14p11, 18q21, 21p11.2, 22p11.1 |
| 32.7 | 1p11 | 11q23, 14p11.2, 18q21.1, 21p11.2 | |||
|
| |||||
| 8 | MDS-RCMD | 64/M | 0 | 1q10/1q21 | 1p10, 19q23 |
| 2.6 | 1q10 | 7p10, 14q10 | |||
| 4.7 | 1q10 | 7p10, 14q10 | |||
| 9.2 | 1q10 | 7p10 | |||
| 11.3 | 1q10 | 7p10, 14q10, 19p10 | |||
|
| |||||
| 9 | post-PV MF | 45/M | 65.2 | 1q11 | 7p10, 9p10, 15p10 |
|
| |||||
| 10 | post-PV MF | 67/F | 248 | 1q11 | 8p23, 17p13 |
| 259 | 1q11 | 8p23, 13p11.1, 15p11.1,17p12, 20p11.2 | |||
Interval: time from diagnosis to detection of jumping translocations.
AML-MRC: acute myeloid leukemia with myelodysplasia related changes; MDS: myelodysplastic syndromes; MF: myelofibrosis; PV: polycythemia vera; RCMD – refractory cytopenia with multilineage dysplasia
Table 3.
Individual treatments and response to therapy.
| Pt. | Disease classification before JT (IPSS)* | Treatment before JT identified (# cycles)+ | Best response | Disease classification at JT | Treatment after JT detection (# cycles) | Best response | Survival (months) after JT detection (Alive - Y/N) |
|---|---|---|---|---|---|---|---|
| 1 | AML | Ida + Ara-C + Pravastatin (1) | Morph CR | AML | none | -- | 1.8 (N) |
| No CyCR | |||||||
|
| |||||||
| 2 | AML | [1] Ida + Ara-C (2) | RD | AML | 5-AZA + Valproic Acid (1) | RD | 1.2 (N) |
| [2] High dose Ara-C (2) | RD | ||||||
| [3] Ida + Ara-C + VP16 (1) | RD | ||||||
| [4] Pixantrone (2) | RD | ||||||
|
| |||||||
| 3 | MDS-RAEB2 (1.5) | [1] 5-AZA (12) | SD | AML | [1] MK-3475 (5.5) | RD | 7.9 (Y) |
| [2] 5-AZA + Sorafenib (30) | CR | [2] FF-10501 [inosine monophosphate dehydrogenase inhibitor] (3) | RD | ||||
| [3] MK-3475* [anti-PD1 antibody] (2.5) | RD | ||||||
|
| |||||||
| 4 | MDS-RAEB2 (2) | Clofarabine + Ida + Ara-C (4) | Morph CR | AML | [1] Cladribine + Ida + Ara-C (1) | [1] PR | 4.6 (Y) |
| [2] Allogeneic SCT | [2] CR/CyCR | ||||||
|
| |||||||
| 5 | MDS-RCMD (0.5) | [1] EPO (3 months) | HI –ER, PlR | MDS-RCMD | SGI-110 (hypomethylating agent) (1) | unknown | 11.2 (N) |
| [2] oral 5-AZA (6) | SD/no HI | ||||||
| [3] ARRY-614 (p38/Tie2 inhibitor) (12) | HI – NeR, PlR | ||||||
|
| |||||||
| 6 | MDS-RAEB1 (1) | [1] Decitabine (14) | CR | MDS-RAEB2 | Fludarabine + Ara-C (2) | DP to AML | 5.5 (N) |
| [2] Clofarabine + Ara-C (9) | CR | ||||||
|
| |||||||
| 7 | MDS-RCMD (0) | [1] EPO (1 month) | No HI | MDS-RCMD | [1] 5-AZA (11) | [1]SD/no HI | 9.0 (N) (died in remission) |
| [2] Lenalidomide (6) | SD/no HI | [2] Allogeneic SCT | [2] CR/CyCR | ||||
|
| |||||||
| 8 | MDS-RCMD (1) | none | -- | MDS-RCMD | 5-AZA + Vorinostat (7) | DP | 13.7 (Y) |
|
| |||||||
| 9 | PV | [1] Hydroxyurea (70) | PR | post PV-MF | Hydroxyurea (1) | unknown | 0.0 (Y) |
|
| |||||||
| 10 | PV, post PV-MF | [1] EPO (6 months) | SD | post PV-MF | Obatoclax (BCL-2 inhibitor) (3) | DP to AML | 12.2 (N) |
| [2] Interferon-α (8 years) | CR | ||||||
International prognostic scoring system (IPSS) at the time of MDS diagnosis.
