

Abstract
Introduction:
Genome-wide association studies linking the α3, β4, and α5 nicotinic acetylcholine receptor (nAChR) subunits to nicotine dependence suggest that α3β4* nAChR may be targets for smoking cessation pharmacotherapies. We previously reported that AT-1001, a selective α3β4* nAChR ligand binds with high affinity to rat α3β4 and human α3β4α5 nAChR, antagonizes epibatidine-induced activation of rat α3β4 nAChR in HEK cells and potently inhibits nicotine self-administration in rats.
Methods:
Two-electrode voltage clamp was used for functional characterization of AT-1001 at recombinant human α3β4 and α4β2 nAChR expressed in Xenopus oocytes.
Results:
Concentration-response curves show that AT-1001 is a partial agonist at human α3β4 nAChR, evoking up to 35% of the maximal acetylcholine (ACh) response (50% effective concentration [EC50] = 0.37 μM). AT-1001 showed very little agonist activity at the α4β2 nAChR, evoking only 6% of the ACh response (EC50 = 1.5 μM). Pre- and co-application of various concentrations of AT-1001 with 50 μM ACh revealed a complex pattern of activation-inhibition by AT-1001 at α3β4 nAChR, which was best fitted by a 2-site equation. At α4β2 nAChR, co-exposure of AT-1001 with ACh only showed inhibition of ACh current with a shallower curve.
Conclusions:
AT-1001 is a partial agonist at the human α3β4 nAChR and causes desensitization at concentrations at which it evokes an inward current, resulting in an overall functional antagonism of α3β4 nAChR. AT-1001 does not significantly activate or desensitize α4β2 nAChR at the same concentrations as at the α3β4 nAChR, but does inhibit ACh responses at α4β2 nAChR at higher concentrations. A combination of these mechanisms may underlie the inhibition of nicotine self-administration by AT-1001, suggesting that AT-1001 and compounds from this class may have clinical potential for smoking cessation pharmacotherapy.
Introduction
Nicotine mediates its addictive effects by interacting with the nicotinic acetylcholine receptors (nAChRs) in the brain, for which the endogenous neurotransmitter is acetylcholine (ACh). The predominant nAChR subtypes in the brain are the homomeric α7 nAChR and the heteromeric nAChR subtype containing the α4 and β2 subunits. Both the α4 and β2 subunits have been definitively shown to be involved in the reinforcing properties of nicotine1,2 and the α4β2 nAChR subtype has been shown to be involved in mesolimbic dopamine release leading to the rewarding effects of nicotine.3–6 Indeed, the clinical success of varenicline, a partial agonist at α4β2 nAChR, as a smoking cessation aid provides validation that targeting neuronal nAChRs is an effective approach to combat nicotine addiction. However, recent advances in the genetic and pharmacological studies of nicotine dependence show that additional nAChR subtypes are contributing to the reinforcing and aversive effects of nicotine. Most prominent among these are the human genetic association studies showing that single nucleotide polymorphisms in the gene cluster CHRNA5/Α3/Β4, encoding for the α3, α5, and β4 nAChR subunits, are closely associated with the risk for heavy smoking, inability to quit, and increased sensitivity to nicotine.7–11 Further, studies in knockout mice show that the β4 nAChR subunit is necessary for nicotine withdrawal because withdrawal is greatly diminished in β4 null mice, but not in β2 null mice.12,13
Unlike the wide distribution of α4β2 nAChR in the brain, the α3 and β4 subunits are expressed in a restricted number of brain areas, mainly the medial habenula and interpeduncular nucleus, major cholinergic tracts in the brain that have recently garnered increasing attention for their involvement in various aspects of nicotine dependence.14–17 Consistent with the genetic and knockout studies, a transgenic mouse model overexpressing the human CHRNA5/Α3/Β4 genomic cluster showed significantly increased β4* nAChR binding in the medial habenula and increased acquisition of nicotine self-administration.18
We recently reported that a potent and selective α3β4 nAChR ligand AT-1001, significantly blocks nicotine self-administration in rats, at relatively low doses (0.75–3mg/kg) given subcutaneously, without affecting nonspecific food responding.19 AT-1001 has nanomolar binding affinity for the rat α3β4 nAChR and also for the human α3β4α5 nAChR transfected into HEK cells.20 In functional assays at the rat α3β4 nAChR, using epibatidine as the agonist, AT-1001 potently antagonized epibatidine-induced current in a reversible manner.19 Here, we report an electrophysiological characterization of the functional profile of AT-1001 at the human α3β4 nAChR expressed in Xenopus oocytes, using the endogenous ligand ACh as the agonist, and compare its effects versus the human α4β2 nAChR. The present results show that AT-1001 is a partial agonist at the human α3β4 nAChR and causes desensitization at the same concentrations at which it activates the α3β4 nAChR. Sustained exposure to AT-1001 inhibits ACh-induced current and function at the α3β4 nAChR. These studies enable a better understanding of the pharmacological effects of AT-1001 in inhibiting the effects of nicotine and its mechanism as a potential smoking cessation medication.
