

Abstract
Epilepsy is a heterogeneous family of neurological disorders that manifest as seizures, i.e. the hypersynchronous activity of large population of neurons. About 30% of epileptic patients do not respond to currently available antiepileptic drugs. Decades of intense research have elucidated the involvement of a number of possible signaling pathways, however, at present we do not have a fundamental understanding of epileptogenesis. In this paper, we review the literature on epilepsy under a wide-angle perspective, a mandatory choice that responds to the recurrent and unanswered question about what is epiphenomenal and what is causal to the disease. While focusing on the involvement of K+ and glutamate/GABA in determining neuronal hyperexcitability, emphasis is given to astrocytic contribution to epileptogenesis, and especially to loss-of-function of astrocytic glutamine synthetase following reactive astrogliosis, a hallmark of epileptic syndromes. We finally introduce the potential involvement of abnormal glycogen synthesis induced by excess glutamate in increasing susceptibility to seizures.
Keywords: epilepsy, potassium, glutamate, GABA, glycogen
1. Introduction
Epilepsy is a group of chronic brain disorders whose clinical manifestation are seizures, which are characterized by recurrence and unpredictability and can range from clinically undetectable episodes to protracted and repeated convulsions. Today, the only available pharmacological treatment strategy aims at seizure control, which is however not effective for about 30% of epileptic patients (Binder and Steinhauser, 2009). Current literature identifies the origin of seizures in neuronal hyperexcitability and resulting hypersynchronous neuronal activity (Fisher et al., 2005). Yet, the etiology is largely unknown for many epilepsies and only few can be satisfactorily described as having a specified genetic, structural or metabolic cause (Korff and Scheffer, 2013). The high percentage of pharmacoresistance has not changed substantially over decades despite the increasing availability of antiepileptic drugs. The lack of knowledge on both epileptogenesis (i.e., the onset of epilepsy) and ictogenesis (i.e., the occurrence of seizures) makes the description of pharmacoresistant epilepsy a puzzle (Pohlmann-Eden and Weaver, 2013). Perhaps the most recurrent question in epilepsy research is whether each identified piece of this puzzle is epiphenomenal or neurobiologically causal.
In the present review, we describe the two historically prevailing hypotheses of epilepsy, namely the potassium (K+) hypothesis (Fertziger and Ranck, 1970; Green, 1964) and the glutamate/γ-aminobutyric acid (GABA) hypothesis (During and Spencer, 1993). These views are not mutually exclusive, and they retain the distinction between synaptic and non-synaptic mechanisms of epileptogenesis. We also advance the hypothesis that the impairment of transmitter cycling and ion homeostasis that accompanies reactive astrocytosis during epileptogenesis is exacerbated by abnormal glycogen metabolism and unavailability of the glycogenolytic response at the onset of seizures.
2. Relevance of neuron-astrocyte signaling in epilepsy
Neurons are the excitable cells of the brain, transmitting and processing electrical signals within specialized neuronal networks. The principal site of neuronal communication is the chemical synapse, a close apposition of membranes belonging to two distinct neurons. These cells are separated by a tiny region of the extracellular space termed synaptic cleft where the presynaptic transmitter release and postsynaptic the receptor-mediated signaling (i.e. neurotransmission) occur. Astrocytes are not excitable cells, still they regulate neuronal excitability by controlling the level of neuroactive compounds in the extracellular space, either indirectly via reuptake or directly by release of transmitter molecules (Dallerac et al., 2013; Newman, 2003). Astrocytic processes ensheat all neuronal elements including but not limited to synapses and axons, with a “coverage” that varies among brain region. For example, in the rat primary visual cortex about 30% of synapses are associated with a perisynaptic astrocyte (Newman, 2004), and the periaxonal astrocytes also exert important homeostatic functions (Sasaki et al., 2011b; Wender et al., 2000). Functional interactions between axons and astrocytes include fast neuron-to-astrocyte K+ flux during neuronal activity, which is especially relevant in cerebral cortex as most cortical grey-matter axons are unmyelinated (see DiNuzzo and Giove, 2012; DiNuzzo et al., 2012, 2013). Astrocytes also form networks of gap junctions coupled cells, which allows trafficking of small molecules via syncytial diffusion. Decades of intense research have unequivocally demonstrated that “virtually every aspects of brain function involves a neuron-glia partnership” (Barres, 2003). Currently established roles of astrocytes include involvement in blood-brain barrier formation and maintenance, stimulation-induced vascular hyperaemic response, water/electrolyte balance, transmitter homeostasis, metabolic interactions with neurons, modulation of neuronal activity and signaling during development and inflammation (reviewed by Ransom and Ransom, 2012). Therefore it is not surprising that astrocytes are nowadays thought to play an important role in the pathogenesis of epilepsy (Binder and Steinhauser, 2009; Carmignoto and Haydon, 2012; de Lanerolle et al., 2010; Devinsky et al., 2013; Seifert et al., 2010; Wetherington et al., 2008).
3. The potassium (K+) hypothesis: depolarization induced by extracellular K+ accumulation
Brain activity depends on generation, summation and propagation of ionic currents along the neuronal plasma membrane. Briefly, at the synapse neurotransmitters bind receptor proteins located postsynaptically thereby increasing membrane ion permeability. Within dendrites, voltage-sensitive ion channels propagate and amplify ionic movements and associated changes in membrane potential. These changes are detected by high density regions of channels on the axon initial segment (AIS), which eventually trigger the propagation of a single action potential along the axonal membrane. The depolarization of presynaptic axon terminal membrane finally causes the exocytotic vesicular release of transmitter molecules. In order to support fast and reliable neuronal signaling, the far-from-equilibrium ion concentration gradients generating membrane potential must be rapidly re-established. This is accomplished by the concerted action of several ion channels and pumps. In this section we briefly describe the changes in activity of the enzymes that might disturb ion homeostasis in epilepsy.
3.1. Depolarization and hyperpolarization produced by ionic movements
Inward Na+ fluxes and outward K+ fluxes are associated with membrane depolarization and hyperpolarization, respectively. Therefore, gain-of-function of Na+ channels (for a comprehensive review, see Eijkelkamp et al., 2012) and/or loss-of-function of K+ channels (for a comprehensive review, see D’Adamo et al., 2013) have proepileptic effects in most cases. Some examples include gain-of-function of voltage-gated Na+ (Nav) channels Nav1.2/1.6 (responsible for action potential generation and propagation) (Sugawara et al., 2001), Nav1.3 (responsible for dendritic postsynaptic potentials propagation) (Estacion et al., 2010; Holland et al., 2008), and loss-of-function of voltage-gated K+ (Kv) channels Kv1.1/1.2 (responsible for action potentials propagation and vesicles release) (Brew et al., 2007; Rho et al., 1999), Kv2.1/8.2 (responsible for dendritic postsynaptic potentials propagation) (Jorge et al., 2011), Kv4.2 (responsible for backpropagating action potentials in dendrites) (Barnwell et al., 2009; Bernard et al., 2004), Kv7.2/7.3 (required for repolarization of AIS) (Peters et al., 2005). There are notable exceptions to this rule, as the effect of these changes is reversed when they target inhibitory neurons. For instance, loss-of-function of Nav1.1 is proepileptic (Yu et al., 2006). This finding is explained by the predominant localization of Nav1.1 channels on the AIS of GABAergic interneurons in neocortex and hippocampus, where they support sustained fast spiking of specified inhibitory neuronal populations (Ogiwara et al., 2007; Yu et al., 2006). Thus, the relationship between the clinical phenotypes and the functional defects may be complex (Ragsdale, 2008).
The amount of charge moved during signal generation and propagation in individual neurons is very small. Yet, in the long run repetitive neuronal activity, as during seizures, turn into sustained changes in both extracellular K+ concentration, which reaches 12–15 mM from a baseline value of 3 mM (Heinemann and Lux, 1977; Pedley et al., 1976), and extracellular Na+ concentration, which decreases by about 10 mM (Hablitz and Heinemann, 1989). Both ion concentration changes are likely mediated by neuronal ion channels, because electrical activity in neurons is associated with similar movements of Na+ and K+ (reviewed by Hertz et al., 2013). The altered extracellular K+ level, in particular, can simultaneously modify the cell membrane potential of many neurons to persistently depolarized values. Such a situation might evolve in “autogenic” paroxysmal discharges (Lebovitz, 1996). Accordingly, conversion of regular firing of pyramidal neurons into burst firing upon elevation of extracellular K+ has been observed in hippocampal slices (Jensen et al., 1994). The role of extracellular K+ is especially important in maintaining hypersynchronous activity of neuronal populations, which is evidenced by the fact that propagation of epileptiform events is untouched by inhibition of chemical neurotransmission (Heinemann et al., 1986; Konnerth et al., 1986; Nelken and Yaari, 1987; Yaari et al., 1986). Increases in extracellular K+ perpetuate and perhaps cause epileptic seizures within a feedback loop with multiple synaptic factors (Fisher et al., 1976; McNamara, 1994). Noticeably, chemical synaptic transmission is not necessary for synchronization of neuronal activity, at least in certain brain regions (reviewed by Dudek et al., 1998).
3.2. Sequestration of neuroactive K+ from extracellular space
Considerable research has demonstrated that astrocytes provide the principal routes to K+ homeostasis, which include both active uptake and spatial buffering of the ion (Kofuji and Newman, 2004). Neuronally released K+ is avidly taken up via the high-capacity astrocytic Na+/K+-activated adenosyntrisphosphatase (NKA), which is associated with the auxiliary protein FXYD7 that diminishes the enzyme affinity for extracellular K+ (Hertz et al., 2013). Within astrocytes K+ is spatially redistributed from sites of high neuronal activity to sites of low K+ concentration through gap junction coupled cells. The latter process is mediated by the astrocyte-specific Kir4.1 channel (Steinhauser et al., 2012). Inward movements of K+ are accompanied by osmotically-driven water fluxes through astrocytic water channel aquaporin-4 (AQP4), leading to transients cell swelling and corresponding shrinkage of extracellular space. Both K+ and water diffuse through the astrocytic syncitium for subsequent Kir4.1- and AQP4-mediated release, respectively.
3.2.1. Astrocytic K+ buffering
Alterations in expression and protein level of Kir4.1 have been reported in surgical specimens of sclerotic hippocampus from temporal lobe epilepsy (TLE) patients (Schroder et al., 2000), and specifically in reactive astrocytes (Bordey and Sontheimer, 1998; Hinterkeuser et al., 2000; Jauch et al., 2002). Down-regulation of astrocytic Kir4.1 has also been found in rat neocortex after blood-brain barrier disruption and serum extravasation (Stewart et al., 2010), a condition leading to albumin-induced ictogenesis (Frigerio et al., 2012). Together, these studies suggests the existence of a close link between Kir channels and electrophysiological changes of astrocytes during epilepsy (Binder and Steinhauser, 2006). However, glial-conditional knockout Kir4.1 mice do not show any evidence of sustained extracellular K+ nor epileptic seizures (Chever et al., 2010).
Spatial redistribution of K+ requires integrity of astrocytic syncitial network as well (Wallraff et al., 2006). Gap-junction coupling through connexins (Cx) 43 and 30 appears to be altered in epilepsy. In the rat kindling model of epilepsy expression of both Cx43 and Cx30 have been found to be initially up-regulated during induction of status epilepticus in a compensatory manner to subside after acquisition of seizures (Akbarpour et al., 2012). However, enhanced Cx43 expression has been observed after kainate-induced status epilepticus (Takahashi et al., 2010) or in specimens from sclerotic hippocampus of TLE patients (Collignon et al., 2006; Fonseca et al., 2002; Naus et al., 1991). It should be noted that gap junctions might have an ambivalent role in epileptogenesis (see Seifert and Steinhauser, 2013). Besides mediating K+ buffering, the gap junctions coupled astrocytic network could indeed sustain epileptiform activity by supporting glucose trafficking (Giaume et al., 2010) or even perpetuate epileptic seizures by spreading Ca2+ signaling and gliotransmission (see below). Accordingly, neuroprotection against epilepsy have been assigned either to enhancement (Samoilova et al., 2008) or suppression (Yoon et al., 2010) of gap junctional communication in astrocytes. Gap junction blockers are not an option, as currently available drugs cannot distinguish between astrocytic and neuronal gap junctions forming astrocytic syncytium and electrical synapses, respectively (Seifert et al., 2010).
A role of AQP4 for K+ homeostasis in epilepsy is evidenced by the fact that increased duration of stimulation-induced seizures are associated with slower K+ transients in the hippocampus of AQP4 knockout mice (Binder et al., 2006). Subcellular expression of AQP4 in brain astrocytes mimics that of Kir4.1, although AQP4 is more strongly expressed in perivascular regions (Nagelhus et al., 2004). However, down-regulation of AQP4 not accompanied by significant changes in Kir4.1 has been reported in hippocampal kainic acid mouse model of epileptogenesis (Lee et al., 2012). Furthermore, perivascular but not parenchymal loss of AQP4 has been observed in hippocampi of TLE patients (Eid et al., 2005). These studies are consistent with the finding that K+ buffering is compromised when perivascular AQP4 is decreased (Amiry-Moghaddam et al., 2003). The interface between astrocyte and vascular endothelium is of particular relevance to epilepsy, especially in sclerotic tissue. Indeed, epileptogenesis is associated with proliferation of microvasculature and extravasation of substances during seizures (for a review, see de Lanerolle et al., 2010).
3.2.2. Active extracellular K+ uptake by astrocytes
Although K+ buffering in astrocytes contributes to cellular K+ homeostasis in the long term, active uptake of neuronally released K+ is essential for fast neuronal signaling (Bay and Butt, 2012). The importance of astrocytic K+ uptake is two-fold: first, by rapidly removing positive charges from the extracellular space it accelerates repolarization of neuronal membrane, and second, it avoids accumulation of interstitial K+ and the resulting shift of the membrane potential towards depolarized values. Net NKA-mediated K+ uptake by astrocytes occurs whenever extracellular K+ increase (Amzica et al., 2002; Ballanyi et al., 1987; Chever et al., 2010; Dufour et al., 2011; Hertz, 1978; Xu et al., 2013). Accordingly, the astrocytic clearance of astrocytic K+ is inhibited by NKA-blocking ouabain alkaloids, but not by other K+ channels inhibitors (Hertz et al., 2013).
That the primary function of NKA in astrocytes is K+ clearance is indicated by the requirement of astrocytic Na+ channels for inward Na+ flux and maintenance of intracellular Na+ level necessary for NKA activity (Sontheimer et al., 1994). As previously mentioned, during seizures there is a decrease in extracellular Na+ due to a large activity-induced influx of the ion into neurons. If from the one side this relieves the inhibitory effect of extracellular Na+ on the astrocytic NKA/FXYD7 complex, from the other side it aggravates the difficulty of astrocytes in securing Na+ for NKA activity (reviewed by DiNuzzo et al., 2013). Eventually, prolonged neuronal hyperactivity results in substantial astrocytic Na+ decrease, as reported during epileptic seizures (Hablitz and Heinemann, 1989). Interestingly, reactive astrocytes from epileptic human and rat hippocampus have enormously increased density of tetrodotoxin-sensitive Nav channels with fast activation and inactivation kinetics (Bordey and Sontheimer, 1998) as well as extracellular Na+ level sensitive Na+ (Nax) channels (Gorter et al., 2010), which can be interpreted as a compensatory mechanism to protect these cells from Na+ loss.