The # of cycles refers to 28-day cycles unless otherwise stated.
Abbreviations: 5-AZA: 5-azacitidine; AML-MRC: AML with myelodysplasia related changes; CR: complete remission; CyCR: cytogenetic complete response; DP: disease progression; EPO: erythropoietin; ER: erythroid response; HI: hematologic improvement; Ida: Idarubicin; JT: jumping translocation; morph CR: morphologic remission; NeR: neutrophil response; PlR: platelet response; PR: partial remission; RAEB2: refractory anemia with excess blasts- 2; RCMD: refractory cytopenia with multilineage dysplasia; RD: resistant disease; SCT: stem cell transplant; SD: stable disease.
The individual treatments and corresponding responses prior to the identification of the JT are shown in table 3. The median number of therapies prior to identification of a JT was 2 (range 0-4), with only one patient not receiving treatment. Treatments prior to the development of a JT were diverse and included cytotoxic chemotherapy, hypomethylating agents, immunomodulatory drugs, targeted small molecule inhibitors and growth factors. The two patients with AML (patient 1, 2) prior to the identification of the JT had poorly responsive disease. Patient 1 was treated with a regimen containing Ara-C and idarubicin and achieved a morphologic CR on a post-induction marrow, yet cytogenetics on that marrow demonstrated acquisition of JT, and the patient relapsed and died shortly thereafter with no additional therapy. Patient 2 was treated with 4 different regimens without response prior to developing a JT. The 6 patients with a preceding diagnosis of MDS (patient 3-8) had a more variable response to therapy prior to identification of a JT, with 4 of these patients achieving a CR, PR or hematologic improvement with therapy. Both patients with post-PV MF had experienced long (>5 years), relatively indolent disease courses with responses to treatment prior to identification of a JT.
Cytogenetic characteristics
The donor and recipient chromosomes involved the jumping translocation are described for each case in table 1 and the karyotype at diagnosis and for the jumping translocation is show in table 2. In all JTs identified, the donor segment was derived from chromosome 1. Donor chromosome breakpoints were identified at: 1q10 (n=2), 1q11 (n=3), 1q21 (n=2), 1q25 (n=1), 1p11 (n=1), 1p22 (n=1). Besides the JTs, additional cytogenetic abnormalities were present in 8 cases (table 2). The additional cytogenetic abnormalities, including del(5q), del(7q), and -17, are generally considered to confer an intermediate or adverse prognosis in the MDS and AML cases even in the absence of a JT 13, 14.
Table 2.
Karyotype at diagnosis and karyotype with jumping translocations.