Materials and methods
Experiments were conducted at human nAChRs expressed in Xenopus laevis oocytes, as described previously.21Briefly, human α3β4 and α4β2 nAChRs were expressed by injecting equal amounts of complementary DNAs encoding α3 and β4 (for α3β4) and α4 and β2 (for α4β2) nAChR, respectively. Currents evoked by ACh or AT-1001 were recorded at room temperature with a holding potential at –80 mV, using a standard two-electrode voltage clamp (HiClamp, Multichannel Systems, Inc.) and analyzed with Matlab (Mathworks Inc.).
To assess agonist properties, oocytes transfected with human α3β4 or α4β2 nAChR were challenged at 2-min intervals with increasing concentrations of AT-1001 for 5 s. An ACh pulse (1,000 μM, 5 s) was applied at the beginning and at the end of each experiment to evaluate the maximal response of the cell to ACh and to normalize the evoked currents. To determine the agonist properties without desensitization, cells expressing α3β4 nAChR were first challenged with a 1,000-μM ACh test pulse, and 5min later, with the highest concentration (100 μM) of AT-1001, followed after a 5-min recovery, by another pulse of 1,000 μM ACh.
To assess antagonist properties, oocytes transfected with human α3β4 or α4β2 nAChR were pre-exposed for 45 s to increasing concentrations of AT-1001 (0.1nM, 1nM, 10nM, 100nM, 1 μM, 10 μM, and 100 μM) and then for 10 s with 50-μM ACh in presence of AT-1001. ACh and AT-1001 were then washed off for 15 s, and the oocyte exposed again to the same AT-1001 concentration for 45 s. In this experimental protocol, the oocyte was exposed once every 2min to a brief pulse of ACh. No desensitization of the receptor was observed with ACh itself with these conditions, as seen from the stable ACh response observed for the lowest concentrations of compound (Figure 3A). This protocol causes cumulative exposure to the compound.
Figure 3.

Effect of sustained exposure to AT-1001 at the human α3β4 and α4β2 nicotinic acetylcholine receptors (nAChRs). (A and C) Current evoked by 50-μM acetylcholine (ACh) test pulses recorded in absence (gray line) or presence of AT-1001 at different concentrations using an experimental protocol that results in a sustained exposure to AT-1001. In this protocol, the cell is first exposed for 45 s to a steady concentration of AT-1001 (0.0001 μM, 0.001 μM, 0.01 μM, 0.1 μM, 1 μM, 10 μM, and 100 μM) and the response evoked by a brief ACh test pulse (50 μM, 10 s) is recorded in presence of the same concentration of compound. ACh and compound are rinsed for 15 s before returning into the same concentration of AT-1001. (A) Note the marked activation of the receptors caused by pre-application of AT-1001 as low as 0.1 μM and current increase observed at 1 μM. (C) Note the small but significant activation of the α4β2 receptors caused by pre-application of AT-1001 at 1 μM and the progressive decline of the ACh responses as a function of the AT-1001 concentration. (B and D) Plot of the peak current evoked by ACh as a function of the concentration of AT-1001 (n = 5–12 for α3β4 and n = 4 for α4β2). (B) The continuous line through the datapoints is the best fit obtained with a two-site equation for α3β4 with f = 0.77, ACh = 50 μM, KA = 225 μM, IC50 = 0.61 μM, and KB = 0.5 μM. (D) Continuous line through the datapoints is the best fit obtained with a Hill equation for α4β2 with an IC50 = 0.77±0.27 μM and nH = 0.27±0.01.
