

Abstract
Introduction:
Resistance to antipsychotic treatment is a significant clinical problem in patients with schizophrenia with approximately 1 in 3 showing limited or no response to repeated treatments with antipsychotic medication. The neurobiological basis for treatment resistance is unknown but recent evidence implicates glutamatergic function in the anterior cingulate cortex. We examined glutamate levels of chronically ill treatment-resistant patients directly compared to treatment-responsive patients.
Methods:
We acquired proton magnetic resonance spectroscopy (1H-MRS) at 3 Tesla from 21 treatment-resistant and 20 treatment-responsive patients. All participants had a DSM-IV diagnosis of schizophrenia. Treatment-resistant patients were classified using the modified Kane criteria. The groups were matched for age, sex, smoking status, and illness duration.
Results:
Glutamate to creatine ratio levels were higher in treatment-resistant patients (Mean [SD] = 1.57 [0.24]) than in treatment-responsive patients (Mean[SD] = 1.38 [0.23]), (T[35] = 2.34, P = .025, 2-tailed), with a large effect size of d = 0.76. A model assuming 2 populations showed a 25% improvement in the fit of the Akaike weights (0.55) over a model assuming 1 population (0.44), producing group values almost identical to actual group means.
Discussion:
Increased anterior cingulate glutamate level is associated with treatment-resistant schizophrenia. This appears to be a stable neurobiological trait of treatment-resistant patients. We discuss possible explanations for glutamatergic dysfunction playing a significant role in resistance to conventional antipsychotic treatments, which are all dopamine-2 receptor blockers. Our findings suggest that glutamatergic treatments may be particularly effective in resistant patients and that 1H-MRS glutamate indices can potentially have clinical use.
Key words: psychosis, antipsychotics, treatment- resistance, NMDA, 1H-MRS, neurochemistry, glutamate, imaging and schizophrenia
Introduction
All current first-line antipsychotics block dopamine-2 (D2) receptors.1,2 However, approximately, a third of patients with schizophrenia show limited if any response to first-line antipsychotic medications, despite high levels of dopamine D2 receptor blockade.3–5 This has been termed as treatment-resistant schizophrenia.3 Although a number of criteria have been developed to define treatment resistance, most clinical guidelines define it as an inadequate response to at least 2 trials of different first-line antipsychotic medications.6,7
It has been proposed that treatment-resistant schizophrenia patients do not respond to dopaminergic antipsychotic medication because their illness is not primarily characterized by a dopaminergic abnormality.8 Supporting this, they do not show the elevation in striatal presynaptic dopamine synthesis capacity, which is reproducibly associated with treatment-responsive schizophrenia.1,9 Additionally, administration of the D2 antagonist haloperidol produced significant metabolic reductions in treatment-resistant patients compared to responsive patients, suggesting that resistant patients might have reduced presynaptic dopamine reserve compared to responsive patients.10 Further support for the idea that treatment resistance is nondopaminergic comes from studies using drugs to deplete presynaptic dopamine stores.11–13 These studies have found no benefit from dopamine depletion in treatment-resistant patients, in contrast to the reduction in psychotic symptoms that is seen in most patients.
Apart from dopaminergic abnormalities, glutamatergic abnormalities are also thought to contribute to the pathophysiology of schizophrenia. Proton magnetic resonance spectroscopy (1H-MRS) studies show that both high-risk and first-episode patients have elevated glutamine and combined glutamate-glutamine (Glx) levels in the anterior cingulate cortex,14–18 while chronic patients show normalized or reduced glutamate-glutamine levels.15,19–26 However, the published studies in chronic schizophrenia have generally focused on patients who were stable and excluded treatment resistance. There is recent evidence that elevated glutamate levels are related to treatment nonresponse.27–29 In a preliminary study, we found that treatment-resistant patients showed increased glutamate levels vs controls, but no difference in striatal dopamine synthesis capacity. In contrast, responders showed increased striatal dopamine synthesis capacity, but no difference in anterior cingulate glutamate levels compared to controls.27 .Elevated glutamate levels were found in nonremitted first episode patients compared to patients in remission in the anterior cingulate cortex28 and left frontal lobe.29 If elevated glutamate levels are found to be a stable neurobiological trait of treatment resistance, they could be used to fast-track such patients to clozapine and/or towards newer glutamatergic compounds under current development. Thus, cortical glutamate levels, as measured with 1H-MRS, could be a potential biomarker for the stratification of treatment-resistant and treatment-responsive patients.
In the present study, we used 1H-MRS to investigate glutamate levels in the anterior cingulate cortex of chronically ill treatment-resistant patients directly compared to treatment-responsive patients. The anterior cingulate appears to be an important region in regards to glutamatergic dysfunction and treatment response30 while clozapine (as the only effective drug for treatment resistance) modulates its metabolic function differentially to haloperidol.31,32 Our preliminary study27 showed that resistant patients had increased anterior cingulate cortex glutamate compared to controls, but lacked enough power to detect differences between the 2 clinical groups. In the present study we used a larger sample that would allow us enough power to detect such differences. Our main hypothesis was that treatment-resistant patients would show significantly increased anterior cingulate cortex glutamate when directly compared to treatment-responsive. As an exploratory analysis we also used finite mixture modeling to assess whether glutamatergic indices as measured by 1H-MRS can potentially be informative as a clinical tool for patient stratification.