| Pt. | Karyotype at diagnosis | Karyotype with jumping translocation |
|---|---|---|
| 1 | 44-48,XX,inv(3)(p25q13.1), del(5)(q13q31),+8,+13[cp20] | 46,XX,del(5)(q13q33)[2]/46,XX,t(1;3)(p22;q21)[1]/46,XX,t(1;5)(p22;q13)[1]/46,XX,der(11)t(1;11)(p22;p15)[1]/46,XX[15] |
| 2 | 46,XX[25] | 46,XX,der(6)t(1;6)(q25;p25)[10]/47,XX,del(2)(q32),der(5)t(1;5)(q25;q35),+mar[1]/46,XX,der(17)t(1;17)(q25;q36)[1]/46,XX[3] |
| 3 | 46,XX[20] | 46,XX,+1,der(1;22)(q10;q10)[3]/46,XX,+1,der(1;13)(q10;q10)[1]/46,XX,+1,der(1;15)(q10;q10)[1]/46,XX[15] |
| 4 | 46,XY[20] | 46,XY,der(7)t(1;7)(q21;q22)[8]/46,XY,der(6)t(1;6)(q21;p25)[2]/46,XY,der(10)t(1;10)(q21;q22)[2]/46,X,der(Y)t(Y;1)(p11.2;q21)[1]/46,XY,der(12)t(1;12)(q21;q15)[1]/46,XY,del(11)(q21q23)[1]/46,XY[5] |
| 5 | 46,XY[20] | 46,X,der(Y)t(Y;1)(q11.23;q11)[2]/46,XY,der(13)t(1;13)(q11;p11.2)[2]/46,XY,der(18)t(1;18)(q11;q23)[2]/45∼46,XY,der(Y)t(Y;1)(q11.23;q11),der(16)t(1;16)(q11;q11.2)[cp4]/46,XY[10] |
| 6 | 46,XX,t(10;21)(q26;q21)[1]/46,XX, del(6)(q13),del(7)(q32),add(13)(q34)[1]/46,XX[5] | 46,XX,t(3;10;21)(q27;q25;q22),der(3)t(1;3)(q21;q25)[5]/46,idem,der(14)t(1;14)(q21;p11.2)[4]/46,idem,der(13)t(1;13)(q21;q34)[3]/46,idem,del(7)(p12)[8] |
| 7 | 46,XX[20] | 46,XX,+1,psu dic(21;1)(p11.2;p11)[7]/46,XX,+1,psu dic(11;1)(q23;p11)[5]/46,XX,+1,psu dic(18;1)(q21.1;p11)[4]/46,XX,+1,psu dic(14;1)(p11.2;p11)[1]/46,XX,del(6)(q13),-9,+mar[1]/44,XX,del(7)(p21),-13,-22[1]/46,XX[1] |
| 8 | 46,XY,+1,der(1;7)(q10;p10)[3]/48,sl,+9,+ 21[14]/46,XY,der(19)t(1;?;19)(q21;?;q13 .3)[2]/46,XY[1] | 46,XY,+1,der(1;7)(q10;p10)[9]/46,XY,+1,der(1;19)(q10;p10)[5]/46,XY,+1,der(1;14)(q10;q10)[4]/47,XY,+1,der(1;7)(q10;p10),+9[1]/46,XY[1] |
| 9 | 47,XY,+9[20] | 47,XY,+9[8]/47,idem,+1,der(1;7)(q10;p10)[3]/47,XY,+der(1;9)(q10;p10)[6]/47,XY,+1,der(1;15)(q10;p10),+9[2]/46,XY[1] |
| 10 | 46,XX[20] | 47,XX,+8,der(17)t(1;17)(q11;p13),del(20)(q13.1)[14]/48,XX,+8,der(15)t(1;15)(q11;p11.1),+der(15)t(1;15)(q11;p11.1),del(20)(q13.1)[2]/47,XX,dup(1)(q11q42),+8,del(20)(q13.1)[1]/47,XX,+der(8)t(1;8)(q11;p23),d/47,XX,+8,der(13)t(1;13)(q11;p11.1),del(20)(q13.1)[1]/47,XX,+8,del(20)(q13.1),der(20)t(1;20)(q11;p11.2)[1] |
After identification, 6 patients had a JT that was observed again at a subsequent bone marrow aspiration, and the median time that the JT persisted between serial samples was 6 months (range 4.7 to 10.8 months). Four patients had a JT identified only on a single bone marrow sample: two patients died shortly after the bone marrow showing a JT (patient 1, 2), one patient had disappearance of the JT with treatment (patient 4) and another patient is alive but has not had another bone marrow aspirate performed (patient 9). Disappearance of the JT translocation was observed in 2 patients; one patient with MDS (patient 7) was treated with high-dose therapy and allogeneic transplant and the other with AML (patient 4) was treated with clofarabine, idarubicin and Ara-C followed by allogeneic transplant. Figure 1 shows a sample chromosomal karyotype for case 4.