Peak inward currents were plotted as a function of the logarithm of the agonist concentration to yield concentration-response curves that are readily fitted by single Hill equations for the α4β2 nAChR, from which pharmacological parameters were derived. A two-site equation, similar to that initially proposed by Cachelin and Rust,22 and adapted by Smulders et al23 was used for the α3β4 nAChR, to take into account a possible open-channel blockade caused by the compound at this nAChR subtype.
| (1) |
Where y is the fraction of the evoked current, f is an arbitrary factor, , B is the concentration of antagonist, KB is the antagonist affinity, cA is , A is the agonist concentration, KA is the agonist affinity, and IC50 is the 50% inhibitory concentration.
Compounds
All chemicals were analytic grade and purchased from Sigma. AT-1001 HCl (N-(2-bromophenyl)-9-methyl-9-azabicyclo[3.3.1] nonan-3-amine hydrochloride)19 was synthesized at Astraea Therapeutics. AT-1001 was prepared as a 10-mM stock solution in dimethyl sulfoxide and diluted in the recording medium on the day of the experiment to obtain the desired test concentration. Acetylcholine HCl (Sigma) was prepared as a 100-mM stock solution in water and diluted to the desired test concentration.
Results
Agonist Properties of AT-1001 at the Human α3β4 and α4β2 nAChR
The results obtained from an activation protocol at human α3β4 and α4β2 nAChR are shown in Figure 1A and C, respectively. Brief exposure to AT-1001 elicited currents at the α3β4 nAChR whose amplitude varied as a function of the concentration of the compound. The response time course and the relative amplitude of the peak current compared to that obtained with 1,000 μM ACh indicate that AT-1001 is a partial agonist with an EC50 = 0.37 μM and evokes at maximum, 35% (n = 4, 10 μM AT-1001) of the maximal ACh-evoked current. The concentration-response curve for AT-1001 reaches a maximum and then progressively declines (Figure 1B). Inhibition of the reference ACh response, together with the shorter response time course observed at higher concentrations of AT-1001, and the rebound observed upon compound removal suggests that this molecule might act also as an open-channel blocker.
Figure 1.

Activation of human α3β4 and α4β2 nicotinic acetylcholine receptor (nAChR) expressed in Xenopus oocytes by AT-1001. (A and C) Typical currents recorded in oocytes expressing the human α3β4 and α4β2 nAChR in response to brief pulses of acetylcholine (ACh) and AT-1001. Oocytes were challenged at 2-min intervals with increasing concentrations of AT-1001 (0.01nM, 0.1nM, 1nM, 30nM, 0.1 μM, 1 μM, 10 μM, and 100 μM) for 5 s. A reference ACh pulse (1,000 μM, 5 s) was applied at the beginning (black square) and at the end (open circle) of each experiment to evaluate the maximal response of the cell to ACh and to normalize the evoked currents. Data were normalized to unity versus the first ACh-evoked response. (B and D) Dose-response concentration activation curves showing a plot of the peak inward current as a function of the logarithm of AT-1001 concentration (n = 4 for α3β4 and n = 4 for α4β2). Filled square indicates the ACh response normalized to unity, whereas the empty circle indicates the amplitude of the ACh-evoked current observed after compound exposure. The differences in fraction of ACh-evoked currents observed between the two receptor subtypes could be due to differences in receptor expression. A continuous line through the datapoints is the best fit obtained with the Hill equation, yielding an EC50 of 0.37±0.1 μM and a Hill coefficient of 2.1±0.29 with a scaling factor of 0.35±0.05 (n = 4) for α3β4 nAChR. Results obtained at α4β2 yielded an EC50 of 1.5±0.2 μM, a Hill coefficient of 1.26±0.03 with a scaling factor of 0.06 (n = 3).