Studies performed on microdissected cerebral glial cells, synaptosomes, mixed glial-neuron cultures and tissue slices have demonstrated that the electrophysiological and metabolic response to stimulation by elevated K+ concentrations as those found during seizures (~12 mM) is primarily astrocytic (Badar-Goffer et al., 1992; Grisar et al., 1979; Henn et al., 1972; Honegger and Pardo, 1999). This notion is consistent with the different catalytic subunit composition of neuronal and astrocytic NKA. Indeed, the astrocytic enzyme exhibits higher transport capacity and lower affinity for extracellular K+ compared with neuronal NKA, the latter being mainly activated by increases in intracellular Na+ and already saturated for K+ even at basal extracellular K+ concentration (see DiNuzzo et al., 2012, 2013). In epileptic tissue, kinetic response to K+ and K+ removal capacity of astrocytic but not neuronal NKA have been found to be down-regulated (Grisar, 1984; Grisar and Delgado-Escueta, 1986; Grisar et al., 1983; Grisar et al., 1992; Guillaume, 1988; Laschet et al., 1991; Laschet et al., 1990). This decrease in enzyme activity seems to be often paralleled by changes in the expression and subunit composition of astrocytic NKA that at first glance are mutually incompatible. For example, increased expression of α2 NKA subunit in astrocytes has been found in rat hippocampus after kainic acid treatment (Anderson and Stahl, 1997). The decline in K+ uptake by astrocytic NKA can be reconciled with the finding of increased expression of α2 NKA subunit by considering that FXYD7 only binds to the α1 subunit of NKA (Beguin et al., 2002). Although these experiments are difficult to generalize, the importance of functional NKA in astrocytes for K+ uptake has led to the hypothesis that astrocytic NKA might play a role in ictogenesis and seizure spread (Guillaume, 1988). The rise in enzyme affinity for K+ and the resulting inability of astrocytic NKA to be fully activated in response to high extracellular K+ (Grisar et al., 1992) would explain the paradoxycal decrease in overall NKA activity sometimes reported during acute seizures, i.e. when signaling and associated energy consumption is enhanced (Fernandes et al., 1996; Vitezic et al., 2008). In the pilocarpine rat model of epilepsy, NKA activity has been shown to be reduced in the acute period but substantially increased in the chronic period (Fernandes et al., 1996; Reime Kinjo et al., 2007). In the same model, it has been recently reported that glucose metabolism and the expression of some enzymes involved in energy metabolism are both decreased in the long-term (Araujo et al., 2014; Hadera et al., 2013). No clear-cut relation has been observed between ATP levels and K+-induced epileptiform activity in epileptogenic rat hippocampus (Marichich and Nasello, 1973). Lack of correlation between changes in glucose consumption and spike-wave discharges has also been reported in rat model of absence epilepsy (Nehlig et al., 1992). In this epilepsy model, normal or decreased glucose metabolism has been found in focally active regions during the ictal period (Nehlig et al., 1993). Furthermore, levels of phosphocreatine and ATP are maintained during many types of epileptic seizures (e.g., Folbergrova et al., 1969), in spite of substantial increase in energy consumption by NKA (Subbalakshmi and Murthy, 1981). Together, the above-mentioned experimental evidence speak against a purely energetic basis for epilepsy. Moreover, changes in enzyme function could result from either a mitochondrial dysnfunction per se or from reduced metabolic demands. In fact, while on one hand a decreased metabolic capacity leads to impaired NKA activity, on the other hand the decreased NKA activity leads to reduced metabolic demand. The partial loss of mitochondrial function observed in the epileptogenic tissue is overtly in favor of the former scenario (Waldbaum and Patel, 2010; Yuen and Sander, 2011). However, it is common knowledge that mitochondrial oxidative phosphorylation is stimulated by ADP (Chance and Williams, 1955), which in the brain is mainly formed as a result of ATP hydrolysis by NKA. In other words, energy production is stimulated by energy consumption, and ADP unavailability to mitochondria is a detrimental event ultimately causing accumulation of reactive oxygen species, mitochondrial damage and apoptosis (see DiNuzzo et al., 2012).
Net K+ uptake by astrocytes is aided by the astrocytic Na+/K+/2Cl− cotransporter 1 (NKCC1). This uptake is substantial only at extracellular K+ concentrations higher than 10–15 mM, as K+ uptake is not affected by the NKCC1 inhibitor furosemide at lower extracellular K+ levels (Walz and Hertz, 1984). Accordingly, activation of NKCC1 requires membrane depolarization and subsequent stimulation of L-type voltage-gated Ca2+ channel (Cav) 1.2 and 1.3 (Cai et al., 2011). Action of NKCC1 is also important in providing Na+ to astrocytes while cotransporting K+ (for a review, see DiNuzzo et al., 2013). The involvement of this astrocytic machinery for K+ uptake in epilepsy awaits experimental investigation.
4. The glutamate/GABA hypothesis: enhancement of excitation and suppression of inhibition
Disruption of the balance between excitation and inhibition is the most obvious mechanism leading to hyperexcitability. In the adult brain, this mechanism entails an enhancement of glutamatergic neurotransmission and/or a suppression of GABAergic neurotransmission. In the present section, we summarize the potential changes in the expression and protein levels of several enzymes involved in chemical synaptic transmission that can increase susceptibility to seizures. Emphasis is given to the release of transmitter molecules by astrocytes.
4.1. Exocytotic and non-exocytotic neuro- and glio-transmission
Changes in vesicular glutamate transporter (VGLUT) and/or vesicular GABA transporter (VGAT) expression and protein level might contribute to altering the balance between excitation and inhibition by changing the quantal content of vesicles. In general, changes in synaptic proteins might parallel synaptic loss or formation (Crevecoeur et al., 2013). For example, in sclerotic hippocampus of TLE patients VGLUT-1 expression has been found to be down-regulated in regions with severe neuronal loss, but strongly up-regulated in regions characterized by formation of new glutamaregic synapses (i.e. after mossy fiber sprouting) (van der Hel et al., 2009). There is no convincing evidence that possibly altered activity of vesicular transmitter transporters per se is epileptogenic.
Synaptic vesicles are released by fusion between vesicle and plasma membranes (i.e. exocytosis) in a Ca2+-dependent manner. In particular, action potential-mediated depolarization of axon terminal activates voltage-gated Ca2+ (Cav) channels. Presynaptic Ca2+ entry initiates the exocytotic process by binding to proteins of the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex. Changes in activity of these proteins might impact seizure susceptibility. As an illustration, up-regulation of putatively presynaptic Cav3.2 expression has been implicated in epileptogenesis in the pilocarpine model of epilepsy (Becker et al., 2008). Recently, increase in SNARE complex has been reported in hippocampal synaptosomes from amygdala-kindled rats (Matveeva et al., 2012a). Genetic manipulation of neurosecretory machinery resulted in attenuation of K+-evoked glutamate release and substantial reduction of kindling-induced epileptogenesis (Matveeva et al., 2012b). Presynaptic transmitter release can be also inhibited by K+ currents without any apparent impairment of Cav channels. For example, hyperpolarization-activated cyclic nucleotide-gated (HCN) K+ channels have been found to suppress the activity of Cav3.2 channels (Huang et al., 2011). However, a role for HCN channels in seizure susceptibility remains to be established (see Benarroch, 2013).
Astrocytes also can release transmitter molecules, a process termed gliotransmission (Santello et al., 2012; Zorec et al., 2012). The astrocytic release of glutamate can occur by exocytotic as well as nonexocytotic mechanisms, while that of GABA appears to be exclusively nonexocytotic (Martineau et al., 2013). Genetic impairment of an astrocytic SNARE domain has been found to reduce status epilepticus and seizure frequency in pilocarpine mouse model of epilepsy (Clasadonte et al., 2013). Several possibilities have been suggested for gliotransmitter release not mediated by vesicular fusion. GABA can in principle be released by reversal of GABA transporter (GAT) proteins (Richerson and Wu, 2003), while this route is unlikely for excitatory amino acid transporter (EAAT) proteins moving glutamate (Longuemare and Swanson, 1997). This discrepancy stems from the different ion movements associated with glutamate and GABA transport. Specifically, both GATs and EAATs capitalize the Na+ gradient by cotransporting 2 or 3 Na+ together with a transmitter molecule. However, glutamate is countertransported with 1 K+, while GABA is cotransported with 1 Cl−. Given the relatively high intracellular Cl− concentration in astrocytes, in principle reversal of GATs could happen under depolarizing conditions (i.e. conditions compatible with Na+ efflux). On the contrary, the K+ concentration inside the cell is at least 10-fold higher than the exterior even during seizures (Lothman et al., 1975; Moody et al., 1974). Recently, GABA release by astrocytes through reversal of GATs has been reported to be directly evoked by glutamate uptake, the latter providing the Na+ driving the glutamate/GABA exchange without ATP usage (Heja et al., 2012). The involvement of astrocytic GABA release in seizure susceptibility has been poorly investigated. However, selective inhibition of glial but not neuronal GATs has been found to exert anticonvulsant effects (Gadea and Lopez-Colome, 2001), suggesting that GATs normally mediate GABA reuptake not release and hence the contribution of GABA release by astrocytes is seemingly of minor importance in epilepsy. Since glutamate accumulates in astrocytes during seizures (see below), it is possible that the uptake of the neurotransmitter is reduced and so is the reversal of astrocytic GATs. GABA appears to be also released through astrocytic Ca2+-activated anion channel (CAAC) bestrophin 1 (Best1) (Lee et al., 2010), although this is certainly not the case in the hippocampus because hippocampal astrocytes do not contain GABA (Yoon et al., 2011). Nonexocytotic release of glutamate can be mediated by volume-sensitive organic anion channel (VSOAC) proteins during astrocyte cell swelling induced by high neuronal activity (Basarsky et al., 1999). However, channel-mediated glutamate release by astrocytes is more likely to occur via Best1 channel (Oh et al., 2012; Park et al., 2013) as well as two-pore domain potassium (K2P) channels (Woo et al., 2012). The K2P2.1 member of the K2P family, also named TWIK-related potassium channel 1 (TREK-1), is involved in the regulation of astrocytic membrane potential though leakage K+ current (for example, see Zhou et al., 2009). Although TREK-1 is apparently permeable to both glutamate and K+, adenoassociated viral delivery of a constitutively active TREK-1 channel has been found to silence hyperexcitable neurons in epileptic rats (Dey et al., 2013). This finding suggest that astrocytic release of glutamate by K2P channels is probably of minor importance in epilepsy. Involvement of Best1 in seizure susceptibility remains to be shown.
4.2. Sensing transmitter molecules
Brain cells possess systems of receptor proteins through which they can sense and respond to extracellular chemical signals. Here we describe the glutamatergic and GABAergic systems, which together account for most of the receptor-mediated signaling and associated transmitter cycling and glucose metabolism in cortical grey matter (Chowdhury et al., 2007). The adrenergic, serotoninergic and dopaminergic system are also potentially relevant to epilepsy, and they are generally thought to exert antiepileptic effects (Giorgi et al., 2004; Starr, 1996; Theodore, 2003). The description of these and other neurotransmitter systems is beyond the scope of the present review.
4.2.1. Glutamate receptors signaling
Glutamate receptor proteins include ionotropic as well as metabotropic receptors. Enhanced glutamatergic activity through excessive stimulation of glutamate receptors might therefore result in neuronal hyperexcitability. It should be noted that the rise in receptor-mediated currents might not necessarily be accompanied by increase in extracellular glutamate concentration. Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainate and N-methyl-D-aspartate (NMDA) receptors are ligand-gated ion channels belonging to the former family of glutamate receptor proteins. Binding of the neurotransmitter to these receptors, which are located on postsynaptic dendrites or spines, causes channel opening and corresponding increase in the permeability of the membrane to Na+, Ca2+ and K+. As a result, the membrane potential experiences a transient depolarization that propagates passively, being eventually reinforced by dendritic Nav and Kv channels, up to the AIS. AMPA and NMDA are the predominant receptor types at excitatory synapses, where they exhibit distinct kinetics in response to glutamate and thus possibly different involvement in epilepsy (Rogawski, 2013). AMPA receptors are of primary importance in initiating seizures because inhibition of AMPA receptors is more effective than inhibition of NMDA receptors in abolishing epileptiform discharges (Hwa and Avoli, 1991; Hwa et al., 1991; Jones and Lambert, 1990; Nakamura et al., 1995; Williamson and Wheal, 1992). Combination of antiepileptic drugs targeting AMPA and NMDA receptors has been shown to prevent seizures in pentylenetetrazole-treated rats (Gmiro and Serdyuk, 2008). Immunoreactivity of AMPA and NMDA receptors subunits has been found to be increased in the hippocampus of TLE patients (Mathern et al., 1998). However, studies on the expression of glutamate receptors should be interpreted carefully. Indeed, paradoxical seizure exacerbation by blockade of glutamate receptors might occur whenever the suppression of these receptors targets inhibitory interneurons (e.g., Maheshwari et al., 2013). Another possible confounding factor, that we mention here but also applies to vesicular transmitter release, is the unsilencing of presynaptic- and/or postsynaptic-silent synapses, an event that might be induced by seizures (Hu et al., 2012; Zhou et al., 2011). Metabotropic glutamate receptor (mGLUR) proteins also have been implicated in epileptogenesis. Activation of these receptors produces membrane depolarization (postsynaptic group I mGLURs), or reduced transmitter release (presynaptic group II and group III mGLURs). Gain-of-function of the former and loss-of-function of the latter have been found to contribute to epileptogenesis in amygdala-kindled rat brain slices (Holmes et al., 1996). Accordingly, stimulation of group I mGLURs has been reported to elicit ictal-like response that convert to long-lasting epileptiform discharges (Wong et al., 2004). As groups II/III mGLURs reduce release probability in both glutamategric and GABAergic neurons, the pro- or anticonvulsant effect of these receptors cannot be easily established in different epilepsy models (Ghauri et al., 1996).
Astrocytes also sense glutamate in a receptor-mediated manner, as these cells are equipped with group II mGLUR3 and possibly group I mGLUR5 (Sun et al., 2013), the latter highly expressed in young brain cells as well as reactive astrocytes (Takano et al., 2014). Elevated levels of astrocytic mGLURs have been found in epilepsy models or in hippocampal specimens from TLE patients (Steinhauser and Seifert, 2002; Tang and Lee, 2001). Activation of mGLURs results in elevation of intracellular Ca2+ and initiation of Ca2+-induced Ca2+-release mechanism in coupled astrocytes. Astrocytic Ca2+ signaling triggers the exocytotic release of glutamate (see above) and ATP (Montana et al., 2006; Zhang et al., 2007). The latter acts on purinergic metabotropic (P2Y) receptors that in turn increase Ca2+ concentration in astrocytes via the phospholipase C pathway, thereby propagating the astrocytic Ca2+ signaling as a Ca2+ wave (Guthrie et al., 1999). In neocortex and hippocampus locally correlated cell groups of few (Sasaki et al., 2011a) to hundreds (Kuga et al., 2011) astrocytes exhibit synchronous Ca2+ oscillations which emerge spontaneously as well as in response to changes in neuronal network activity. During epileptiform activity, Ca2+ signaling in astrocytes is enhanced resulting in increased frequency of Ca2+ oscillations, which can be reduced by anticonvulsant drugs (Fellin and Haydon, 2005; Tian et al., 2005). Accordingly, attenuation of astrocytic Ca2+ signaling is neuroprotective and reduce glutamate gliotransmission and excitotoxicity after pilocarpine-induced status epilepticus (Ding et al., 2007). The role of this excitatory loop underlying astrocyte hyperactivity in promoting excessive astrocyte-neuron coupling and approach of neurons to seizure threshold is a very promising field for epilepsy research (Gomez-Gonzalo et al., 2010). ATP also stimulates ionotropic purinergic (P2X) receptors located on both neurons and astrocytes (Lalo et al., 2014; Verkhratsky et al., 2012). These receptors are permeable mainly to Na+ and Ca2+ and thus exert a depolarizing action on cell membrane potential. Extracellular ATP is however rapidly degraded by ectonucleotidases to adenosine (Wink et al., 2006), which acts on adenosine purinergic (P1) receptors located on presynaptic neurons (Burnstock, 2013). These receptors might promote both heterosynaptic depression (A1 receptors) or facilitation (A2 receptors) depending on brain areas and cell types (for example, see Bannon et al., 2013), which should preclude generalization of the effect of purinergic neurotransmission in epilepsy. However, adenosine has been shown to exert anticonvulsant effects, for example in rat amygdala kindling model (Dragunow and Goddard, 1984). This and other studies have indicated that the involvement of adenosine in brain hyperexcitability has mainly to do with the role of A1 receptors in preventing seizure spread (for a review, see Boison, 2011).