Figure 1.
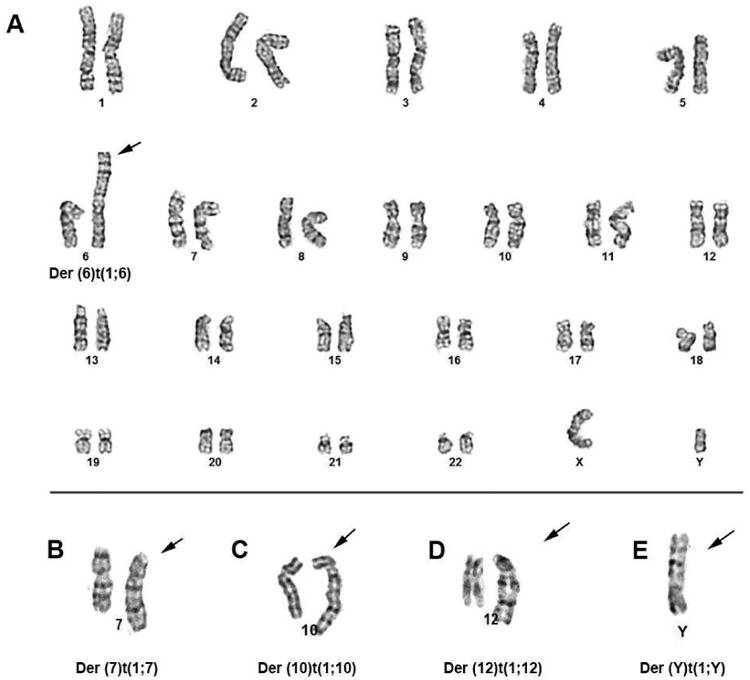
Case #4. A: karyotype, 46,XY,der(6)t(1;6)(q11;p25); B – E: derivative chromosomes of 7, 10, 12, Y from jumping translocation of t(1;v). For more details see Table 1 and 2.
Response to treatment
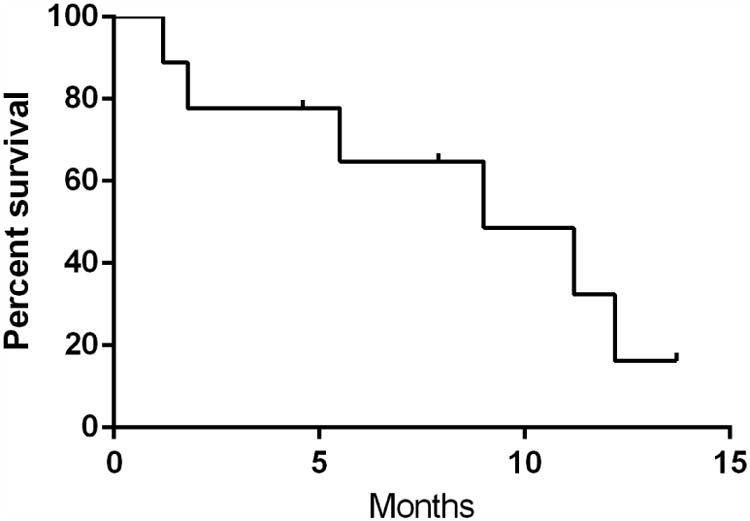
Nine patients received disease-directed treatment after identification of a JT, which is described in table 3. One patient (patient 1) with AML had a jumping translocation identified after induction chemotherapy and relapsed and died prior to subsequent treatment. Treatments were heterogeneous and six patients were enrolled on clinical trials involving investigational therapy. Overall, the response to treatment after identification of a JT was poor. None of the four patients with AML achieved a complete remission (CR) with treatment. One patient with AML achieved a partial remission (PR: >50% reduction in blasts) and received an allogeneic stem cell transplant and is currently alive in remission approximately 3 months after transplant. Three patients with MDS received therapy with hypomethylating agents (azacitidine, n=2; SGI 110, n=1). One of these patients (patient 5) received only one cycle of therapy and did not have another bone marrow exam, although he died related to MDS 11 months later. Neither of the other 2 patients with MDS (patients 7, 8) had a hematologic response, bone marrow morphologic response or cytogenetic response according to IWG 2006 criteria 8. One of these patients (patient 7) later received high-dose therapy with lenalidomide, melphalan and fludarabine followed by allogeneic cord-blood transplant, which was given as part of a clinical trial. This patient had a bone marrow aspirate performed on day 34 after transplant which showed donor engraftment without clear evidence of MDS, although the patient died on day 73 from complications related to graft vs. host disease. One patient with MDS-RAEB2 (patient 6) received treatment with fludarabine and Ara-C and then progressed to AML and subsequently died. One patient with post-PV MF (patient 10) progressed to AML while being treated with an experimental BCL-2 inhibitor. The other patient with post-PV MF (patient 9) was recently diagnosed as having progressed to myelofibrosis and is alive and continuing treatment with hydroxyurea. The median overall survival after identification of a JT was 9 months (95% CI 2.5-15.5 months) (figure 2).