To better evaluate the agonist activity of AT-1001 in naive oocytes, cells were exposed only once to AT-1001. In this experimental protocol (Figure 2), the response of the cell to 1mM of ACh was first measured and, after a 5-min wash, the cell was exposed to one pulse of AT-1001 (100 μM) followed by another ACh test pulse (1mM) applied after 5-min recovery. As shown in Figure 2, the amplitude of current evoked by 100 μM of AT-1001 reaches only 23% of the current evoked by ACh and the subsequent ACh response was profoundly reduced. These data confirm that AT-1001 elicits only a partial agonist response at α3β4 nAChR and causes inhibition of the subsequent responses, possibly by a combination of desensitization and/or open-channel blockade.
Figure 2.
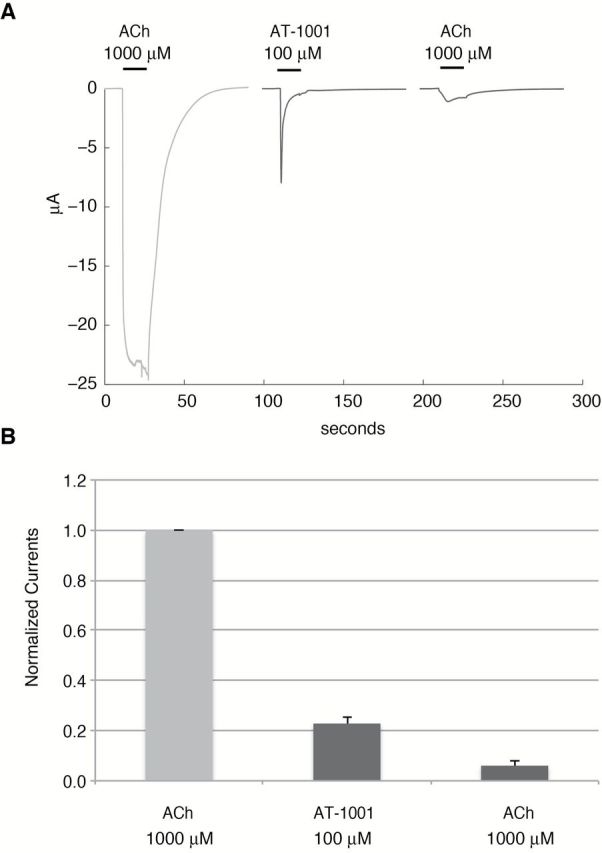
AT-1001 a partial agonist at the human α3β4 receptors. (A and B) (A) Typical currents evoked by a brief pulse (10 s) of acetylcholine (ACh), AT-1001, and ACh were recorded in cell at 5-min intervals, which is sufficient to allow full recovery of the response under control conditions. As seen from the trace, 100 μM AT-1001 evokes only a fraction of the ACh current and causes inhibition of the subsequent ACh response. (B) Histograms illustrating the response, normalized to unity versus the current evoked by ACh. Bars indicate the SEM obtained for eight cells.
At the human α4β2 nAChR, AT-1001 revealed a different profile as shown in Figure 1C and D. AT-1001 only minimally activates the human α4β2 nAChR, evoking a maximum current approximately 6% of the ACh response, with a lower potency (EC50 = 1.5 μM) than at the human α3β4 nAChR. The smaller amplitude of the ACh-evoked currents observed at the α4β2 nAChR (Figure 1C) compared with that at α3β4 nAChR (Figure 1A) is attributed to a lower level of receptor expression. Although the EC50 of AT-1001 at human α4β2 nAChR is only 4-fold lower than at human α3β4 nAChR, the much lower maximal stimulation at the human α4β2 nAChR suggests that AT-1001 exerts minimal activation of the human α4β2 nAChR and that its partial agonist activity is selective for the α3β4 nAChR.