4.2.2. GABA receptors signaling
Similarly to glutamate, the effect of GABA is mediated by ionotropic (GABAA) receptors and metabotropic (GABAB) receptors. Both receptors induce membrane hyperpolarization, the former through Cl- influx and the latter through G-protein coupled Kir channels. GABAA-mediated Cl− currents also provide a shunt mechanism that nullifies concomitant depolarizations. Loss-of-function of GABAA receptors has been associated with epileptic syndromes in both humans and rodents (Arain et al., 2012; Laschet et al., 2007; Reid et al., 2013; Zhou et al., 2013), but GABAA receptor-mediated conductance might also paradoxically generate synchronous depolarizing events that engage neuronal networks during ictogenesis (Avoli and de Curtis, 2011). The general finding that GABAB receptor agonists exacerbate seizures, while GABAB receptor antagonists suppress seizures (Han et al., 2012) is counterintuitive as well. Similarly, reports of GABAergic neurons immunoreactivity are not uniform, and different studies reported decreased, unaffected or even increased inhibitory neuronal density depending on brain region, interneuron type or receptor isoform examined (reviewed by de Lanerolle and Lee, 2005). It should be realized that important subtypes of GABAergic interneurons in the cortex and hippocampus inhibit other inhibitory neurons, thus gain-of-function of these disinhibitory neurons might result in aggravation of seizures (for example, see Wittner et al., 2005).
In rat neocortex, GABAergic neurotransmitter cycling flux is 8–13% of glutamatergic flux at P10 and 18–22% at P30 (Chowdhury et al., 2007; Patel et al., 2005). This increase parallels the rapid expansion of GABAergic synapses per neurons (Micheva and Beaulieu, 1996) and the shift in the action of GABA at its ionotropic receptors from depolarizing to hyperpolarizing (Stein et al., 2004). The effect of GABA depends on the Cl− concentration gradient between intra- and extracellular compartments, which is largely due to the relative activity of inward NKCC1 and outward K+/Cl− cotrasporter 2 (KCC2) (Alvarez-Leefmans et al., 2001; Delpire and Mount, 2002; Gamba, 2005). Early in development neuronal cells express high levels of the ubiquitous NKCC1 and very low levels of the neuron-specific KCC2, a pattern of expression resulting in relative high Cl− concentration in neurons and depolarizing outward Cl− flux upon increase in membrane permeability after GABA action. The transition to the adult brain is accompanied by progressive up-regulation of KCC2 expression and down-regulation of NKCC1 expression in neurons, which brings about a substantial decrease in intracellular Cl− concentration and GABA-induced hyperpolarizing inward Cl− flux (Clayton et al., 1998; Plotkin et al., 1997; Rivera et al., 1999; Wang et al., 2002; Yamada et al., 2004). The features of the immature brain are important, as some molecular mechanisms involved in normal brain development, such as those described above, are recapitulated during epileptogenesis (Elliott and Lowenstein, 2004; Elliott et al., 2003). In particular, astroglial reactivity is associated with this reversion to an immature stage through the expression of early developmental genes and growth factors (Ridet et al., 1997). During epileptogenesis, elevations in neuronal Cl− concentration attributable to down-regulation of KCC2 and up-regulation of NKCC1 impairs GABA receptor mediated inhibition or even converts it to excitation (Blaesse et al., 2009; Payne et al., 2003). Defective neuronal Cl− extrusion due to imbalance of these cation-chloride cotransporters is thought to be an important factor in epileptogenesis, because it might disturb the ratio of excitatory and inhibitory inputs in cortical neurons (e.g., Viitanen et al., 2010).
Astrocytes are gabaergic/gabaceptive cells expressing both GABAA and GABAB receptors (Lee et al., 2011). Both receptors results in increased Ca2+ concentration in astrocytes. The GABAA-mediated effect is rapid and involves membrane depolarization due to the relatively high Cl− level in these cells (Yoon et al., 2012) and subsequent opening of voltage-gated Ca2+ channels (Meier et al., 2008). GABAB receptor activation trigger a delayed Ca2+ release from intracellular stores (Meier et al., 2008). In either case, astrocytic Ca2+ signaling eventually modulates neuronal activity via gliotransmission. Accordingly, activation of astrocytic GABAB receptors has been reported to potentiate neuronal inhibitory synaptic transmission in a Ca2+-dependent manner (Kang et al., 1998). The elucidation of the possible epileptogenic effects of GABAergic signaling in astrocytes remains to be investigated.
4.3. Terminating transmitter action
Postsynaptic action of neurotransmitter must be rapidly terminated to ensure high fidelity encoding of information. This is particularly important for glutamate in order to avoid excitotoxicity, which can result in neuronal damage and eventually apoptotic cell death. The foremost process to stop receptor-mediated signaling is reuptake of synaptically-released transmitter molecules. Once taken up, the fate of glutamate and GABA inside cells is important as well to maintain concentration gradient and transmitter homeostasis.
4.3.1. Reuptake of synaptically released glutamate and GABA
Since glutamate uptake stops postsynaptic currents, any impairment of glutamate removal from extracellular space can potentially lead to neuronal hyperexcitation. Similarly, in principle it is possible that neuronal hyperactivity could be generated by intensification of reuptake of inhibitory neurotransmitter. Changes in the expression and activity of glutamate and GABA transporters cannot be interpreted without considering concomitant changes in cell density and the specific distribution of transporter proteins in neurons and astrocytes.
The situation is relatively straightforward for glutamate, where most if not all reuptake occurs via astrocytic EAATs (Coulter and Eid, 2012). Expression levels and activity of these transporters have been found to be increased, decreased or unchanged depending on the brain region examined or epilepsy model (Guo et al., 2010; Mathern et al., 1999; Proper et al., 2002; Sarac et al., 2009; Tessler et al., 1999). The relevant changes do not however make much sense in damaged tissue, such as the sclerotic hippocampus, where substantial neuronal loss is accompanied by astrocytic proliferation. Indeed, reduction of glutamate transporters is possibly an effect of reduced synaptic activity (Yang et al., 2009), and as such secondary to epilepsy-induced neuronal cell loss. In sclerotic tissue, the conflicting experimental results leave open the question of whether the (relatively small) alterations in EAAT activity are of relevance to epilepsy (Seifert and Steinhauser, 2013).
Contrary to glutamate, GABA is taken up by both presynaptic neurons and astrocytes via GATs (Pow et al., 2005). Thus, for example a decrease in neuronal GAT possibly implies reduced inhibitory GABAergic input (i.e. less GABA terminals), which facilitates hyperexcitability. The loss of GATs observed in epileptogenic hippocampus removed from TLE patients (Lee et al., 2006) or the reduction of GAT in the brain of genetically epilepsy-prone rats (Akbar et al., 1998) should be interpreted along this line of thought. On the other hand, up-regulation of GATs have been found in tremor rat hippocampus (Mao et al., 2010), in the hippocampus of rats after amygdalar FeCl3 injection-induced seizures (Ueda and Willmore, 2000), or in amygdala-kindled rat brain (Hirao et al., 1998). These finding can possibly be explained by an increased action of GABA reuptake and a resulting reduction of extracellular GABA level. This is consistent with the anticonvulsive effect of GAT inhibitors (see Madsen et al., 2010), which are surprisingly observed also in immature hippocampus (Sharopov et al., 2013). The relative neuronal versus astrocytic contribution to GABA reuptake as well as any cell-specific impairment of GATs is presently unknown. Moreover, it is not clear whether changes in GAT expression are compensatory or concausal to the pathology (Dalby, 2003).
4.3.2. Intracellular metabolism of glutamate and GABA
Reuptake of transmitter molecules is interconnected with their metabolism in the intracellular compartment. Indeed, effective catabolism and transport of neurotransmitters avoid their accumulation in cytosol and/or extracellular space. This is especially relevant for astrocyte-mediated reuptake of glutamate (note that also GABA enters the glutamate pool after astrocytic reuptake and transamination). Within astrocytes, a fraction of glutamate is channeled into tricarboxylic acid (TCA) cycle, while the remaining is converted to glutamine by the astrocyte-specific enzyme glutamine synthetase (GS) (reviewed by Mangia et al., 2012). Glutamine is released into extracellular space from where it is taken up by neuronal sodium-coupled neutral amino acid transporter (SNAT) proteins and subsequently converted back to glutamate by the neuron-specific enzyme phosphate activated glutaminase (PAG). The relative proportion of glutamate metabolized versus that cycling in neurons as glutamine rises as the level of the amino acid increases (McKenna et al., 1996). Metabolization of glutamate can proceed via transamination by alanine aminotransferase or branched-chain aminotransferase. Normally, glutamate participates in the malate-aspartate shuttle, which requires action of aspartate aminotransferase as well as aspartate-glutamate carrier. Within mitochondria, glutamate can be further processed through glutamate dehydrogenase (GDH). Changes in activity of one of these enzymes can potentially enhance excitation or suppress inhibition. The synthesis of glutamine by GS deserves a special consideration and will be examined below. Unfortunately, there are few and often inconsistent studies that examined the changes of enzymes participating in glutamate metabolism. For example, in kainate model of epilepsy GDH expression remained unaltered between prior and after onset of seizures (Hammer et al., 2008), whereas it apparently decreased relative to substrate flow through TCA cycle in sclerotic hippocampus from TLE patients (Malthankar-Phatak et al., 2006). Changes in other enzymes involved in glutamate metabolism during epilepsy have not yet been investigated in details.
GABAergic synapses are exquisitely sensitive to availability of glutamine, which is primarily provided by astrocytes. After impairment of astrocytic glutamate cyling, failure of GABAergic signaling occurs within minutes (Fricke et al., 2007; Liang et al., 2006; Yang and Cox, 2011), whereas glutamatergic signaling is maintained for hours (Kam and Nicoll, 2007; Ortinski et al., 2010). However, during seizures glutamine uptake appears to primarily support glutamatergic activity, as inhibition of SNAT-mediated neuronal glutamine transport reduces epileptiform activity in rat kainate epilepsy model (Kanamori and Ross, 2013).
5. How excess glutamate can impair K+ homeostasis
So far, we have described the experimental evidence associating dysfunction of a number of identified proteins to neuronal hyperexcitability and epileptogenesis. It should be clear that isolating an individual mechanism to establish a causal relation is an extremely difficult, if not impossible, task due to the existence of many feedback loops in neuronal-glial activity. Nonetheless, in the following we propose a potential mechanism relating excitotoxic glutamate level to persistently elevated extracellular K+ concentration.
5.1. Dysfunction of glutamine synthetase and glutamate cycling
Reactive astrogliosis and neuronal loss are primary hallmarks of epileptogenesis. Hypertrophy and proliferation of reactive astrocytes and infiltration of activated microglia result in phenotipic changes in neurons, which are secondary to astrocyte- and microglia-mediated release of several growth factors. For example, the release of brain-derived neurotrophic factor by reactive astrocytes causes a down-regulation in the expression of neuronal KCC2 as well as an up-regulation in the expression of neuronal NKCC1 (Rivera et al., 2002; Rivera et al., 2004). These alterations are responsible for the relatively high intracellular chloride concentration that depolarizes the membrane upon stimulation of ionotropic GABA receptors, resulting in reduced inhibition and neuronal hyperexcitability (see above). Neuronal cell death initiated by excitotoxicity is thought to be exacerbated by the loss of astrocyte-neuron cell-cell contact following the deterioration in extracellular matrix proteins (Eid et al., 2013b). These events trigger a number of changes in astrocytic gene expression that have been found to increase protein nitration, oxidative stress and also β-amyloid deposits that ultimately lead to reduction in astrocytic GS (Eid et al., 2008b). In the kainate rat model of epilepsy it has been found that GS is upregulated in latent phase of epileptogenesis and down-regulated in chronic phase after the onset of recurrent seizures (Hammer et al., 2008). This finding might explain the slightly increased GS expression reported in mouse hippocampus after kainic acid-induced epileptogenesis (Lee et al., 2012) or the unaltered GS protein levels observed in pentylenetetrazol epilepsy model (Bidmon et al., 2008). The latter study, however, supported the notion that GS undergoes posttranscriptional modifications (e.g., protein nitration) with loss-of-function not necessarily accompanied by reduced expression (see also Eid et al., 2013a). Dysfunction of GS along with decreased extracellular and intracellular glutamine levels has been consistently found in sclerotic hippocampus (Cavus et al., 2005; Eid et al., 2004; Petroff et al., 2003; Petroff et al., 2002; van der Hel et al., 2005).
Although it is commonly acknowledged that decrease in GS activity is proconvulsant, it should be realized that the variations in expression and protein level of GS could not translate in obvious changes of neurotransmission and extracellular transmitter level. Indeed, neuronal glutamate release is strongly dependent on the continuous availability of glutamine (Tani et al., 2010). This notion suggests that suppression of GS activity might impair glutamatergic neurotransmission and thus mitigate the rise in extracellular glutamate (Devinsky et al., 2013). Suppressed GS activity and failure of glutamate-glutamine cycle also depletes synaptic GABA (Ortinski et al., 2010). Thus, dysfunction of GS produce local synaptic perturbations either towards excitation or inhibition, both likely accompanied by reduced neurotransmission. Extracellular glutamate has been found to increase by 20% in epileptic rat hippocampus (Walker et al., 1995) or by 30-40% in kainate- or pilocarpine rodent epilepsy model (Liu et al., 1997). It is likely that the most of glutamate accumulates in astrocytes during hypoactivity of GS (Danbolt, 2001; Petroff et al., 2002). Indeed, the Na+-dependent high-affinity and high-capacity EAATs (Schousboe, 1981) are capable of maintaining inward transmitter flux in astrocytes even at the high K+ level attainable during seizures (Claudio et al., 2002 and references therein). It is noted that astrocytic proliferation is not accompanied by substantial loss of EAATs expression and activity (see above). Paradoxically, the concentration of glutamate has been found to increase several-fold even in sclerotic hippocampus characterized by 60–80% of neuronal loss and doubled glial density (Cavus et al., 2005; Kim et al., 2004; Petroff et al., 2003). After inhibition of GS by the xenobiotic L-methionine-SR-sulfoximine (MSO), glutamate has been found to increase four-fold in hippocampal slice astrocytes (Laake et al., 1995). In GS-deficient hippocampal formation of laboratory rats the concentration of glutamate in astrocytes has been found to be increased by ~50% (Perez et al., 2012). In normal animals, GS inhibition leads to acute decrease in extracellular and neuronal glutamate concentration (Bottcher et al., 2003; Paulsen and Fonnum, 1989; Rothstein and Tabakoff, 1984). That sustained epileptiform discharges can occur without any increase in extracellular glutamate has been directly demonstrated in picrotoxin- or bicuculline-injected rats (Millan et al., 1991). Drop in synaptosomal glutamate and, by inference, in glutamatergic neurotransmission have been reported in rat brain after chronic MSO administration (Somers and Beckstead, 1990) without energy failure (Subbalakshmi and Murthy, 1981). Uncoupling between glutamate and neuronal hyperactivity is further suggested by the fact that the tissue concentration of the transmitter remains elevated long after cessation of seizures (During and Spencer, 1993). Together, these arguments suggest that reduction in GS activity exerts its proconvulsant effects by another route than increased neuronal glutamatergic neurotransmission (see also Kam and Nicoll, 2007) and that the increase in glutamate is probably secondary to the epileptogenic action of MSO.
A critical role of GS impairment in initiating epilepsy is suggested by the fact that recurrent seizure activity develops after pharmacological inhibition of GS (Eid et al., 2008a). Indeed, systemic administration MSO causes severe seizures in laboratory animals, which were first observed decades ago (Folbergrova et al., 1969; Szegedy, 1978) and then repeatedly utilized as a model of TLE (Eid et al., 2008a; Wang et al., 2009), as the neuropathological features of MSO-treated animals resemble human mesial temporal sclerosis (see Eid et al., 2008b).