Figure 2. Survival after identification of jumping translocation.
Discussion
The occurrence of jumping translocations in hematologic malignancies is rare and the clinicopathologic characteristics and the response to therapy in these patients are poorly defined. Our series of patients provides important insights into this unusual genetic aberration. While evidently rare, the incidence of JTs may be under-estimated in our series as cases that were not explicitly reported as a JT would not have been identified. In our JT series, chromosome 1 was the only donor chromosome identified, and this has been most frequently reported in other cases of myeloid malignancy associated with a JT 1, 3, 15. Chromosome 1 is the largest human chromosome and alterations involving chromosome 1 are frequently found in many types of cancer 16. The donor breakpoints we identified frequently involved areas of centromeric DNA (1q10, 1q11, 1p11) 17. Another donor breakpoint, 1q21, is an area that encompasses several genes but also contains a large area of repetitive DNA, which may predispose to crossover events 16. Chromosomes 3 and 11 have also been identified as donor chromosomes in previous reports of myeloid neoplasms associated with JT 3, 5, 6. The tendency for chromosome 1 to act as a donor chromosome in these events is poorly understood, but this bias suggests a predisposing mechanism or survival advantage associated with this change. It has been reported that the gain or amplification of the cyclin-dependent kinases regulatory subunit 1B (CKS1B) gene located on 1q21 can override the DNA damage response barrier, promote tumor development and is associated with an adverse prognosis in multiple myeloma 18, 19.
Chromosome 1 also contains large areas of constitutive heterochromatin which may predispose to centromeric instability and fusion events 16. Furthermore, DNA excision repair at areas of heterochromatin on chromosome 1 has been reported to be inefficient relative to other areas 20. Sawyer et al have reported that JTs involving chromosome 1 are associated with decondensation of pericentric DNA 21. They propose a model suggesting that decondensation may lead to partial endoreplication of the 1q segment resulting in a triradial chromosome 1, which predisposes to cross-over events or translocation of one chromatid to an area of telomeric DNA on another chromosome. Shortening of telomeres on recipient chromosomes has been reported in some cases of JT which may promote fusion with a donor chromosome 15, 22, 23, although this is not universally described 24.
Three patients (patients 1, 6, 9) in our series had a common cytogenetic abnormality present at diagnosis and at the time of identification of a JT. This finding suggests that, for these patients, the acquisition of JTs occurred in one of the original clones. For example, patient 1 initially had del(5)(q13q33) and this abnormality persisted 1.4 months later when a JT was identified. Interestingly, chromosome 5 also acted as a recipient chromosome and the location of the breakpoint was identical to site of the deletion. Patient 9, had an additional chromosome 9 present at diagnosis and when a JT was identified, and this chromosome also acted as a recipient chromosome. The subsequent involvement of the abnormal/extra chromosome as a recipient chromosome in these cases suggests these abnormalities were unstable. Patient 6 initially had t(10;21)(q26;q21) and later had t(3;10;21) (q27;q25;q22) present at the time a JT was identified, also suggesting an evolutionary relationship between these clones.