Antagonist Properties of AT-1001 at the Human α3β4 and α4β2 nAChR
Inhibition experiments using a pre- and co-application protocol of increasing concentrations of AT-1001 with ACh revealed that AT-1001 interacts in a complex manner with human α3β4 nAChR. Typical currents observed during this protocol are shown in Figure 3A, and the dose-response inhibition curve shown in Figure 3B. As seen in Figure 3A, the low concentrations of AT-1001 (0.1–10nM) show no effect on the ACh response (first gray trace, Figure 3A). However, there is a marked inhibition of α3β4 nAChR for AT-1001 concentrations at and above 1 μM, and complete suppression of the ACh-evoked currents for concentrations 10 μM or above. But AT-1001 concentrations ranging from 100 to 1,000nM appear to activate the human α3β4 nAChR, as seen by the enhancement of the ACh response at these concentrations (Figure 3A). Moreover, AT-1001 causes an initial potentiation of the ACh-evoked currents in a narrow concentration range (0.1–1 μM), and subsequently, potently and steeply inhibits the ACh-evoked response at the α3β4 nAChR at concentrations above 1 μM, with a 50% inhibition concentration between 0.3 and 1 μM. These data show that AT-1001 causes two distinct actions at the human α3β4 nAChR, with partial activation at lower concentrations and inhibition of the ACh response at higher concentrations.
To better visualize the inhibition experimental protocol and its outcome at the α3β4 nAChR, responses recorded for three concentrations of AT-1001 are superimposed and represented with a higher resolution timescale in Figure 4. The 0.01-μM AT-1001 does not evoke an inward current and does not affect the current evoked by 50-μM ACh (green trace). However, exposure to 0.1 and 1 μM of AT-1001 evokes a significant and sustained inward current. The progressive decline of the inward current observed at 1 μM is indicative of a progressive desensitization of the receptors by AT-1001. These data clearly illustrate the potentiation and inhibition of the ACh-evoked current observed at 1 μM in Figure 3A that is used to evaluate the fraction of active receptors and is fitted to a two-site equation (Figure 3B).
Figure 4.

Experimental protocol used for the determination of sustained exposure to AT-1001. Traces recorded in presence of three concentrations of AT-1001 are superimposed. As described for Figure 3, the cell is first exposed for 45 s to a steady concentration of AT-1001 (here 0.01, 0.1, and 1 μM) and current recorded. The response evoked by a brief acetylcholine (ACh) test pulse (50 μM, 10 s) is then recorded in presence of the same concentration of compound. The bars above the current traces indicate timing of AT-1001 and ACh exposures. ACh and compound are rinsed for 15 s before returning into the same concentration of AT-1001. Arrows indicate the measurement of current amplitude for these three concentrations. Note that returning into 1 μM of AT-1001 causes an inward current corresponding to the activation of the α3β4 at this concentration. The small amplitude of the current during the second exposure is attributed to receptor desensitization, as shown by the dashed line that is in continuation of the response evoked by the first exposure (at the 1 μM of AT-1001).
At the α4β2 nAChR, however, AT-1001 acts as an antagonist (Figure 3C and D), showing a shallow concentration-inhibition curve (IC50 = 0.8 μM) and almost complete inhibition at higher concentrations up to 100 μM (Figures 3C and D).