5.2. Synthesis of abnormal glycogen and accumulation of extracellular K+
In GS-deficient hippocampus of laboratory rats, the increase in glutamate concentration seems to occur mostly in astrocytes (Perez et al., 2012). This finding is consistent with the dependence of neuronal glutamate exocytosis on astrocyte-derived glutamine (Tani et al., 2010) and with the high affinity and capacity of astrocytes for inward glutamate transport even at the high K+ attainable during seizures (see Claudio et al., 2002). Elevation in astrocytic glutamate concentration and subsequent entry of glutamate into TCA cycle has been correlated with incorporation of glutamate carbons into glycogen via the gluconeogenetic pathway (reviewed by DiNuzzo et al., 2011). In particular, de novo synthesis of glycogen paralleled by activation of the gluconeogenic enzyme fructose-1,6-bisphosphatase (FBPase) has been repeatedly reported after MSO administration both in intact brain (Delorme and Hevor, 1985; Hevor et al., 1986) and in astrocyte-enriched cell cultures (Verge and Hevor, 1995). Gluconeogenic enzymes including pyruvate carboxylase and FBPase are further stimulated by high K+ (Kaufman and Driscoll, 1992; Verge and Hevor, 1995).
Glycogen synthesis appears to be defective under conditions associated with elevated glutamate and K+. Specifically, often but not always MSO-treated animals accumulated abnormal glycogen particles resembling aggregates of insoluble polyglucosans (Delorme and Hevor, 1985; Phelps, 1975). Those animals that did not synthesize abnormal glycogen were seizure-resistant (Bernard-Helary et al., 2000), a feature that turned out to depend on the capacity of metabolizing glycogen (Bernard-Helary et al., 2000; Folbergrova et al., 1996). Aggregates of insoluble polyglucosans in both astrocytes and neurons are also an hallmark of Lafora disease, one of the most severe form of myoclonus epilepsy (Tagliabracci et al., 2008; Valles-Ortega et al., 2011). Inclusions of polyglucosan bodies have long been reported in astrocytes of epileptic patients and often correlated with seizure duration (reviewed by DiNuzzo et al., 2014), although a causal link between polyglucosans accumulation and predisposition to seizures remains to be demonstrated. The resistance of glycogen to hydrolysis is relevant to epilepsy, as rapid mobilization of glycogen has recently been reported to sustain uptake of extracellular K+ by astrocytes (Choi et al., 2012; Xu et al., 2013). Importantly, inhibition of glycogenolysis has been found to completely abolish astrocytic K+ uptake, even in the presence of glucose (Xu et al., 2013). Astrocytes are the primary cells responsible for clearance of neuronally-released K+ into extracellular space (Hertz et al., 2013) and impairment of active astrocytic K+ uptake has been suggested as an important causal factor for epileptic seizures (D’Ambrosio, 2004). Although the regulatory mechanisms underlying K+-induced glycogenolysis are not fully understood (DiNuzzo et al., 2013), it is conceivable that unmetabolizable glycogen contributes to the rise in extracellular K+ observed in epileptic tissue (DiNuzzo et al., 2014).
6. Concluding remarks
In this review, we have outlined the mechanisms that potentially have a role in seizure susceptibility and epileptogenesis. Although incomplete and largely undetailed, our description shows that virtually any process involved in the neuro-glial signaling machinery has been found to be altered in the epileptic brain. Thus, glutamatergic and GABAergic signaling with the associated increases in extracellular K+ all contribute to epileptogenesis via one or more of the pathways described above. We have put forward the view that during epileptogenesis excess glutamatergic neurotransmission leads to reactive astrogliosis, loss-of-function of glutamine synthetase and impaired astrocytic glutamate reuptake. These events initiate a mechanism that induces the gluconeogenesis-mediated synthesis of aberrant glycogen, which ultimately results in unavailability of the glycogenolytic response necessary for astrocytes to actively take up extracellular K+, thus eventually culminating in the epileptic crisis.
Figure 1. Schematic illustration of metabolic pathways of neuron-astrocyte signaling relevant to epileptic seizures.
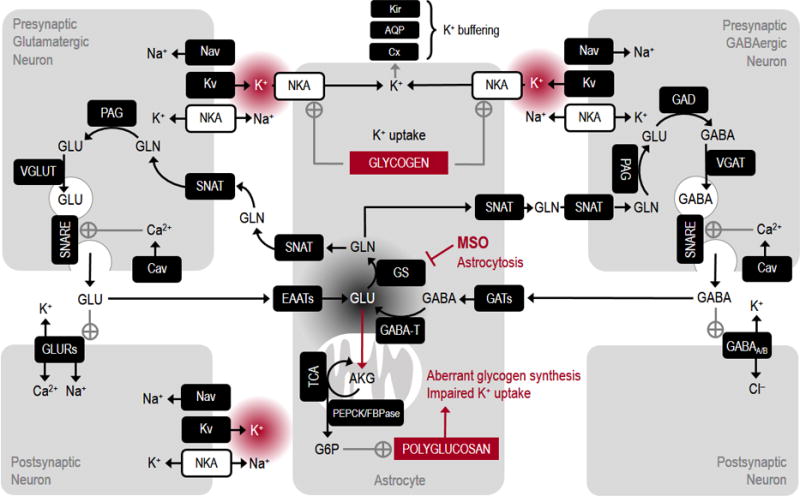
Neuronal electrical activity includes generation and propagation of axonal action potentials through opening of depolarizing Nav and hyperpolarizing Kv. Arrival of traveling depolarization at axon terminal triggers opening of Cav and entry of Ca2+, which in turn activates the secretory machinery of vesicle exocytosis. Relased transmitter (either glutamate or GABA in glutamatergic or GABAergic neurons, respectively) binds to postsynaptic receptors (GLURs or GABAA/B) thereby producing depolarizing excitatory or hyperpolarizing inhibitory ion fluxes on postsynaptic neurons. Depolarization is eventually amplified and propagated in dendrites by Nav and Kv. Transmitter molecules are readily taken up mainly by astrocytes via the relative transporter proteins (EAATs or GATs) and then returned back to neurons in the glutamate-glutamine cycle (note that GABA is converted to glutamate by astrocytic GABA-T). Cycling of neurotransmitter strictly requires the action of astrocyte-specific GS. Inhibition of GS by MSO and/or loss-of-function during epilepsy-induced astrocytosis determines the depletion of presynaptic transmitter molecules and accumulation of glutamate in astrocytes. High glutamate levels in these cells result in excessive channeling of glutamate in TCA cycle and dowstreaming stimulation of gluconeogenic pathway (e.g. PEPCK and FBPase). The activation of FBPase is correlated with new synthesis of aberrant glycogen molecules called polyglucosans, which are partly insoluble and thus inaccessible to degrading enzymes. Ion gradients are primarily re-established by action of NKA in both neurons and astrocytes. However, astrocytic NKA is poised for reuptake of neuronally released K+, a process requiring glycogenolysis. Thus, defective glycogenolysis due to accumulation of polyglucosans impairs astrocytic K+ uptake. Excess extracellular K+ shifts neuronal membrane potential towards chronically depolarized values and possibly initiates and perpetuates the epileptic seizure. Note that in the figure, the stimulation of polyglucosans synthesis by glucose-6-phosphate (G6P) is present only for illustration purposes, as the exact mechanisms leading to polyglucosans synthesis are presently unknown. AQP, aquaporin; Cav, voltage-gated Ca2+ channel; Cx, connexin; EAATs, excitatory amino acid transporters; FBPase, fructose-1,6-bisphosphatase; GAD, glutamate decarboxylase; GABAA/B, GABA receptors; GABA-T, GABA transaminase; GATs, GABA transporters; GLURs, glutamate receptors; GS, glutamine synthetase; Kir, inward rectifier K+ channel; Kv, voltage-gated K+ channel; MSO, L-methionine-SR-sulfoximine; Nav, voltage-gated Na+ channel; NKA, Na+-K+-activated adenosintrisphosphatase; PAG, phosphate-activated glutaminase; PEPCK, phosphoenolpyruvate carboxykinase; SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptor; SNAT, sodium-coupled neutral amino acid transporter; TCA, tricarboxylic acid cycle; VGAT, vesicular GABA transporter; VGLUT, vesicular glutamate transporter.
Acknowledgments
The author S.M. thanks the grant KL2 RR033182 from the National Insititute of Health (NIH) to the University of Minnesota Clinical and Translational Science Institute (CTSI) for support.
Abbreviations
- AIS
axon initial segment
- AMPA
alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AQP4
aquaporin-4
- Best1
bestrophin 1 channel
- Cav
voltage-gated Ca2+ channel
- Cx
connexin
- EAAT
excitatory amino acid transporter
- FBPase
fructose-1,6-bisphosphatase
- GABA
γ-aminobutyric acid
- GABA-T
GABA transaminase
- GAT
GABA transporter
- GDH
glutamate dehydrogenase
- GS
glutamine synthetase
- HCN
hyperpolarization-activated cyclic nucleotide-gated K+ channel
- K2P
two-pore domain K+ channel
- KCC2
K+/Cl− cotransporter 2
- Kir
inward rectifier K+ channel
- Kv
voltage-gated K+ channel
- mGLUR
metabotropic glutamate receptor
- MSO
L-methionine-SR-sulfoximine
- Nav
voltage-gated Na+ channel
- Nax
extracellular Na+ level sensitive Na+ channel
- NKA
Na+-K+-activated adenosintrisphosphatase
- NKCC1
Na+/K+/2Cl− cotransporter 1
- NMDA
N-methyl-D-aspartate
- PAG
phosphate activated glutaminase
- P1(A1/A2)
purinergic adenosine receptor
- P2X
ionotropic purinergic ATP receptor
- P2Y
metabotropic purinergic ATP receptor
- SNARE
soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- SNAT
sodium-coupled neutral amino acid transporter
- TCA
tricarboxylic acid
- TLE
temporal lobe epilepsy
- TREK-1
TWIK-related K+ channel 1
- VGAT
vesicular GABA transporter
- VGLUT
vesicular glutamate transporter
- VSOAC
volume-sensitive organic anion channel
Footnotes
Disclosure/Conflict of interests
The authors declare no conflict of interest.
References
- Akbar MT, Rattray M, Williams RJ, Chong NW, Meldrum BS. Reduction of GABA and glutamate transporter messenger RNAs in the severe-seizure genetically epilepsy-prone rat. Neuroscience. 1998;85:1235–1251. doi: 10.1016/s0306-4522(97)00684-2. [DOI] [PubMed] [Google Scholar]
- Akbarpour B, Sayyah M, Babapour V, Mahdian R, Beheshti S, Kamyab AR. Expression of connexin 30 and connexin 32 in hippocampus of rat during epileptogenesis in a kindling model of epilepsy. Neurosci Bull. 2012;28:729–736. doi: 10.1007/s12264-012-1279-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Leefmans FJ, Leon-Olea M, Mendoza-Sotelo J, Alvarez FJ, Anton B, Garduno R. Immunolocalization of the Na(+)-K(+)-2Cl(−) cotransporter in peripheral nervous tissue of vertebrates. Neuroscience. 2001;104:569–582. doi: 10.1016/s0306-4522(01)00091-4. [DOI] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Williamson A, Palomba M, Eid T, de Lanerolle NC, Nagelhus EA, Adams ME, Froehner SC, Agre P, Ottersen OP. Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc Natl Acad Sci U S A. 2003;100:13615–13620. doi: 10.1073/pnas.2336064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amzica F, Massimini M, Manfridi A. Spatial buffering during slow and paroxysmal sleep oscillations in cortical networks of glial cells in vivo. J Neurosci. 2002;22:1042–1053. doi: 10.1523/JNEUROSCI.22-03-01042.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson WR, Stahl WL. Alpha 2 mRNA of Na+K+ ATPase is increased in astroctyes of rat hippocampus after treatment with kainic acid. Neurochem Int. 1997;31:549–556. doi: 10.1016/s0197-0186(97)00006-5. [DOI] [PubMed] [Google Scholar]
- Arain FM, Boyd KL, Gallagher MJ. Decreased viability and absence-like epilepsy in mice lacking or deficient in the GABAA receptor alpha1 subunit. Epilepsia. 2012;53:e161–165. doi: 10.1111/j.1528-1167.2012.03596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo B, Torres L, Stein M, Cabral FR, Herai R, Okamoto O, Cavalheiro E. Decreased expression of proteins involved in energy metabolism in the hippocampal granular layer of rats submitted to the pilocarpine epilepsy model. Neuroci Lett. 2014;561:46–51. doi: 10.1016/j.neulet.2013.12.040. [DOI] [PubMed] [Google Scholar]
- Avoli M, de Curtis M. GABAergic synchronization in the limbic system and its role in the generation of epileptiform activity. Prog Neurobiol. 2011;95:104–132. doi: 10.1016/j.pneurobio.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badar-Goffer RS, Ben-Yoseph O, Bachelard HS, Morris PG. Neuronal-glial metabolism under depolarizing conditions. A 13C-n.m.r. study. Biochem J. 1992;282(Pt 1):225–230. doi: 10.1042/bj2820225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballanyi K, Grafe P, ten Bruggencate G. Ion activities and potassium uptake mechanisms of glial cells in guinea-pig olfactory cortex slices. J Physiol. 1987;382:159–174. doi: 10.1113/jphysiol.1987.sp016361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannon N, Zhang P, Ilin V, Chistiakova M, Volgushev M. Modulation of synaptic transmission by adenosine in layer 2/3 of the rat visual cortex in vitro. Neuroscience. 2013 doi: 10.1016/j.neuroscience.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnwell LF, Lugo JN, Lee WL, Willis SE, Gertz SJ, Hrachovy RA, Anderson AE. Kv4.2 knockout mice demonstrate increased susceptibility to convulsant stimulation. Epilepsia. 2009;50:1741–1751. doi: 10.1111/j.1528-1167.2009.02086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA. What is a glial cell? Glia. 2003;43:4–5. doi: 10.1002/glia.10252. [DOI] [PubMed] [Google Scholar]
- Basarsky TA, Feighan D, MacVicar BA. Glutamate release through volume-activated channels during spreading depression. J Neurosci. 1999;19:6439–6445. doi: 10.1523/JNEUROSCI.19-15-06439.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bay V, Butt AM. Relationship between glial potassium regulation and axon excitability: a role for glial Kir4.1 channels. Glia. 2012;60:651–660. doi: 10.1002/glia.22299. [DOI] [PubMed] [Google Scholar]
- Becker AJ, Pitsch J, Sochivko D, Opitz T, Staniek M, Chen CC, Campbell KP, Schoch S, Yaari Y, Beck H. Transcriptional upregulation of Cav3.2 mediates epileptogenesis in the pilocarpine model of epilepsy. J Neurosci. 2008;28:13341–13353. doi: 10.1523/JNEUROSCI.1421-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beguin P, Crambert G, Monnet-Tschudi F, Uldry M, Horisberger JD, Garty H, Geering K. FXYD7 is a brain-specific regulator of Na,K-ATPase alpha 1-beta isozymes. EMBO J. 2002;21:3264–3273. doi: 10.1093/emboj/cdf330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch EE. HCN channels: function and clinical implications. Neurology. 2013;80:304–310. doi: 10.1212/WNL.0b013e31827dec42. [DOI] [PubMed] [Google Scholar]
- Bernard-Helary K, Lapouble E, Ardourel M, Hevor T, Cloix JF. Correlation between brain glycogen and convulsive state in mice submitted to methionine sulfoximine. Life Sci. 2000;67:1773–1781. doi: 10.1016/s0024-3205(00)00756-6. [DOI] [PubMed] [Google Scholar]
- Bernard C, Anderson A, Becker A, Poolos NP, Beck H, Johnston D. Acquired dendritic channelopathy in temporal lobe epilepsy. Science. 2004;305:532–535. doi: 10.1126/science.1097065. [DOI] [PubMed] [Google Scholar]
- Bidmon HJ, Gorg B, Palomero-Gallagher N, Schleicher A, Haussinger D, Speckmann EJ, Zilles K. Glutamine synthetase becomes nitrated and its activity is reduced during repetitive seizure activity in the pentylentetrazole model of epilepsy. Epilepsia. 2008;49:1733–1748. doi: 10.1111/j.1528-1167.2008.01642.x. [DOI] [PubMed] [Google Scholar]