Six patients initially had a diploid karyotype. Serial molecular testing was not performed in these patients so somatic mutations could not be used to determine the relationship between the original and later clones for any case. Three patients with an initial diploid karyotype acquired other cytogenetic abnormalities prior to acquiring a JT. Patient 2 had cytogenetic analysis performed on a bone marrow aspirate 10 weeks prior to the acquisition of a JT that showed a deletion of chromosome 1 (del(1)(p13)). In this case, it seems plausible that this deletion could represent a transition stage, occurring prior to replication and fusion of the deleted segment onto the recipient chromosomes. Patient 3 and 10 were initially diploid and acquired del(6)(q13q23) and del(20)(q11.2) respectively, although neither of these chromosomes were later involved in a JT. Patient 8 was the only patient that had a JT identified at the time of diagnosis, with a translocation identified between chromosome 1 and chromosome 7 as well as a translocation involving chromosome 1, an unknown chromosome and chromosome 19. Interestingly, the breakpoint of chromosome 1 differed for these two translocations (1q10 and 1q21), although only the 1q10 fragment was involved in subsequent JTs. As well, the translocation between chromosome 1 and 19 was lost on the subsequent karyotype but recurred on a karyotype 11 months after diagnosis, although the unknown chromosome fragment was not involved in this later translocation.
The clinical characteristics of our cohort reinforce the available evidence that JTs are strongly associated with myelodysplasia. Our series of 10 patients included 4 patients with MDS and 4 patients with AML; the 4 patients with AML either had preceding MDS or a diagnosis of AML with MDS related changes. One patient with post-PV MF also had significant dysplasia in all myeloid lineages, and was classified as having a probable overlapping diagnosis of MDS and post-PV MF on several bone marrow examinations. Of note, the median time to identification of a JT was over 2 years from the date of diagnosis, which suggests that this is a late event during the disease course. In contrast, a recent study reporting on the acquisition of other cytogenetic abnormalities in MDS reported a median time from diagnosis to development of an additional abnormality of 8 months (range 1-77 months) (Jabbour, AJH, 2014). However, two patients (patient 1, patient 8) in our series developed a JT shortly after diagnosis (<3 months) and interestingly both had complex cytogenetic abnormalities at diagnosis. Our findings are consistent with a previous study by Najfeld et al on a series of MDS (n=16) and MPN (n=7) patients that also reported that the development of a JT appears to be a late event 4. They reported 8 patients (MDS=5, MPN=3) had a JT identified at the time of diagnosis and the remainder evolved over time. The median time to development of JT for the cohort that evolved was 34 and 47 months in patients with MDS (n=11) and MPN (n=4) respectively. The late acquisition of a JT suggests the development of chromosomal instability and dysfunctional DNA repair over time related to disease progression and/or to treatment.
Most of our patients had received treatment prior to the development of a JT, and some cases of JTs may have originated from DNA breakage occurring during chemotherapy and treatments affecting DNA repair. No predisposing sensitivity to DNA damage such as a diagnosis of Fanconi's anemia or dyskeratosis congenita was identified in any patient, although no definitive conclusions about this relationship can be ascertained in this analysis due to the small sample size, heterogeneous nature of treatments, and retrospective nature of this review. Nagai et al (2010) have previously reported on a case of a persistent JT that was present in an AML patient after achieving remission, suggesting the abnormality could be related to prior therapy 25. The patient's initial karyotype was 46, X, −X,+10 (17/20 cells) and she attained a first CR with anthracycline, Ara-C and etoposide therapy. She relapsed 3 years later and was treated with a similar regimen again achieving CR. Her first bone marrow to reveal a JT was 2 years after achieving the 2nd CR and the karyotype showed a translocation between 1q21 and 7q36. Subsequent bone marrow aspirates showed further translocations of 1q21 with chromosome 13 and the X chromosome. The JT persisted for 15 years on repeat bone marrow examinations despite remaining in remission with no evidence of therapy related MDS. The authors of this case speculated that the JT occurred in non-leukemic cells and might be therapy-related.