Discussion
AT-1001 belongs to a series of small-molecule nAChR ligands designed as selective ligands for the α3β4 nAChR,24 for which no selective ligands were previously reported. AT-1001 has single-digit nanomolar affinity at the α3β4 nAChR and potently inhibits nicotine self-administration in rat.19 AT-1001 has been shown to have equally high binding affinity for the human α3α5β4 nAChR transfected into HEK cells in radioligand displacement assays using [3H]-epibatidine.20 In electrophysiological assays at rat α3β4 nAChR using epibatidine as the agonist, AT-1001 antagonized epibatidine-induced current at 10nM, consistent with its inhibition of epibatidine-induced calcium flux at the rat α3β4 nAChR.19 To assess the functional activity and selectivity of AT-1001 at human nAChRs, we determined the functional profile of AT-1001 at human α3β4 and α4β2 nAChRs expressed in Xenopus oocytes. In these experiments, we used the endogenous ligand ACh as the agonist, instead of epibatidine, especially because epibatidine is known to evoke responses with a slower onset and offset, as well as a smaller amplitude than ACh-evoked responses at the α4β2 nAChRs.25
The functional characterization reported here shows that AT-1001 is a low-efficacy partial agonist at the human α3β4 nAChR with an EC50 of 0.37 μM and a 35% agonist efficacy at 10 μM compared with ACh. To avoid the possible reduction of the response by cumulative exposure to AT-1001, effects of a single test pulse at 100 μM AT-1001 were determined and compared with ACh. As shown in Figure 2, the amplitude of the response reached only 23% of the ACh-evoked current and exhibited a fast desensitization profile during agonist exposure. These data confirmed that AT-1001 acts as a partial agonist at the α3β4 receptors. The marked reduction of the ACh-evoked current observed after exposure to AT-1001 concentrations of 1 μM and above, suggests that at high concentrations AT-1001 causes desensitization or acts as an open-channel blocker of the α3β4 nAChR. Inhibition experiments with relatively short-lasting drug challenges showed that at concentrations above 1 μM, AT-1001 fully antagonizes the ACh response at α3β4 nAChRs, but interestingly, at concentrations between 0.1 and 1 μM, caused potentiation of the ACh-evoked response (Figure 4). Similar effects were previously reported for cholinergic drugs at α4β2 nAChRs.23,26 The AT-1001 concentration-inhibition curve at α3β4 nAChR (Figure 3B) therefore has a complex shape that is better fitted by a two-site equation (Equation 1) that was initially developed to take into account the bell-shaped inhibition curve observed with some antagonists at the α3β4 nAChR.22 This equation was subsequently elaborated further to take into account the possibility that the test compound might also act as an open-channel blocker.23
The present data also allow further insight into the functional selectivity of AT-1001 for the human α3β4 nAChR versus the human α4β2 nAChR. AT-1001 binds with about 100-fold higher affinity to rat α3β4 and human α3β4α5 nAChR than to rat α4β2 nAChRs.19,20 The current functional studies at the human nAChR show that although AT-1001 is only about 4-fold more potent as a partial agonist at human α3β4 nAChR (EC50 = 0.37 μM) than at human α4β2 nAChRs (EC50 = 1.5 μM), its very low intrinsic activity (6%) at the human α4β2 nAChR indicates that AT-1001 does not significantly activate the human α4β2 nAChR and is thus a functionally selective α3β4 nAChR partial agonist at the human nAChR.
Given that AT-1001 shows a low, but measurable level of activation of the human α3β4 nAChR at lower doses, its complex effect on ACh-induced current in the antagonist protocol (Figure 3A) might result from the combined activation and desensitization of the receptor. Although it could be argued that the 45-s exposure time designed in the experimental protocol is insufficient to reach full equilibrium, it should be noted that for each concentration, the cell is exposed for 95 s and that the overall cumulative protocol lasts up to 30min. Nonetheless, these data clearly indicate that AT-1001 causes two distinct actions at the human α3β4 nAChR, activation at lower doses and inhibition/desensitization at doses 1 μM and above.
These data have implications for the mechanism that could underlie the effect of AT-1001 on nicotine self-administration. Instead of acting as a full antagonist without any agonist effect,19 AT-1001 likely partially activates the α3β4 nAChR. But because AT-1001 causes desensitization at nearly the same concentrations as it activates the α3β4 nAChR, the predominant effect of AT-1001 at α3β4 nAChRs is a reduction in α3β4 nAChR function. Therefore, the inhibition of nicotine self-administration by AT-1001 is possibly mediated two mechanisms: partial agonism and desensitization, resulting in an overall functional antagonism of α3β4 nAChR. Given the binding selectivity of AT-1001 for the α3β4 versus the α4β2 nAChR,19 it is likely, that at low doses, the interactions with α3β4 nAChRs represent the main mechanisms through which AT-1001 inhibits nicotine self-administration in rats. Nonetheless, it is possible that effects at α4β2 nAChRs could play a role in the in vivo activity and that AT-1001 might also inhibit, but not activate, a small proportion of α4β2 nAChRs.