- Binder DK, Steinhauser C. Functional changes in astroglial cells in epilepsy. Glia. 2006;54:358–368. doi: 10.1002/glia.20394. [DOI] [PubMed] [Google Scholar]
- Binder DK, Steinhauser C. Role of Astrocyte Dysfunction in Epilepsy. In: Schwartzkroin PA, editor. Encyclopedia of Basic Epilepsy Research. Vol. 1. Academic Press; Oxford: 2009. pp. 412–417. [Google Scholar]
- Binder DK, Yao X, Zador Z, Sick TJ, Verkman AS, Manley GT. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia. 2006;53:631–636. doi: 10.1002/glia.20318. [DOI] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Boison D. Methylxanthines, seizures, and excitotoxicity. Handb Exp Pharmacol. 2011:251–266. doi: 10.1007/978-3-642-13443-2_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H. Properties of human glial cells associated with epileptic seizure foci. Epilepsy Res. 1998;32:286–303. doi: 10.1016/s0920-1211(98)00059-x. [DOI] [PubMed] [Google Scholar]
- Bottcher T, Goiny M, Bering J, Domhof S, Nau R, Ungerstedt U. Regional differences in glutamine synthetase inhibition by L-methionine sulfoximine: a microdialysis study in the rabbit brain. Exp Brain Res. 2003;150:194–200. doi: 10.1007/s00221-003-1401-0. [DOI] [PubMed] [Google Scholar]
- Brew HM, Gittelman JX, Silverstein RS, Hanks TD, Demas VP, Robinson LC, Robbins CA, McKee-Johnson J, Chiu SY, Messing A, Tempel BL. Seizures and reduced life span in mice lacking the potassium channel subunit Kv1.2, but hypoexcitability and enlarged Kv1 currents in auditory neurons. J Neurophysiol. 2007;98:1501–1525. doi: 10.1152/jn.00640.2006. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Introduction to purinergic signalling in the brain. Adv Exp Med Biol. 2013;986:1–12. doi: 10.1007/978-94-007-4719-7_1. [DOI] [PubMed] [Google Scholar]
- Cai L, Du T, Song D, Li B, Hertz L, Peng L. Astrocyte ERK phosphorylation precedes K(+)-induced swelling but follows hypotonicity-induced swelling. Neuropathology. 2011;31:250–264. doi: 10.1111/j.1440-1789.2010.01172.x. [DOI] [PubMed] [Google Scholar]
- Carmignoto G, Haydon PG. Astrocyte calcium signaling and epilepsy. Glia. 2012;60:1227–1233. doi: 10.1002/glia.22318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavus I, Kasoff WS, Cassaday MP, Jacob R, Gueorguieva R, Sherwin RS, Krystal JH, Spencer DD, Abi-Saab WM. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann Neurol. 2005;57:226–235. doi: 10.1002/ana.20380. [DOI] [PubMed] [Google Scholar]
- Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. III. The steady state. J Biol Chem. 1955;217:409–427. [PubMed] [Google Scholar]
- Chever O, Djukic B, McCarthy KD, Amzica F. Implication of Kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional Kir4.1 knock-out mice. J Neurosci. 2010;30:15769–15777. doi: 10.1523/JNEUROSCI.2078-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HB, Gordon GR, Zhou N, Tai C, Rungta RL, Martinez J, Milner TA, Ryu JK, McLarnon JG, Tresguerres M, Levin LR, Buck J, MacVicar BA. Metabolic communication between astrocytes and neurons via bicarbonate-responsive soluble adenylyl cyclase. Neuron. 2012;75:1094–1104. doi: 10.1016/j.neuron.2012.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury GM, Patel AB, Mason GF, Rothman DL, Behar KL. Glutamatergic and GABAergic neurotransmitter cycling and energy metabolism in rat cerebral cortex during postnatal development. J Cereb Blood Flow Metab. 2007;27:1895–1907. doi: 10.1038/sj.jcbfm.9600490. [DOI] [PubMed] [Google Scholar]
- Clasadonte J, Dong J, Hines DJ, Haydon PG. Astrocyte control of synaptic NMDA receptors contributes to the progressive development of temporal lobe epilepsy. Proc Natl Acad Sci U S A. 2013;110:17540–17545. doi: 10.1073/pnas.1311967110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudio OI, Berrios N, Garcia M, Casasnovas R, Ortiz JG. Veratridine, but not elevated K+, inhibits excitatory amino acid transporter activity in rat hippocampal slices. Epilepsia. 2002;43(Suppl 5):184–187. doi: 10.1046/j.1528-1157.43.s.5.5.x. [DOI] [PubMed] [Google Scholar]
- Clayton GH, Owens GC, Wolff JS, Smith RL. Ontogeny of cation-Cl-cotransporter expression in rat neocortex. Brain Res Dev Brain Res. 1998;109:281–292. doi: 10.1016/s0165-3806(98)00078-9. [DOI] [PubMed] [Google Scholar]
- Collignon F, Wetjen NM, Cohen-Gadol AA, Cascino GD, Parisi J, Meyer FB, Marsh WR, Roche P, Weigand SD. Altered expression of connexin subtypes in mesial temporal lobe epilepsy in humans. J Neurosurg. 2006;105:77–87. doi: 10.3171/jns.2006.105.1.77. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Eid T. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia. 2012;60:1215–1226. doi: 10.1002/glia.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crevecoeur J, Kaminski RM, Rogister B, Foerch P, Vandenplas C, Neveux M, Mazzuferi M, Kroonen J, Poulet C, Martin D, Sadzot B, Rikir E, Klitgaard H, Moonen G, Deprez M. Expression pattern of synaptic vesicle protein 2 (SV2) isoforms in patients with temporal lobe epilepsy and hippocampal sclerosis. Neuropathol Appl Neurobiol. 2013 doi: 10.1111/nan.12054. [DOI] [PubMed] [Google Scholar]
- D’Adamo MC, Catacuzzeno L, Di Giovanni G, Franciolini F, Pessia M. K channelepsy: progress in the neurobiology of potassium channels and epilepsy. Front Cell Neurosci. 2013;7:134. doi: 10.3389/fncel.2013.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambrosio R. The role of glial membrane ion channels in seizures and epileptogenesis. Pharmacol Ther. 2004;103:95–108. doi: 10.1016/j.pharmthera.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Dalby NO. Inhibition of gamma-aminobutyric acid uptake: anatomy, physiology and effects against epileptic seizures. Eur J Pharmacol. 2003;479:127–137. doi: 10.1016/j.ejphar.2003.08.063. [DOI] [PubMed] [Google Scholar]
- Dallerac G, Chever O, Rouach N. How do astrocytes shape synaptic transmission? Insights from electrophysiology. Front Cell Neurosci. 2013;7:159. doi: 10.3389/fncel.2013.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- de Lanerolle NC, Lee TS. New facets of the neuropathology and molecular profile of human temporal lobe epilepsy. Epilepsy Behav. 2005;7:190–203. doi: 10.1016/j.yebeh.2005.06.003. [DOI] [PubMed] [Google Scholar]
- de Lanerolle NC, Lee TS, Spencer DD. Astrocytes and epilepsy. Neurotherapeutics. 2010;7:424–438. doi: 10.1016/j.nurt.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme P, Hevor TK. Glycogen particles in methionine sulfoximine epileptogenic rodent brain and liver after the administration of methionine and actinomycin D. Neuropathol Appl Neurobiol. 1985;11:117–128. doi: 10.1111/j.1365-2990.1985.tb00009.x. [DOI] [PubMed] [Google Scholar]
- Delpire E, Mount DB. Human and murine phenotypes associated with defects in cation-chloride cotransport. Annu Rev Physiol. 2002;64:803–843. doi: 10.1146/annurev.physiol.64.081501.155847. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci. 2013;36:174–184. doi: 10.1016/j.tins.2012.11.008. [DOI] [PubMed] [Google Scholar]
- Dey D, Eckle VS, Vitko I, Sullivan KA, Lasiecka ZM, Winckler B, Stornetta RL, Williamson JM, Kapur J, Perez-Reyes E. A potassium leak channel silences hyperactive neurons and ameliorates status epilepticus. Epilepsia. 2013 doi: 10.1111/epi.12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, Coulter DA, Carmignoto G, Haydon PG. Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J Neurosci. 2007;27:10674–10684. doi: 10.1523/JNEUROSCI.2001-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNuzzo M, Giove F. Activity-dependent energy budget for neocortical signaling: effect of short-term synaptic plasticity on the energy expended by spiking and synaptic activity. J Neurosci Res. 2012;90:2094–2102. doi: 10.1002/jnr.23098. [DOI] [PubMed] [Google Scholar]
- DiNuzzo M, Mangia S, Maraviglia B, Giove F. The role of astrocytic glycogen in supporting the energetics of neuronal activity. Neurochem Res. 2012;37:2432–2438. doi: 10.1007/s11064-012-0802-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNuzzo M, Mangia S, Maraviglia B, Giove F. Regulatory mechanisms for glycogenolysis and K+ uptake in brain astrocytes. Neurochem Int. 2013;63:458–464. doi: 10.1016/j.neuint.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNuzzo M, Mangia S, Maraviglia B, Giove F. Does abnormal glycogen structure contribute to increased susceptibility to seizures in epilepsy? Metab Brain Dis. 2014 doi: 10.1007/s11011-014-9524-5. in print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNuzzo M, Maraviglia B, Giove F. Why does the brain (not) have glycogen? Bioessays. 2011;33:319–326. doi: 10.1002/bies.201000151. [DOI] [PubMed] [Google Scholar]
- Dragunow M, Goddard GV. Adenosine modulation of amygdala kindling. Exp Neurol. 1984;84:654–665. doi: 10.1016/0014-4886(84)90212-7. [DOI] [PubMed] [Google Scholar]
- Dudek FE, Yasumura T, Rash JE. ‘Non-synaptic’ mechanisms in seizures and epileptogenesis. Cell Biol Int. 1998;22:793–805. doi: 10.1006/cbir.1999.0397. [DOI] [PubMed] [Google Scholar]
- Dufour S, Dufour P, Chever O, Vallee R, Amzica F. In vivo simultaneous intra- and extracellular potassium recordings using a micro-optrode. J Neurosci Methods. 2011;194:206–217. doi: 10.1016/j.jneumeth.2010.10.004. [DOI] [PubMed] [Google Scholar]
- During MJ, Spencer DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. 1993;341:1607–1610. doi: 10.1016/0140-6736(93)90754-5. [DOI] [PubMed] [Google Scholar]
- Eid T, Ghosh A, Wang Y, Beckstrom H, Zaveri HP, Lee TS, Lai JC, Malthankar-Phatak GH, de Lanerolle NC. Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain. 2008a;131:2061–2070. doi: 10.1093/brain/awn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Lee TS, Thomas MJ, Amiry-Moghaddam M, Bjornsen LP, Spencer DD, Agre P, Ottersen OP, de Lanerolle NC. Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the human epileptogenic hippocampus. Proc Natl Acad Sci U S A. 2005;102:1193–1198. doi: 10.1073/pnas.0409308102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Lee TS, Wang Y, Perez E, Drummond J, Lauritzen F, Bergersen LH, Meador-Woodruff JH, Spencer DD, de Lanerolle NC, McCullumsmith RE. Gene expression of glutamate metabolizing enzymes in the hippocampal formation in human temporal lobe epilepsy. Epilepsia. 2013a;54:228–238. doi: 10.1111/epi.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Thomas MJ, Spencer DD, Runden-Pran E, Lai JC, Malthankar GV, Kim JH, Danbolt NC, Ottersen OP, de Lanerolle NC. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet. 2004;363:28–37. doi: 10.1016/s0140-6736(03)15166-5. [DOI] [PubMed] [Google Scholar]
- Eid T, Tu N, Lee TS, Lai JC. Regulation of astrocyte glutamine synthetase in epilepsy. Neurochem Int. 2013b;63:670–681. doi: 10.1016/j.neuint.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Williamson A, Lee TS, Petroff OA, de Lanerolle NC. Glutamate and astrocytes–key players in human mesial temporal lobe epilepsy? Epilepsia. 2008b;49(Suppl 2):42–52. doi: 10.1111/j.1528-1167.2008.01492.x. [DOI] [PubMed] [Google Scholar]
- Eijkelkamp N, Linley JE, Baker MD, Minett MS, Cregg R, Werdehausen R, Rugiero F, Wood JN. Neurological perspectives on voltage-gated sodium channels. Brain. 2012;135:2585–2612. doi: 10.1093/brain/aws225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott RC, Lowenstein DH. Gene expression profiling of seizure disorders. Neurochem Res. 2004;29:1083–1092. doi: 10.1023/b:nere.0000023595.12396.1b. [DOI] [PubMed] [Google Scholar]
- Elliott RC, Miles MF, Lowenstein DH. Overlapping microarray profiles of dentate gyrus gene expression during development- and epilepsy-associated neurogenesis and axon outgrowth. J Neurosci. 2003;23:2218–2227. doi: 10.1523/JNEUROSCI.23-06-02218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estacion M, Gasser A, Dib-Hajj SD, Waxman SG. A sodium channel mutation linked to epilepsy increases ramp and persistent current of Nav1.3 and induces hyperexcitability in hippocampal neurons. Exp Neurol. 2010;224:362–368. doi: 10.1016/j.expneurol.2010.04.012. [DOI] [PubMed] [Google Scholar]
- Fellin T, Haydon PG. Do astrocytes contribute to excitation underlying seizures? Trends Mol Med. 2005;11:530–533. doi: 10.1016/j.molmed.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Fernandes MJ, Naffah-Mazzacoratti MG, Cavalheiro EA. Na+K+ ATPase activity in the rat hippocampus: a study in the pilocarpine model of epilepsy. Neurochem Int. 1996;28:497–500. doi: 10.1016/0197-0186(95)00153-0. [DOI] [PubMed] [Google Scholar]
- Fertziger AP, Ranck JB., Jr Potassium accumulation in interstitial space during epileptiform seizures. Exp Neurol. 1970;26:571–585. doi: 10.1016/0014-4886(70)90150-0. [DOI] [PubMed] [Google Scholar]
- Fisher RS, Pedley TA, Moody WJ, Jr, Prince DA. The role of extracellular potassium in hippocampal epilepsy. Arch Neurol. 1976;33:76–83. doi: 10.1001/archneur.1976.00500020004002. [DOI] [PubMed] [Google Scholar]
- Fisher RS, van Emde Boas W, Blume W, Elger C, Genton P, Lee P, Engel J., Jr Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE) Epilepsia. 2005;46:470–472. doi: 10.1111/j.0013-9580.2005.66104.x. [DOI] [PubMed] [Google Scholar]
- Folbergrova J, Katsura KI, Siesjo BK. Glycogen accumulated in the brain following insults is not degraded during a subsequent period of ischemia. J Neurol Sci. 1996;137:7–13. doi: 10.1016/0022-510x(96)82226-x. [DOI] [PubMed] [Google Scholar]
- Folbergrova J, Passonneau JV, Lowry OH, Schulz DW. Glycogen, ammonia and related metabolities in the brain during seizures evoked by methionine sulphoximine. J Neurochem. 1969;16:191–203. doi: 10.1111/j.1471-4159.1969.tb05937.x. [DOI] [PubMed] [Google Scholar]
- Fonseca CG, Green CR, Nicholson LF. Upregulation in astrocytic connexin 43 gap junction levels may exacerbate generalized seizures in mesial temporal lobe epilepsy. Brain Res. 2002;929:105–116. doi: 10.1016/s0006-8993(01)03289-9. [DOI] [PubMed] [Google Scholar]
- Fricke MN, Jones-Davis DM, Mathews GC. Glutamine uptake by System A transporters maintains neurotransmitter GABA synthesis and inhibitory synaptic transmission. J Neurochem. 2007;102:1895–1904. doi: 10.1111/j.1471-4159.2007.04649.x. [DOI] [PubMed] [Google Scholar]
- Frigerio F, Frasca A, Weissberg I, Parrella S, Friedman A, Vezzani A, Noe FM. Long-lasting pro-ictogenic effects induced in vivo by rat brain exposure to serum albumin in the absence of concomitant pathology. Epilepsia. 2012;53:1887–1897. doi: 10.1111/j.1528-1167.2012.03666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadea A, Lopez-Colome AM. Glial transporters for glutamate, glycine, and GABA: II. GABA transporters. J Neurosci Res. 2001;63:461–468. doi: 10.1002/jnr.1040. [DOI] [PubMed] [Google Scholar]
- Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev. 2005;85:423–493. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- Ghauri M, Chapman AG, Meldrum BS. Convulsant and anticonvulsant actions of agonists and antagonists of group III mGluRs. Neuroreport. 1996;7:1469–1474. doi: 10.1097/00001756-199606170-00005. [DOI] [PubMed] [Google Scholar]
- Giaume C, Koulakoff A, Roux L, Holcman D, Rouach N. Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat Rev Neurosci. 2010;11:87–99. doi: 10.1038/nrn2757. [DOI] [PubMed] [Google Scholar]
- Giorgi FS, Pizzanelli C, Biagioni F, Murri L, Fornai F. The role of norepinephrine in epilepsy: from the bench to the bedside. Neurosci Biobehav Rev. 2004;28:507–524. doi: 10.1016/j.neubiorev.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Gmiro VE, Serdyuk SE. Combined blockade of AMPA and NMDA receptors in the brain of rats prevents pentylenetetrazole-induced clonic and tonic-clonic seizures without ataxia. Bull Exp Biol Med. 2008;145:728–730. doi: 10.1007/s10517-008-0194-3. [DOI] [PubMed] [Google Scholar]
- Gomez-Gonzalo M, Losi G, Chiavegato A, Zonta M, Cammarota M, Brondi M, Vetri F, Uva L, Pozzan T, de Curtis M, Ratto GM, Carmignoto G. An excitatory loop with astrocytes contributes to drive neurons to seizure threshold. PLoS Biol. 2010;8:e1000352. doi: 10.1371/journal.pbio.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorter JA, Zurolo E, Iyer A, Fluiter K, van Vliet EA, Baayen JC, Aronica E. Induction of sodium channel Na(x) (SCN7A) expression in rat and human hippocampus in temporal lobe epilepsy. Epilepsia. 2010;51:1791–1800. doi: 10.1111/j.1528-1167.2010.02678.x. [DOI] [PubMed] [Google Scholar]
- Green JD. The Hippocampus. Physiol Rev. 1964;44:561–608. doi: 10.1152/physrev.1964.44.4.561. [DOI] [PubMed] [Google Scholar]
- Grisar T. Glial and neuronal Na+-K+ pump in epilepsy. Ann Neurol. 1984;16(Suppl):S128–134. doi: 10.1002/ana.410160719. [DOI] [PubMed] [Google Scholar]
- Grisar T, Delgado-Escueta AV. Astroglial contribution in human temporal lobe epilepsy: K+ activation of Na+,K+-ATPase in bulk isolated glial cells and synaptosomes. Brain Res. 1986;364:1–11. doi: 10.1016/0006-8993(86)90981-9. [DOI] [PubMed] [Google Scholar]
- Grisar T, Franck G, Delgado-Escueta AV. Glial contribution to seizure: K+ activation of (Na+, K+)-ATPase in bulk isolated glial cells and synaptosomes of epileptogenic cortex. Brain Res. 1983;261:75–84. doi: 10.1016/0006-8993(83)91285-4. [DOI] [PubMed] [Google Scholar]
- Grisar T, Frere JM, Franck G. Effect of K+ ions on kinetic properties of the (Na+, K+)-ATPase (EC 3.6.1.3) of bulk isolated glial cells, perikarya and synaptosomes from rabbit brain cortex. Brain Res. 1979;165:87–103. doi: 10.1016/0006-8993(79)90047-7. [DOI] [PubMed] [Google Scholar]
- Grisar T, Guillaume D, Delgado-Escueta AV. Contribution of Na+,K(+)-ATPase to focal epilepsy: a brief review. Epilepsy Res. 1992;12:141–149. doi: 10.1016/0920-1211(92)90034-q. [DOI] [PubMed] [Google Scholar]
- Guillaume D. Brain cortical (Na+ K+)-ATPase in epilepsy. A biochemical study in animals and humans. Acta Neurol Belg. 1988;88:257–280. [PubMed] [Google Scholar]
- Guo F, Sun F, Yu JL, Wang QH, Tu DY, Mao XY, Liu R, Wu KC, Xie N, Hao LY, Cai JQ. Abnormal expressions of glutamate transporters and metabotropic glutamate receptor 1 in the spontaneously epileptic rat hippocampus. Brain Res Bull. 2010;81:510–516. doi: 10.1016/j.brainresbull.2009.10.008. [DOI] [PubMed] [Google Scholar]
- Guthrie PB, Knappenberger J, Segal M, Bennett MV, Charles AC, Kater SB. ATP released from astrocytes mediates glial calcium waves. J Neurosci. 1999;19:520–528. doi: 10.1523/JNEUROSCI.19-02-00520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hablitz JJ, Heinemann U. Alterations in the microenvironment during spreading depression associated with epileptiform activity in the immature neocortex. Brain Res Dev Brain Res. 1989;46:243–252. doi: 10.1016/0165-3806(89)90288-5. [DOI] [PubMed] [Google Scholar]
- Hadera MG, Smeland OB, McDonald TS, Tan KN, Sonnewald U, Borges K. Triheptanoin partially restores levels of tricarboxylic acid cycle intermediates in the mouse pilocarpine model of epilepsy. J Neurochem. 2013 doi: 10.1111/jnc.12610. [DOI] [PubMed] [Google Scholar]
- Hammer J, Alvestad S, Osen KK, Skare O, Sonnewald U, Ottersen OP. Expression of glutamine synthetase and glutamate dehydrogenase in the latent phase and chronic phase in the kainate model of temporal lobe epilepsy. Glia. 2008;56:856–868. doi: 10.1002/glia.20659. [DOI] [PubMed] [Google Scholar]
- Han HA, Cortez MA, Snead OC. GABAB Receptor and Absence Epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. National Center for Biotechnology Information (US); 2012. [PubMed] [Google Scholar]
- Rogawski Michael A, Antonio V Delgado-Escueta, Jeffrey L Noebels, Massimo Avoli, Richard W Olsen., Bethesda (MD)
- Heinemann U, Konnerth A, Pumain R, Wadman WJ. Extracellular calcium and potassium concentration changes in chronic epileptic brain tissue. Adv Neurol. 1986;44:641–661. [PubMed] [Google Scholar]
- Heinemann U, Lux HD. Ceiling of stimulus induced rises in extracellular potassium concentration in the cerebral cortex of cat. Brain Res. 1977;120:231–249. doi: 10.1016/0006-8993(77)90903-9. [DOI] [PubMed] [Google Scholar]
- Heja L, Nyitrai G, Kekesi O, Dobolyi A, Szabo P, Fiath R, Ulbert I, Pal-Szenthe B, Palkovits M, Kardos J. Astrocytes convert network excitation to tonic inhibition of neurons. BMC Biol. 2012;10:26. doi: 10.1186/1741-7007-10-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henn FA, Haljamae H, Hamberger A. Glial cell function: active control of extracellular K + concentration. Brain Res. 1972;43:437–443. doi: 10.1016/0006-8993(72)90399-x. [DOI] [PubMed] [Google Scholar]
- Hertz L. An intense potassium uptake into astrocytes, its further enhancement by high concentrations of potassium, and its possible involvement in potassium homeostasis at the cellular level. Brain Res. 1978;145:202–208. doi: 10.1016/0006-8993(78)90812-0. [DOI] [PubMed] [Google Scholar]
- Hertz L, Xu J, Song D, Yan E, Gu L, Peng L. Astrocytic and neuronal accumulation of elevated extracellular K(+) with a 2/3 K(+)/Na(+) flux ratio-consequences for energy metabolism, osmolarity and higher brain function. Front Comput Neurosci. 2013;7:114. doi: 10.3389/fncom.2013.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevor TK, Delorme P, Beauvillain JC. Glycogen synthesis and immunocytochemical study of fructose-1,6-biphosphatase in methionine sulfoximine epileptogenic rodent brain. J Cereb Blood Flow Metab. 1986;6:292–297. doi: 10.1038/jcbfm.1986.51. [DOI] [PubMed] [Google Scholar]
- Hinterkeuser S, Schroder W, Hager G, Seifert G, Blumcke I, Elger CE, Schramm J, Steinhauser C. Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur J Neurosci. 2000;12:2087–2096. doi: 10.1046/j.1460-9568.2000.00104.x. [DOI] [PubMed] [Google Scholar]
- Hirao T, Morimoto K, Yamamoto Y, Watanabe T, Sato H, Sato K, Sato S, Yamada N, Tanaka K, Suwaki H. Time-dependent and regional expression of GABA transporter mRNAs following amygdala-kindled seizures in rats. Brain Res Mol Brain Res. 1998;54:49–55. doi: 10.1016/s0169-328x(97)00323-9. [DOI] [PubMed] [Google Scholar]
- Holland KD, Kearney JA, Glauser TA, Buck G, Keddache M, Blankston JR, Glaaser IW, Kass RS, Meisler MH. Mutation of sodium channel SCN3A in a patient with cryptogenic pediatric partial epilepsy. Neurosci Lett. 2008;433:65–70. doi: 10.1016/j.neulet.2007.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes KH, Keele NB, Shinnick-Gallagher P. Loss of mGluR-mediated hyperpolarizations and increase in mGluR depolarizations in basolateral amygdala neurons in kindling-induced epilepsy. J Neurophysiol. 1996;76:2808–2812. doi: 10.1152/jn.1996.76.4.2808. [DOI] [PubMed] [Google Scholar]
- Honegger P, Pardo B. Separate neuronal and glial Na+,K+-ATPase isoforms regulate glucose utilization in response to membrane depolarization and elevated extracellular potassium. J Cereb Blood Flow Metab. 1999;19:1051–1059. doi: 10.1097/00004647-199909000-00013. [DOI] [PubMed] [Google Scholar]
- Hu Y, Jiang L, Chen H. Change of presynaptic vesicle cycling in the hippocampus after status convulsion. Neurosciences (Riyadh) 2012;17:82–84. [PubMed] [Google Scholar]
- Huang Z, Lujan R, Kadurin I, Uebele VN, Renger JJ, Dolphin AC, Shah MM. Presynaptic HCN1 channels regulate Cav3.2 activity and neurotransmission at select cortical synapses. Nat Neurosci. 2011;14:478–486. doi: 10.1038/nn.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwa GG, Avoli M. The involvement of excitatory amino acids in neocortical epileptogenesis: NMDA and non-NMDA receptors. Exp Brain Res. 1991;86:248–256. doi: 10.1007/BF00228949. [DOI] [PubMed] [Google Scholar]
- Hwa GG, Avoli M, Oliver A, Villemure JG. Bicuculline-induced epileptogenesis in the human neocortex maintained in vitro. Exp Brain Res. 1991;83:329–339. doi: 10.1007/BF00231156. [DOI] [PubMed] [Google Scholar]
- Jauch R, Windmuller O, Lehmann TN, Heinemann U, Gabriel S. Effects of barium, furosemide, ouabaine and 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS) on ionophoretically-induced changes in extracellular potassium concentration in hippocampal slices from rats and from patients with epilepsy. Brain Res. 2002;925:18–27. doi: 10.1016/s0006-8993(01)03254-1. [DOI] [PubMed] [Google Scholar]
- Jensen MS, Azouz R, Yaari Y. Variant firing patterns in rat hippocampal pyramidal cells modulated by extracellular potassium. J Neurophysiol. 1994;71:831–839. doi: 10.1152/jn.1994.71.3.831. [DOI] [PubMed] [Google Scholar]
- Jones RS, Lambert JD. Synchronous discharges in the rat entorhinal cortex in vitro: site of initiation and the role of excitatory amino acid receptors. Neuroscience. 1990;34:657–670. doi: 10.1016/0306-4522(90)90172-z. [DOI] [PubMed] [Google Scholar]
- Jorge BS, Campbell CM, Miller AR, Rutter ED, Gurnett CA, Vanoye CG, George AL, Jr, Kearney JA. Voltage-gated potassium channel KCNV2 (Kv8.2) contributes to epilepsy susceptibility. Proc Natl Acad Sci U S A. 2011;108:5443–5448. doi: 10.1073/pnas.1017539108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam K, Nicoll R. Excitatory synaptic transmission persists independently of the glutamate-glutamine cycle. J Neurosci. 2007;27:9192–9200. doi: 10.1523/JNEUROSCI.1198-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamori K, Ross BD. Electrographic seizures are significantly reduced by in vivo inhibition of neuronal uptake of extracellular glutamine in rat hippocampus. Epilepsy Res. 2013;107:20–36. doi: 10.1016/j.eplepsyres.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Kaufman EE, Driscoll BF. Carbon dioxide fixation in neuronal and astroglial cells in culture. J Neurochem. 1992;58:258–262. doi: 10.1111/j.1471-4159.1992.tb09304.x. [DOI] [PubMed] [Google Scholar]
- Kim DW, Lee SK, Yun CH, Kim KK, Lee DS, Chung CK, Chang KH. Parietal lobe epilepsy: the semiology, yield of diagnostic workup, and surgical outcome. Epilepsia. 2004;45:641–649. doi: 10.1111/j.0013-9580.2004.33703.x. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129:1045–1056. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konnerth A, Heinemann U, Yaari Y. Nonsynaptic epileptogenesis in the mammalian hippocampus in vitro. I. Development of seizurelike activity in low extracellular calcium. J Neurophysiol. 1986;56:409–423. doi: 10.1152/jn.1986.56.2.409. [DOI] [PubMed] [Google Scholar]
- Korff CM, Scheffer IE. Epilepsy classification: a cycle of evolution and revolution. Curr Opin Neurol. 2013;26:163–167. doi: 10.1097/WCO.0b013e32835ee58e. [DOI] [PubMed] [Google Scholar]
- Kuga N, Sasaki T, Takahara Y, Matsuki N, Ikegaya Y. Large-scale calcium waves traveling through astrocytic networks in vivo. J Neurosci. 2011;31:2607–2614. doi: 10.1523/JNEUROSCI.5319-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laake JH, Slyngstad TA, Haug FM, Ottersen OP. Glutamine from glial cells is essential for the maintenance of the nerve terminal pool of glutamate: immunogold evidence from hippocampal slice cultures. J Neurochem. 1995;65:871–881. doi: 10.1046/j.1471-4159.1995.65020871.x. [DOI] [PubMed] [Google Scholar]
- Lalo U, Palygin O, Rasooli-Nejad S, Andrew J, Haydon PG, Pankratov Y. Exocytosis of ATP from astrocytes modulates phasic and tonic inhibition in the neocortex. PLoS Biol. 2014;12:e1001747. doi: 10.1371/journal.pbio.1001747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laschet J, Guillaume D, Grisar T, Vergniolle-Burette M, Minet A. Effect of milacemide on audiogenic seizures and cortical (Na+, K+)-ATPase of DBA/2J mice. Epilepsia. 1991;32:151–156. doi: 10.1111/j.1528-1157.1991.tb05628.x. [DOI] [PubMed] [Google Scholar]
- Laschet J, Guillaume D, Vergniolle-Burette M, Grisar T. Milacemide stimulates deficient glial Na+, K(+)-ATPase in freezing-induced epileptogenic cortex of cats. Brain Res. 1990;517:168–174. doi: 10.1016/0006-8993(90)91022-9. [DOI] [PubMed] [Google Scholar]
- Laschet JJ, Kurcewicz I, Minier F, Trottier S, Khallou-Laschet J, Louvel J, Gigout S, Turak B, Biraben A, Scarabin JM, Devaux B, Chauvel P, Pumain R. Dysfunction of GABAA receptor glycolysis-dependent modulation in human partial epilepsy. Proc Natl Acad Sci U S A. 2007;104:3472–3477. doi: 10.1073/pnas.0606451104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebovitz RM. Quantitative examination of dynamic interneuronal coupling via single-spike extracellular potassium ion transients. J Theor Biol. 1996;180:11–25. doi: 10.1006/jtbi.1996.0074. [DOI] [PubMed] [Google Scholar]
- Lee DJ, Hsu MS, Seldin MM, Arellano JL, Binder DK. Decreased expression of the glial water channel aquaporin-4 in the intrahippocampal kainic acid model of epileptogenesis. Exp Neurol. 2012;235:246–255. doi: 10.1016/j.expneurol.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Schwab C, McGeer PL. Astrocytes are GABAergic cells that modulate microglial activity. Glia. 2011;59:152–165. doi: 10.1002/glia.21087. [DOI] [PubMed] [Google Scholar]
- Lee S, Yoon BE, Berglund K, Oh SJ, Park H, Shin HS, Augustine GJ, Lee CJ. Channel-mediated tonic GABA release from glia. Science. 2010;330:790–796. doi: 10.1126/science.1184334. [DOI] [PubMed] [Google Scholar]