Most importantly, our results suggest that patients with myeloid neoplasms associated with a JT have poor responses to chemotherapy and shortened survival. None of the patients we identified had a durable response to conventional chemotherapy or hypomethylating agents, although 2 patients achieved remission with intensive chemotherapy and allogeneic transplant however the follow-up period for these patients is short. Furthermore, JTs appear to be associated with a high risk for transformation to AML with 2 patients having a JT present at the time of transformation and 2 patients progressing to AML shortly after identification. This increased risk has been similarly described in another cohort of patients with MDS and MPN associated with a JT, where approximately 25% of patients transformed to AML at a median of 8-9 months from identification 4. Similarly, Jabbour et al report that the acquisition of other cytogenetic abnormalities in lower-risk MDS is associated with a higher risk of transformation to AML (RR 2.6, 95% CI 1.5-4.5) and a median overall survival of only 17 months 26. Although our group is heterogeneous, the median overall survival is very low and appears to be most similar to patients with AML and a complex karyotype or very high risk MDS (IPSS-R) where the median survival is estimated to be less than 1 year 13, 14. While we cannot determine the independent prognostic value of JTs due to the relatively small and heterogeneous nature of our study, our findings suggest an overall poor prognosis associated with JT acquisition, in terms of treatment resistance, disease progression and short overall survival. Future studies examining the prognostic impact of cytogenetic abnormalities in AML and MDS should consider studying jumping translocations as distinct entities to further elucidate their independent prognostic value.
Conclusion
Our findings indicate that JTs are rare events in myeloid malignancies and that chromosome 1 is most frequently observed as the donor chromosome. In our series, the acquisition of a JT occurred late in the disease course and appears to be associated with the finding of dysplasia on bone marrow morphology. Most patients were resistant to treatment with chemotherapy after identification and the overall survival was short, suggesting JTs are associated with a poor prognosis.
Clinical Practice Point.
The acquisition of a jumping translocation in myeloid malignancies may indicate a poor prognosis with increased risk of transformation to AML and resistance to chemotherapy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berger R, Bernard OA. Jumping translocations. Genes, chromosomes & cancer. 2007;46:717–723. doi: 10.1002/gcc.20456. [DOI] [PubMed] [Google Scholar]
- 2.Lejeune JMC, Prieur M, Van den Akker J. Translocation sauteuse (5p;15q), (8q;15q), (12q;15q) Ann Géné t. 1979;22:210–213. [PubMed] [Google Scholar]
- 3.Bernard M, Lemee F, Picard F, et al. Jumping translocation in acute leukemia of myelomonocytic lineage: a case report and review of the literature. Leukemia. 2000;14:119–122. doi: 10.1038/sj.leu.2401637. [DOI] [PubMed] [Google Scholar]
- 4.Najfeld V, Tripodi J, Scalise A, et al. Jumping translocations of the long arms of chromosome 1 in myeloid malignancies is associated with a high risk of transformation to acute myeloid leukaemia. British journal of haematology. 2010;151:288–291. doi: 10.1111/j.1365-2141.2010.08355.x. [DOI] [PubMed] [Google Scholar]
- 5.Zou YS, Ouahchi K, Lu Y, et al. Jumping translocations of 3q21 in an acute monocytic leukemia (M5) patient reveal mechanisms of multistage telomere shortening in pathogenesis of AML. Leukemia research. 2012;36:e31–33. doi: 10.1016/j.leukres.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Fan YS, Rizkalla K, William BF, Engel CJ. Jumping translocations of 11q in acute myeloid leukemia and 1q in follicular lymphoma. Cancer genetics and cytogenetics. 2000;118:35–41. doi: 10.1016/s0165-4608(99)00149-1. [DOI] [PubMed] [Google Scholar]
- 7.Swerdlow SH, C E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008. [Google Scholar]