In this respect, it is interesting to compare the possible mechanisms of the α3β4 partial agonist AT-1001 with that of the α4β2 partial agonist varenicline. Varenicline mainly inhibits the functional activity of its main target, the α4β2 nAChR, while activating only a minor fraction,27 while desensitization of a small fraction of an off-target receptor, the α3β4* nAChR, may contribute to its effect.28 AT-1001 appears to represent an intriguing “opposite” mechanism, in that its main effect is inhibition/desensitization of the α3β4* nAChR, with a possible contribution by inhibition of a minor fraction of α4β2* nAChR. With both α3β4* and α4β2* nAChR involved in nicotine reward and withdrawal, AT-1001 and related compounds seem promising candidates for nicotine addiction pharmacotherapy.
Funding
This work was supported by the National Institutes of Health (R43DA033744 to NZ).
Declaration of Interests
None declared.
Acknowledgments
We thank Hans Rollema (Rollema Biomedical Consulting) for his insightful comments and guidance on this manuscript.
References
- 1. Picciotto MR, Zoli M, Rimondini R, et al. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–177. [DOI] [PubMed] [Google Scholar]
- 2. Tapper AR, McKinney SL, Nashmi R, et al. Nicotine activation of alpha4* receptors: sufficient for reward, tolerance, and sensitization. Science. 2004;306:1029–1032. [DOI] [PubMed] [Google Scholar]
- 3. Corrigall WA, Franklin KB, Coen KM, Clarke PB. The mesolimbic dopaminergic system is implicated in the reinforcing effects of nicotine. Psychopharmacology. 1992;107:285–289. [DOI] [PubMed] [Google Scholar]
- 4. Maskos U, Molles BE, Pons S, et al. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature. 2005;436:103–107. [DOI] [PubMed] [Google Scholar]
- 5. McCallum SE, Parameswaran N, Bordia T, Fan H, McIntosh JM, Quik M. Differential regulation of mesolimbic alpha 3/alpha 6 beta 2 and alpha 4 beta 2 nicotinic acetylcholine receptor sites and function after long-term oral nicotine to monkeys. J Pharmacol Exp Ther. 2006;318:381–388. [DOI] [PubMed] [Google Scholar]
- 6. Graupner M, Maex R, Gutkin B. Endogenous cholinergic inputs and local circuit mechanisms govern the phasic mesolimbic dopamine response to nicotine. PLoS Comput Biol. 2013;9:e1003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weiss RB, Baker TB, Cannon DS, et al. A candidate gene approach identifies the CHRNA5-A3-B4 region as a risk factor for age-dependent nicotine addiction. PLoS Genet. 2008;4:e1000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berrettini W, Yuan X, Tozzi F, et al. Alpha-5/alpha-3 nicotinic receptor subunit alleles increase risk for heavy smoking. Mol Psychiatry. 2008;13:368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saccone NL, Wang JC, Breslau N, et al. The CHRNA5-CHRNA3-CHRNB4 nicotinic receptor subunit gene cluster affects risk for nicotine dependence in African-Americans and in European-Americans. Cancer Res. 2009;69:6848–6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Caporaso N, Gu F, Chatterjee N, et al. Genome-wide and candidate gene association study of cigarette smoking behaviors. PLoS One. 2009;4:e4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schlaepfer IR, Hoft NR, Collins AC, et al. The CHRNA5/A3/B4 gene cluster variability as an important determinant of early alcohol and tobacco initiation in young adults. Biol Psychiatry. 2008;63:1039–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Salas R, Pieri F, De Biasi M. Decreased signs of nicotine withdrawal in mice null for the beta4 nicotinic acetylcholine receptor subunit. J Neurosci. 2004;24:10035–10039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stoker AK, Olivier B, Markou A. Role of α7- and β4-containing nicotinic acetylcholine receptors in the affective and somatic aspects of nicotine withdrawal: studies in knockout mice. Behav Genet. 