- Lee TS, Bjornsen LP, Paz C, Kim JH, Spencer SS, Spencer DD, Eid T, de Lanerolle NC. GAT1 and GAT3 expression are differently localized in the human epileptogenic hippocampus. Acta Neuropathol. 2006;111:351–363. doi: 10.1007/s00401-005-0017-9. [DOI] [PubMed] [Google Scholar]
- Liang SL, Carlson GC, Coulter DA. Dynamic regulation of synaptic GABA release by the glutamate-glutamine cycle in hippocampal area CA1. J Neurosci. 2006;26:8537–8548. doi: 10.1523/JNEUROSCI.0329-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Stafstrom CE, Sarkisian MR, Yang Y, Hori A, Tandon P, Holmes GL. Seizure-induced glutamate release in mature and immature animals: an in vivo microdialysis study. Neuroreport. 1997;8:2019–2023. doi: 10.1097/00001756-199705260-00043. [DOI] [PubMed] [Google Scholar]
- Longuemare MC, Swanson RA. Net glutamate release from astrocytes is not induced by extracellular potassium concentrations attainable in brain. J Neurochem. 1997;69:879–882. doi: 10.1046/j.1471-4159.1997.69020879.x. [DOI] [PubMed] [Google Scholar]
- Lothman E, Lamanna J, Cordingley G, Rosenthal M, Somjen G. Responses of electrical potential, potassium levels, and oxidative metabolic activity of the cerebral neocortex of cats. Brain Res. 1975;88:15–36. doi: 10.1016/0006-8993(75)90943-9. [DOI] [PubMed] [Google Scholar]
- Madsen KK, White HS, Schousboe A. Neuronal and non-neuronal GABA transporters as targets for antiepileptic drugs. Pharmacol Ther. 2010;125:394–401. doi: 10.1016/j.pharmthera.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Maheshwari A, Nahm WK, Noebels JL. Paradoxical proepileptic response to NMDA receptor blockade linked to cortical interneuron defect in stargazer mice. Front Cell Neurosci. 2013;7:156. doi: 10.3389/fncel.2013.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malthankar-Phatak GH, de Lanerolle N, Eid T, Spencer DD, Behar KL, Spencer SS, Kim JH, Lai JC. Differential glutamate dehydrogenase (GDH) activity profile in patients with temporal lobe epilepsy. Epilepsia. 2006;47:1292–1299. doi: 10.1111/j.1528-1167.2006.00543.x. [DOI] [PubMed] [Google Scholar]
- Mangia S, Giove F, Dinuzzo M. Metabolic pathways and activity-dependent modulation of glutamate concentration in the human brain. Neurochem Res. 2012;37:2554–2561. doi: 10.1007/s11064-012-0848-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Guo F, Yu J, Min D, Wang Z, Xie N, Chen T, Shaw C, Cai J. Up-regulation of GABA transporters and GABA(A) receptor alpha1 subunit in tremor rat hippocampus. Neurosci Lett. 2010;486:150–155. doi: 10.1016/j.neulet.2010.09.033. [DOI] [PubMed] [Google Scholar]
- Marichich ES, Nasello AG. Epilepsy and adenosinetriphosphate (ATP): effect of electrical stimulation and high potassium perfusion on hippocampal ATP contents. Brain Res. 1973;57:409–416. doi: 10.1016/0006-8993(73)90146-7. [DOI] [PubMed] [Google Scholar]
- Martineau M, Shi T, Puyal J, Knolhoff AM, Dulong J, Gasnier B, Klingauf J, Sweedler JV, Jahn R, Mothet JP. Storage and uptake of D-serine into astrocytic synaptic-like vesicles specify gliotransmission. J Neurosci. 2013;33:3413–3423. doi: 10.1523/JNEUROSCI.3497-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathern GW, Mendoza D, Lozada A, Pretorius JK, Dehnes Y, Danbolt NC, Nelson N, Leite JP, Chimelli L, Born DE, Sakamoto AC, Assirati JA, Fried I, Peacock WJ, Ojemann GA, Adelson PD. Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology. 1999;52:453–472. doi: 10.1212/wnl.52.3.453. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Pretorius JK, Mendoza D, Lozada A, Leite JP, Chimelli L, Fried I, Sakamoto AC, Assirati JA, Adelson PD. Increased hippocampal AMPA and NMDA receptor subunit immunoreactivity in temporal lobe epilepsy patients. J Neuropathol Exp Neurol. 1998;57:615–634. doi: 10.1097/00005072-199806000-00008. [DOI] [PubMed] [Google Scholar]
- Matveeva EA, Davis VA, Whiteheart SW, Vanaman TC, Gerhardt GA, Slevin JT. Kindling-induced asymmetric accumulation of hippocampal 7S SNARE complexes correlates with enhanced glutamate release. Epilepsia. 2012a;53:157–167. doi: 10.1111/j.1528-1167.2011.03345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matveeva EA, Price DA, Whiteheart SW, Vanaman TC, Gerhardt GA, Slevin JT. Reduction of vesicle-associated membrane protein 2 expression leads to a kindling-resistant phenotype in a murine model of epilepsy. Neuroscience. 2012b;202:77–86. doi: 10.1016/j.neuroscience.2011.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna MC, Sonnewald U, Huang X, Stevenson J, Zielke HR. Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J Neurochem. 1996;66:386–393. doi: 10.1046/j.1471-4159.1996.66010386.x. [DOI] [PubMed] [Google Scholar]
- McNamara JO. Cellular and molecular basis of epilepsy. J Neurosci. 1994;14:3413–3425. doi: 10.1523/JNEUROSCI.14-06-03413.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier SD, Kafitz KW, Rose CR. Developmental profile and mechanisms of GABA-induced calcium signaling in hippocampal astrocytes. Glia. 2008;56:1127–1137. doi: 10.1002/glia.20684. [DOI] [PubMed] [Google Scholar]
- Micheva KD, Beaulieu C. Quantitative aspects of synaptogenesis in the rat barrel field cortex with special reference to GABA circuitry. J Comp Neurol. 1996;373:340–354. doi: 10.1002/(SICI)1096-9861(19960923)373:3<340::AID-CNE3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Millan MH, Obrenovitch TP, Sarna GS, Lok SY, Symon L, Meldrum BS. Changes in rat brain extracellular glutamate concentration during seizures induced by systemic picrotoxin or focal bicuculline injection: an in vivo dialysis study with on-line enzymatic detection. Epilepsy Res. 1991;9:86–91. doi: 10.1016/0920-1211(91)90017-a. [DOI] [PubMed] [Google Scholar]
- Montana V, Malarkey EB, Verderio C, Matteoli M, Parpura V. Vesicular transmitter release from astrocytes. Glia. 2006;54:700–715. doi: 10.1002/glia.20367. [DOI] [PubMed] [Google Scholar]
- Moody WJ, Futamachi KJ, Prince DA. Extracellular potassium activity during epileptogenesis. Exp Neurol. 1974;42:248–263. doi: 10.1016/0014-4886(74)90023-5. [DOI] [PubMed] [Google Scholar]
- Nagelhus EA, Mathiisen TM, Ottersen OP. Aquaporin-4 in the central nervous system: cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience. 2004;129:905–913. doi: 10.1016/j.neuroscience.2004.08.053. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Abe S, Akazawa K. AMPA-, but not NMDA-, receptor blocker, NBQX, prevents seizure induction in magnesium-deficient rats. Magnes Res. 1995;8:55–56. [PubMed] [Google Scholar]
- Naus CC, Bechberger JF, Paul DL. Gap junction gene expression in human seizure disorder. Exp Neurol. 1991;111:198–203. doi: 10.1016/0014-4886(91)90007-y. [DOI] [PubMed] [Google Scholar]
- Nehlig A, Vergnes M, Marescaux C, Boyet S. Mapping of cerebral energy metabolism in rats with genetic generalized nonconvulsive epilepsy. J Neural Transm Suppl. 1992;35:141–153. doi: 10.1007/978-3-7091-9206-1_10. [DOI] [PubMed] [Google Scholar]
- Nehlig A, Vergnes M, Marescaux C, Boyet S. Cerebral energy metabolism in rats with genetic absence epilepsy is not correlated with the pharmacological increase or suppression of spike-wave discharges. Brain Res. 1993;618:1–8. doi: 10.1016/0006-8993(93)90421-i. [DOI] [PubMed] [Google Scholar]
- Nelken I, Yaari Y. The role of interstitial potassium in the generation of low-calcium hippocampal seizures. Isr J Med Sci. 1987;23:124–131. [PubMed] [Google Scholar]
- Newman EA. New roles for astrocytes: regulation of synaptic transmission. Trends Neurosci. 2003;26:536–542. doi: 10.1016/S0166-2236(03)00237-6. [DOI] [PubMed] [Google Scholar]
- Newman EA. Glia and synaptic transmission. In: Kettenmann H, Ransom B, editors. Neuroglia. 2nd. Oxford University Press; New York: 2004. pp. 355–366. [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, Furuichi T, Hensch TK, Yamakawa K. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–5914. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SJ, Han KS, Park H, Woo DH, Kim HY, Traynelis SF, Lee CJ. Protease activated receptor 1-induced glutamate release in cultured astrocytes is mediated by Bestrophin-1 channel but not by vesicular exocytosis. Mol Brain. 2012;5:38. doi: 10.1186/1756-6606-5-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, Haydon PG, Coulter DA. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci. 2010;13:584–591. doi: 10.1038/nn.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Han KS, Oh SJ, Jo S, Woo J, Yoon BE, Lee CJ. High glutamate permeability and distal localization of Best1 channel in CA1 hippocampal astrocyte. Mol Brain. 2013;6:54. doi: 10.1186/1756-6606-6-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AB, de Graaf RA, Mason GF, Rothman DL, Shulman RG, Behar KL. The contribution of GABA to glutamate/glutamine cycling and energy metabolism in the rat cortex in vivo. Proc Natl Acad Sci U S A. 2005;102:5588–5593. doi: 10.1073/pnas.0501703102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen RE, Fonnum F. Role of glial cells for the basal and Ca2+-dependent K+-evoked release of transmitter amino acids investigated by microdialysis. J Neurochem. 1989;52:1823–1829. doi: 10.1111/j.1471-4159.1989.tb07263.x. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Pedley TA, Fisher RS, Futamachi KJ, Prince DA. Regulation of extracellular potassium concentration in epileptogenesis. Fed Proc. 1976;35:1254–1259. [PubMed] [Google Scholar]
- Perez EL, Lauritzen F, Wang Y, Lee TS, Kang D, Zaveri HP, Chaudhry FA, Ottersen OP, Bergersen LH, Eid T. Evidence for astrocytes as a potential source of the glutamate excess in temporal lobe epilepsy. Neurobiol Dis. 2012;47:331–337. doi: 10.1016/j.nbd.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci. 2005;8:51–60. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- Petroff OA, Errante LD, Kim JH, Spencer DD. N-acetyl-aspartate, total creatine, and myo-inositol in the epileptogenic human hippocampus. Neurology. 2003;60:1646–1651. doi: 10.1212/01.wnl.0000068020.85450.8b. [DOI] [PubMed] [Google Scholar]
- Petroff OA, Errante LD, Rothman DL, Kim JH, Spencer DD. Neuronal and glial metabolite content of the epileptogenic human hippocampus. Ann Neurol. 2002;52:635–642. doi: 10.1002/ana.10360. [DOI] [PubMed] [Google Scholar]
- Phelps CH. An ultrastructural study of methionine sulphoximine-induced glycogen accumulation in astrocytes of the mouse cerebral cortex. J Neurocytol. 1975;4:479–490. doi: 10.1007/BF01261377. [DOI] [PubMed] [Google Scholar]
- Plotkin MD, Snyder EY, Hebert SC, Delpire E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA’s excitatory role in immature brain. J Neurobiol. 1997;33:781–795. doi: 10.1002/(sici)1097-4695(19971120)33:6<781::aid-neu6>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Pohlmann-Eden B, Weaver DF. The puzzle(s) of pharmacoresistant epilepsy. Epilepsia. 2013;54(Suppl 2):1–4. doi: 10.1111/epi.12174. [DOI] [PubMed] [Google Scholar]
- Pow DV, Sullivan RK, Williams SM, Scott HL, Dodd PR, Finkelstein D. Differential expression of the GABA transporters GAT-1 and GAT-3 in brains of rats, cats, monkeys and humans. Cell Tissue Res. 2005;320:379–392. doi: 10.1007/s00441-004-0928-0. [DOI] [PubMed] [Google Scholar]
- Proper EA, Hoogland G, Kappen SM, Jansen GH, Rensen MG, Schrama LH, van Veelen CW, van Rijen PC, van Nieuwenhuizen O, Gispen WH, de Graan PN. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain. 2002;125:32–43. doi: 10.1093/brain/awf001. [DOI] [PubMed] [Google Scholar]
- Ragsdale DS. How do mutant Nav1.1 sodium channels cause epilepsy? Brain Res Rev. 2008;58:149–159. doi: 10.1016/j.brainresrev.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Ransom BR, Ransom CB. Astrocytes: multitalented stars of the central nervous system. Methods Mol Biol. 2012;814:3–7. doi: 10.1007/978-1-61779-452-0_1. [DOI] [PubMed] [Google Scholar]
- Reid CA, Kim T, Phillips AM, Low J, Berkovic SF, Luscher B, Petrou S. Multiple molecular mechanisms for a single GABAA mutation in epilepsy. Neurology. 2013;80:1003–1008. doi: 10.1212/WNL.0b013e3182872867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reime Kinjo E, Arida RM, Mara de Oliveira D, da Silva Fernandes MJ. The Na+/K+ATPase activity is increased in the hippocampus after multiple status epilepticus induced by pilocarpine in developing rats. Brain Res. 2007;1138:203–207. doi: 10.1016/j.brainres.2006.12.068. [DOI] [PubMed] [Google Scholar]
- Rho JM, Szot P, Tempel BL, Schwartzkroin PA. Developmental seizure susceptibility of kv1.1 potassium channel knockout mice. Dev Neurosci. 1999;21:320–327. doi: 10.1159/000017381. [DOI] [PubMed] [Google Scholar]
- Richerson GB, Wu Y. Dynamic equilibrium of neurotransmitter transporters: not just for reuptake anymore. J Neurophysiol. 2003;90:1363–1374. doi: 10.1152/jn.00317.2003. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J Cell Biol. 2002;159:747–752. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipila S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA. AMPA receptors as a molecular target in epilepsy therapy. Acta Neurol Scand Suppl. 2013:9–18. doi: 10.1111/ane.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Tabakoff B. Alteration of striatal glutamate release after glutamine synthetase inhibition. J Neurochem. 1984;43:1438–1446. doi: 10.1111/j.1471-4159.1984.tb05406.x. [DOI] [PubMed] [Google Scholar]
- Samoilova M, Wentlandt K, Adamchik Y, Velumian AA, Carlen PL. Connexin 43 mimetic peptides inhibit spontaneous epileptiform activity in organotypic hippocampal slice cultures. Exp Neurol. 2008;210:762–775. doi: 10.1016/j.expneurol.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Santello M, Cali C, Bezzi P. Gliotransmission and the tripartite synapse. Adv Exp Med Biol. 2012;970:307–331. doi: 10.1007/978-3-7091-0932-8_14. [DOI] [PubMed] [Google Scholar]
- Sarac S, Afzal S, Broholm H, Madsen FF, Ploug T, Laursen H. Excitatory amino acid transporters EAAT-1 and EAAT-2 in temporal lobe and hippocampus in intractable temporal lobe epilepsy. Apmis. 2009;117:291–301. doi: 10.1111/j.1600-0463.2009.02443.x. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Kuga N, Namiki S, Matsuki N, Ikegaya Y. Locally synchronized astrocytes. Cereb Cortex. 2011a;21:1889–1900. doi: 10.1093/cercor/bhq256. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Matsuki N, Ikegaya Y. Action-potential modulation during axonal conduction. Science. 2011b;331:599–601. doi: 10.1126/science.1197598. [DOI] [PubMed] [Google Scholar]
- Schousboe A. Transport and metabolism of glutamate and GABA in neurons are glial cells. Int Rev Neurobiol. 1981;22:1–45. doi: 10.1016/s0074-7742(08)60289-5. [DOI] [PubMed] [Google Scholar]