- 8.Cheson BD, Greenberg PL, Bennett JM, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108:419–425. doi: 10.1182/blood-2005-10-4149. [DOI] [PubMed] [Google Scholar]
- 9.Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2003;21:4642–4649. doi: 10.1200/JCO.2003.04.036. [DOI] [PubMed] [Google Scholar]
- 10.Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood. 2013;121:4778–4781. doi: 10.1182/blood-2013-01-478891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tefferi A, Cervantes F, Mesa R, et al. Revised response criteria for myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European LeukemiaNet (ELN) consensus report. Blood. 2013;122:1395–1398. doi: 10.1182/blood-2013-03-488098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaffer L, McGowan-Jordan J, Schmid M. International system for human cytogenetic nomenclature (ISCN 2013) Recommendations of the international standing commitee on human cytogenetic nomenclature. Karger; Basel: 2013. [Google Scholar]
- 13.Grimwade D, Walker H, Oliver F, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood. 1998;92:2322–2333. [PubMed] [Google Scholar]
- 14.Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–2465. doi: 10.1182/blood-2012-03-420489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manola KN, Georgakakos VN, Stavropoulou C, et al. Jumping translocations in hematological malignancies: a cytogenetic study of five cases. Cancer genetics and cytogenetics. 2008;187:85–94. doi: 10.1016/j.cancergencyto.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 16.Gregory SG, Barlow KF, McLay KE, et al. The DNA sequence and biological annotation of human chromosome 1. Nature. 2006;441:315–321. doi: 10.1038/nature04727. [DOI] [PubMed] [Google Scholar]
- 17.NCBI. NCBI Homo sapiens Annotation Release 106 Chromosome 1. 20142014 [Google Scholar]
- 18.Chang H, Jiang N, Jiang H, et al. CKS1B nuclear expression is inversely correlated with p27Kip1 expression and is predictive of an adverse survival in patients with multiple myeloma. Haematologica. 2010;95:1542–1547. doi: 10.3324/haematol.2010.022210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liberal V, Martinsson-Ahlzen HS, Liberal J, et al. Cyclin-dependent kinase subunit (Cks) 1 or Cks2 overexpression overrides the DNA damage response barrier triggered by activated oncoproteins. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2754–2759. doi: 10.1073/pnas.1102434108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Surralles J, Darroudi F, Natarajan AT. Low level of DNA repair in human chromosome 1 heterochromatin. Genes, chromosomes & cancer. 1997;20:173–184. [PubMed] [Google Scholar]
- 21.Sawyer JR, Tricot G, Mattox S, Jagannath S, Barlogie B. Jumping translocations of chromosome 1q in multiple myeloma: evidence for a mechanism involving decondensation of pericentromeric heterochromatin. Blood. 1998;91:1732–1741. [PubMed] [Google Scholar]
- 22.Wan TS, Ma SK, Chow EY, Li YH, Lin SY, Chan LC. Pathogenesis of jumping translocations: a molecular cytogenetics study. Leukemia research. 2004;28:1075–1079. doi: 10.1016/j.leukres.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 23.Hatakeyama S, Fujita K, Mori H, Omine M, Ishikawa F. Shortened telomeres involved in a case with a jumping translocation at 1q21. Blood. 1998;91:1514–1519. [PubMed] [Google Scholar]
- 24.Hatakeyama S, Osawa M, Omine M, Ishikawa F. JTB: a novel membrane protein gene at 1q21 rearranged in a jumping translocation. Oncogene. 1999;18:2085–2090. doi: 10.1038/sj.onc.1202510. [DOI] [PubMed] [Google Scholar]
- 25.Nagai S, Nannya Y, Takahashi T, Kurokawa M. Jumping translocation involving 1q21 during long-term complete remission of acute myeloid leukemia. Annals of hematology. 2010;89:741–742. doi: 10.1007/s00277-009-0855-y. [DOI] [PubMed] [Google Scholar]
- 26.Jabbour E, Takahashi K, Wang X, et al. Acquisition of cytogenetic abnormalities in patients with IPSS defined lower-risk myelodysplastic syndrome is associated with poor prognosis and transformation to acute myelogenous leukemia. American journal of hematology. 2013;88:831–837. doi: 10.1002/ajh.23513. [DOI] [PMC free article] [PubMed] [Google Scholar]