2012;42:423–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grady SR, Moretti M, Zoli M, et al. Rodent habenulo-interpeduncular pathway expresses a large variety of uncommon nAChR subtypes, but only the alpha3beta4* and alpha3beta3beta4* subtypes mediate acetylcholine release. J Neurosci. 2009;29:2272–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frahm S, Slimak MA, Ferrarese L, et al. Aversion to nicotine is regulated by the balanced activity of β4 and α5 nicotinic receptor subunits in the medial habenula. Neuron. 2011;70:522–535. [DOI] [PubMed] [Google Scholar]
- 16. Fowler CD, Lu Q, Johnson PM, Marks MJ, Kenny PJ. Habenular α5 nicotinic receptor subunit signalling controls nicotine intake. Nature. 2011;471:597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Görlich A, Antolin-Fontes B, Ables JL, et al. Reexposure to nicotine during withdrawal increases the pacemaking activity of cholinergic habenular neurons. Proc Natl Acad Sci USA. 2013;110:17077–17082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gallego X, Molas S, Amador-Arjona A, et al. Overexpression of the CHRNA5/A3/B4 genomic cluster in mice increases the sensitivity to nicotine and modifies its reinforcing effects. Amino Acids. 2012;43:897–909. [DOI] [PubMed] [Google Scholar]
- 19. Toll L, Zaveri NT, Polgar WE, et al. AT-1001: a high affinity and selective α3β4 nicotinic acetylcholine receptor antagonist blocks nicotine self-administration in rats. Neuropsychopharmacology. 2012;37:1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu J, Perry DC, Bupp JE, et al. [¹²⁵I]AT-1012, a new high affinity radioligand for the α3β4 nicotinic acetylcholine receptors. Neuropharmacology. 2014;77:193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hogg RC, Bandelier F, Benoit A, Dosch R, Bertrand D. An automated system for intracellular and intranuclear injection. J Neurosci Methods. 2008;169:65–75. [DOI] [PubMed] [Google Scholar]
- 22. Cachelin AB, Rust G. Unusual pharmacology of (+)-tubocurarine with rat neuronal nicotinic acetylcholine receptors containing beta 4 subunits. Mol Pharmacol. 1994;46:1168–1174. [PubMed] [Google Scholar]
- 23. Smulders CJ, Zwart R, Bermudez I, van Kleef RG, Groot-Kormelink PJ, Vijverberg HP. Cholinergic drugs potentiate human nicotinic alpha4beta2 acetylcholine receptors by a competitive mechanism. Eur J Pharmacol. 2005;509:97–108. [DOI] [PubMed] [Google Scholar]
- 24. Zaveri N, Jiang F, Olsen C, Polgar W, Toll L. Novel α3β4 nicotinic acetylcholine receptor-selective ligands. Discovery, structure-activity studies, and pharmacological evaluation. J Med Chem. 2010;53:8187–8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Buisson B, Vallejo YF, Green WN, Bertrand D. The unusual nature of epibatidine responses at the alpha4beta2 nicotinic acetylcholine receptor. Neuropharmacology. 2000;39:2561–2569. [DOI] [PubMed] [Google Scholar]
- 26. Zwart R, van Kleef RG, Gotti C, Smulders CJ, Vijverberg HP. Competitive potentiation of acetylcholine effects on neuronal nicotinic receptors by acetylcholinesterase-inhibiting drugs. J Neurochem. 2000;75:2492–2500. [DOI] [PubMed] [Google Scholar]
- 27. Rollema H, Shrikhande A, Ward KM, et al. Pre-clinical properties of the alpha4beta2 nicotinic acetylcholine receptor partial agonists varenicline, cytisine and dianicline translate to clinical efficacy for nicotine dependence. Br J Pharmacol. 2010;160:334–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rollema H, Russ C, Lee TC, Hurst RS, Bertrand D. Functional interactions of varenicline and nicotine with nAChR subtypes implicated in cardiovascular control. Nicotine Tob Res. 2014;16:733–742. [DOI] [PubMed] [Google Scholar]