- Schroder W, Hinterkeuser S, Seifert G, Schramm J, Jabs R, Wilkin GP, Steinhauser C. Functional and molecular properties of human astrocytes in acute hippocampal slices obtained from patients with temporal lobe epilepsy. Epilepsia. 2000;41(Suppl 6):S181–184. doi: 10.1111/j.1528-1157.2000.tb01578.x. [DOI] [PubMed] [Google Scholar]
- Seifert G, Carmignoto G, Steinhauser C. Astrocyte dysfunction in epilepsy. Brain Res Rev. 2010;63:212–221. doi: 10.1016/j.brainresrev.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Seifert G, Steinhauser C. Neuron-astrocyte signaling and epilepsy. Exp Neurol. 2013;244:4–10. doi: 10.1016/j.expneurol.2011.08.024. [DOI] [PubMed] [Google Scholar]
- Sharopov S, Chen R, Sun H, Kolbaev SN, Kirischuk S, Luhmann HJ, Kilb W. Inhibition of different GABA transporter systems is required to attenuate epileptiform activity in the CA3 region of the immature rat hippocampus. Epilepsy Res. 2013 doi: 10.1016/j.eplepsyres.2013.11.019. [DOI] [PubMed] [Google Scholar]
- Somers DL, Beckstead RM. Chronic methionine sulfoximine administration reduces synaptosomal aspartate and glutamate in rat striatum. Neurosci Lett. 1990;115:335–340. doi: 10.1016/0304-3940(90)90478-r. [DOI] [PubMed] [Google Scholar]
- Sontheimer H, Fernandez-Marques E, Ullrich N, Pappas CA, Waxman SG. Astrocyte Na+ channels are required for maintenance of Na+/K(+)-ATPase activity. J Neurosci. 1994;14:2464–2475. doi: 10.1523/JNEUROSCI.14-05-02464.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr MS. The role of dopamine in epilepsy. Synapse. 1996;22:159–194. doi: 10.1002/(SICI)1098-2396(199602)22:2<159::AID-SYN8>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Stein V, Hermans-Borgmeyer I, Jentsch TJ, Hubner CA. Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol. 2004;468:57–64. doi: 10.1002/cne.10983. [DOI] [PubMed] [Google Scholar]
- Steinhauser C, Seifert G. Glial membrane channels and receptors in epilepsy: impact for generation and spread of seizure activity. Eur J Pharmacol. 2002;447:227–237. doi: 10.1016/s0014-2999(02)01846-0. [DOI] [PubMed] [Google Scholar]
- Steinhauser C, Seifert G, Bedner P. Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia. 2012;60:1192–1202. doi: 10.1002/glia.22313. [DOI] [PubMed] [Google Scholar]
- Stewart TH, Eastman CL, Groblewski PA, Fender JS, Verley DR, Cook DG, D’Ambrosio R. Chronic dysfunction of astrocytic inwardly rectifying K+ channels specific to the neocortical epileptic focus after fluid percussion injury in the rat. J Neurophysiol. 2010;104:3345–3360. doi: 10.1152/jn.00398.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbalakshmi GY, Murthy CR. Effects of methionine sulfoximine on cerebral ATPase. Biochem Pharmacol. 1981;30:2127–2130. doi: 10.1016/0006-2952(81)90232-x. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Tsurubuchi Y, Agarwala KL, Ito M, Fukuma G, Mazaki-Miyazaki E, Nagafuji H, Noda M, Imoto K, Wada K, Mitsudome A, Kaneko S, Montal M, Nagata K, Hirose S, Yamakawa K. A missense mutation of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci U S A. 2001;98:6384–6389. doi: 10.1073/pnas.111065098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, McConnell E, Pare JF, Xu Q, Chen M, Peng W, Lovatt D, Han X, Smith Y, Nedergaard M. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science. 2013;339:197–200. doi: 10.1126/science.1226740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegedy L. Enzyme histochemical studies in the rat hippocampus, cerebellar cortex and brainstem motor nuclei during methionine sulphoximine convulsions. Cell Mol Biol Incl Cyto Enzymol. 1978;23:43–53. [PubMed] [Google Scholar]
- Tagliabracci VS, Girard JM, Segvich D, Meyer C, Turnbull J, Zhao X, Minassian BA, Depaoli-Roach AA, Roach PJ. Abnormal metabolism of glycogen phosphate as a cause for Lafora disease. J Biol Chem. 2008;283:33816–33825. doi: 10.1074/jbc.M807428200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi DK, Vargas JR, Wilcox KS. Increased coupling and altered glutamate transport currents in astrocytes following kainic-acid-induced status epilepticus. Neurobiol Dis. 2010;40:573–585. doi: 10.1016/j.nbd.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, He W, Han X, Wang F, Xu Q, Wang X, Oberheim Bush NA, Cruz N, Dienel GA, Nedergaard M. Rapid manifestation of reactive astrogliosis in acute hippocampal brain slices. Glia. 2014;62:78–95. doi: 10.1002/glia.22588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang FR, Lee WL. Expression of the group II and III metabotropic glutamate receptors in the hippocampus of patients with mesial temporal lobe epilepsy. J Neurocytol. 2001;30:137–143. doi: 10.1023/a:1011939223872. [DOI] [PubMed] [Google Scholar]
- Tani H, Dulla CG, Huguenard JR, Reimer RJ. Glutamine is required for persistent epileptiform activity in the disinhibited neocortical brain slice. J Neurosci. 2010;30:1288–1300. doi: 10.1523/JNEUROSCI.0106-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessler S, Danbolt NC, Faull RL, Storm-Mathisen J, Emson PC. Expression of the glutamate transporters in human temporal lobe epilepsy. Neuroscience. 1999;88:1083–1091. doi: 10.1016/s0306-4522(98)00301-7. [DOI] [PubMed] [Google Scholar]
- Theodore WH. Does Serotonin Play a Role in Epilepsy? Epilepsy Curr. 2003;3:173–177. doi: 10.1046/j.1535-7597.2003.03508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian GF, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, Kang J, Nedergaard M. An astrocytic basis of epilepsy. Nat Med. 2005;11:973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda Y, Willmore LJ. Molecular regulation of glutamate and GABA transporter proteins by valproic acid in rat hippocampus during epileptogenesis. Exp Brain Res. 2000;133:334–339. doi: 10.1007/s002210000443. [DOI] [PubMed] [Google Scholar]
- Valles-Ortega J, Duran J, Garcia-Rocha M, Bosch C, Saez I, Pujadas L, Serafin A, Canas X, Soriano E, Delgado-Garcia JM, Gruart A, Guinovart JJ. Neurodegeneration and functional impairments associated with glycogen synthase accumulation in a mouse model of Lafora disease. EMBO Mol Med. 2011;3:667–681. doi: 10.1002/emmm.201100174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hel WS, Notenboom RG, Bos IW, van Rijen PC, van Veelen CW, de Graan PN. Reduced glutamine synthetase in hippocampal areas with neuron loss in temporal lobe epilepsy. Neurology. 2005;64:326–333. doi: 10.1212/01.WNL.0000149636.44660.99. [DOI] [PubMed] [Google Scholar]
- van der Hel WS, Verlinde SA, Meijer DH, de Wit M, Rensen MG, van Gassen KL, van Rijen PC, van Veelen CW, de Graan PN. Hippocampal distribution of vesicular glutamate transporter 1 in patients with temporal lobe epilepsy. Epilepsia. 2009;50:1717–1728. doi: 10.1111/j.1528-1167.2009.02054.x. [DOI] [PubMed] [Google Scholar]
- Verge V, Hevor TK. Regulation of fructose-1,6-bisphosphatase activity in primary cultured astrocytes. Neurochem Res. 1995;20:1049–1056. doi: 10.1007/BF00995559. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Pankratov Y, Lalo U, Nedergaard M. P2X receptors in neuroglia. Wiley Interdiscip Rev Membr Transp Signal 1. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viitanen T, Ruusuvuori E, Kaila K, Voipio J. The K+-Cl cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J Physiol. 2010;588:1527–1540. doi: 10.1113/jphysiol.2009.181826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitezic D, Pelcic JM, Zupan G, Vitezic M, Ljubicic D, Simonic A. NA+, K+-ATPase activity in the brain of the rats with kainic acid-induced seizures: influence of lamotrigine. Psychiatr Danub. 2008;20:269–276. [PubMed] [Google Scholar]
- Waldbaum S, Patel M. Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res. 2010;88:23–45. doi: 10.1016/j.eplepsyres.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MC, Galley PT, Errington ML, Shorvon SD, Jefferys JG. Ascorbate and glutamate release in the rat hippocampus after perforant path stimulation: a “dialysis electrode” study. J Neurochem. 1995;65:725–731. doi: 10.1046/j.1471-4159.1995.65020725.x. [DOI] [PubMed] [Google Scholar]
- Wallraff A, Kohling R, Heinemann U, Theis M, Willecke K, Steinhauser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 2006;26:5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz W, Hertz L. Intense furosemide-sensitive potassium accumulation in astrocytes in the presence of pathologically high extracellular potassium levels. J Cereb Blood Flow Metab. 1984;4:301–304. doi: 10.1038/jcbfm.1984.42. [DOI] [PubMed] [Google Scholar]
- Wang C, Shimizu-Okabe C, Watanabe K, Okabe A, Matsuzaki H, Ogawa T, Mori N, Fukuda A, Sato K. Developmental changes in KCC1, KCC2, and NKCC1 mRNA expressions in the rat brain. Brain Res Dev Brain Res. 2002;139:59–66. doi: 10.1016/s0165-3806(02)00536-9. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zaveri HP, Lee TS, Eid T. The development of recurrent seizures after continuous intrahippocampal infusion of methionine sulfoximine in rats: a video-intracranial electroencephalographic study. Exp Neurol. 2009;220:293–302. doi: 10.1016/j.expneurol.2009.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender R, Brown AM, Fern R, Swanson RA, Farrell K, Ransom BR. Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J Neurosci. 2000;20:6804–6810. doi: 10.1523/JNEUROSCI.20-18-06804.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetherington J, Serrano G, Dingledine R. Astrocytes in the epileptic brain. Neuron. 2008;58:168–178. doi: 10.1016/j.neuron.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson R, Wheal HV. The contribution of AMPA and NMDA receptors to graded bursting activity in the hippocampal CA1 region in an acute in vitro model of epilepsy. Epilepsy Res. 1992;12:179–188. doi: 10.1016/0920-1211(92)90039-v. [DOI] [PubMed] [Google Scholar]
- Wink MR, Braganhol E, Tamajusuku AS, Lenz G, Zerbini LF, Libermann TA, Sevigny J, Battastini AM, Robson SC. Nucleoside triphosphate diphosphohydrolase-2 (NTPDase2/CD39L1) is the dominant ectonucleotidase expressed by rat astrocytes. Neuroscience. 2006;138:421–432. doi: 10.1016/j.neuroscience.2005.11.039. [DOI] [PubMed] [Google Scholar]
- Wittner L, Eross L, Czirjak S, Halasz P, Freund TF, Magloczky Z. Surviving CA1 pyramidal cells receive intact perisomatic inhibitory input in the human epileptic hippocampus. Brain. 2005;128:138–152. doi: 10.1093/brain/awh339. [DOI] [PubMed] [Google Scholar]
- Wong RK, Chuang SC, Bianchi R. Plasticity mechanisms underlying mGluR-induced epileptogenesis. Adv Exp Med Biol. 2004;548:69–75. doi: 10.1007/978-1-4757-6376-8_5. [DOI] [PubMed] [Google Scholar]
- Woo DH, Han KS, Shim JW, Yoon BE, Kim E, Bae JY, Oh SJ, Hwang EM, Marmorstein AD, Bae YC, Park JY, Lee CJ. TREK-1 and Best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell. 2012;151:25–40. doi: 10.1016/j.cell.2012.09.005. [DOI] [PubMed] [Google Scholar]
- Xu J, Song D, Xue Z, Gu L, Hertz L, Peng L. Requirement of Glycogenolysis for Uptake of Increased Extracellular K(+) in Astrocytes: Potential Implications for K (+) Homeostasis and Glycogen Usage in Brain. Neurochem Res. 2013;38:472–485. doi: 10.1007/s11064-012-0938-3. [DOI] [PubMed] [Google Scholar]
- Yaari Y, Konnerth A, Heinemann U. Nonsynaptic epileptogenesis in the mammalian hippocampus in vitro. II. Role of extracellular potassium. J Neurophysiol. 1986;56:424–438. doi: 10.1152/jn.1986.56.2.424. [DOI] [PubMed] [Google Scholar]
- Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ, Fukuda A. Cl− uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol. 2004;557:829–841. doi: 10.1113/jphysiol.2004.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Cox CL. Attenuation of inhibitory synaptic transmission by glial dysfunction in rat thalamus. Synapse. 2011;65:1298–1308. doi: 10.1002/syn.20964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gozen O, Watkins A, Lorenzini I, Lepore A, Gao Y, Vidensky S, Brennan J, Poulsen D, Won Park J, Li Jeon N, Robinson MB, Rothstein JD. Presynaptic regulation of astroglial excitatory neurotransmitter transporter GLT1. Neuron. 2009;61:880–894. doi: 10.1016/j.neuron.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon BE, Jo S, Woo J, Lee JH, Kim T, Kim D, Lee CJ. The amount of astrocytic GABA positively correlates with the degree of tonic inhibition in hippocampal CA1 and cerebellum. Mol Brain. 2011;4:42. doi: 10.1186/1756-6606-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon BE, Woo J, Lee CJ. Astrocytes as GABA-ergic and GABA-ceptive cells. Neurochem Res. 2012;37:2474–2479. doi: 10.1007/s11064-012-0808-z. [DOI] [PubMed] [Google Scholar]
- Yoon JJ, Green CR, O’Carroll SJ, Nicholson LF. Dose-dependent protective effect of connexin43 mimetic peptide against neurodegeneration in an ex vivo model of epileptiform lesion. Epilepsy Res. 2010;92:153–162. doi: 10.1016/j.eplepsyres.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- Yuen AW, Sander JW. Impaired mitochondrial energy production: the basis of pharmacoresistance in epilepsy. Med Hypotheses. 2011;77:536–540. doi: 10.1016/j.mehy.2011.06.025. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Chen G, Zhou W, Song A, Xu T, Luo Q, Wang W, Gu XS, Duan S. Regulated ATP release from astrocytes through lysosome exocytosis. Nat Cell Biol. 2007;9:945–953. doi: 10.1038/ncb1620. [DOI] [PubMed] [Google Scholar]
- Zhou C, Huang Z, Ding L, Deel ME, Arain FM, Murray CR, Patel RS, Flanagan CD, Gallagher MJ. Altered cortical GABAA receptor composition, physiology, and endocytosis in a mouse model of a human genetic absence epilepsy syndrome. J Biol Chem. 2013;288:21458–21472. doi: 10.1074/jbc.M112.444372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Lippman JJ, Sun H, Jensen FE. Hypoxia-induced neonatal seizures diminish silent synapses and long-term potentiation in hippocampal CA1 neurons. J Neurosci. 2011;31:18211–18222. doi: 10.1523/JNEUROSCI.4838-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Xu G, Xie M, Zhang X, Schools GP, Ma L, Kimelberg HK, Chen H. TWIK-1 and TREK-1 are potassium channels contributing significantly to astrocyte passive conductance in rat hippocampal slices. J Neurosci. 2009;29:8551–8564. doi: 10.1523/JNEUROSCI.5784-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorec R, Araque A, Carmignoto G, Haydon PG, Verkhratsky A, Parpura V. Astroglial excitability and gliotransmission: an appraisal of Ca2+ as a signalling route. ASN Neuro. 2012;4 doi: 10.1042/AN20110061. [DOI] [PMC free article] [PubMed] [Google Scholar]