Methods
Subjects
The study was approved by the Institute of Psychiatry Research Ethics Committee. All subjects gave written informed consent to participate. Forty-one patients diagnosed with schizophrenia (as defined by DSM-IV criteria) took part in the study. All patients were recruited and scanned within 14 months. Twenty-one patients were classified as treatment resistant using the modified Kane criteria.33 These require that patients had previously received at least 2 sequential trials of different antipsychotics, each of at least 6 weeks’ duration at a daily dose of at least 400mg of chlorpromazine (CPZ) equivalents,34 but had shown inadequate response, including persistent psychotic symptoms. This was operationalized as having a rating of at least moderate severity on 1 or more items on the positive symptom subscale of the Positive and Negative Syndrome Scale (PANSS),35 and having a score < 59 on the Global Assessment of Functioning (corresponding to at least moderate functional impairment) with no periods of symptomatic or functional improvement over at least the preceding 6 months.
Twenty patients were classified as treatment responsive. These patients had responded to antipsychotic treatment and met the Remission in Schizophrenia Working Group criteria for treatment remission.36 They scored ≤ 3 on all items of the PANSS (corresponding to mild severity or questionable/no symptoms) and had not experienced a symptomatic relapse in the 6 months prior to the study. All treatment-resistant and 12 treatment-responsive patients were recruited from the South London and Maudsley NHS Trust. Eight treatment-responsive patients were recruited from the Central and North West London NHS Foundation Trust. One resistant patient and 2 responsive patients were scanned that were also included in the Demjaha et al27 study.
Exclusion criteria were presence of a neurological condition, comorbid psychiatric or physical illness, substance dependency/abuse (except nicotine), pregnancy, lack of capacity, contradictions to having an MRI scan, current or previous use of clozapine, and nonadherence to antipsychotic treatment. Adherence to medication was determined by clinical interview, reviewing pharmacy, and medical records. Patients were excluded if there was evidence of nonadherence at any point in the 6 months prior to the scan. To control for possible interactions of nonantipsychotic medications with the glutamatergic system, patients were also excluded if they had received additional psychotropic medications at any point 6 months prior to the scan. CPZ equivalents were determined as described in the Maudsley Prescribing Guidelines.37 Symptoms at the time of scanning were assessed using the PANSS scale.35
Data Acquisition and Analysis
Scanning was conducted on a Philips 3 Tesla Intera MRI scanner, using the same scanning procedures reported elsewhere.38 An initial localizer scan was followed by acquisition of a whole-brain 3D-MPRAGE scan (TR = 9.6ms, TE = 4.5ms, flip angle 8°, slice thickness = 1.2mm, 0.94mm × 0.94mm in plane resolution, 150 slices). PRESS (Point RESolved Spectroscopy) data were then acquired (TE = 35ms; TR = 3000ms; 64 averages), utilizing the default “Excite” water suppression routine. Autoshimming was performed using a second-order pencil beam shim. The 3D-MPRAGE scan was used to position the 1H-MRS voxels. An anterior cingulate voxel (20×20×20mm) was prescribed from the midline sagittal slice, with the centre of the voxel positioned 13mm above the anterior section of the genu of corpus callosum at 90o to the AC-PC line (also see supplementary material). Before the acquisition of the 1H-MRS scan, 2 structural MRI and 4 functional MRI sequences were also acquired (data not reported here).
Data were analyzed using LCModel version 6.3.39 A standard basis set of 16 metabolites was used (comprising L -alanine, aspartate, creatine, phosphocreatine, GABA, glucose, glutamine, glutamate, glycerophosphocholine, glycine, myoinositol, L-lactate, N-acetyl aspartate, N-acetylaspartylglutamate, phosphocholine, and taurine), including simulated lipids and macromolecules as part of LCModel basis set that was acquired with the same field strength (3T), localization sequence (PRESS) and echo time (35ms). Model metabolites and concentrations employed in the basis set are fully detailed in the LCModel manual (http://www.s-provencher.com/pub/LCModel/manual/manual.pdf). Metabolite concentration estimates were expressed in ratio to total creatine (Cr) which is calculated as Cr plus phosphocreatine within LCModel. NAA was expressed as N-acetyl aspartate plus N-acetylaspartylglutamate, and choline as glycerylphosphorylcholine plus phosphocholinen (but not choline itself as not typically quantifiable with these scanning parameters). Only metabolite concentration estimates associated with Cramer-Rao lower bounds (CRLB) <20% as reported by LCModel were included in the analysis. Additionally signal-to-noise ratio (S/N) ≥8 and linewidth of <0.1 ppm were required for inclusion.
Statistical Analysis
Group differences in metabolite data, demographic and clinical data were assessed with an independent-sample t-test or Pearson’s χ 2 as appropriate using Matlab 7.9 and SPSS 21 .software. Statistical significance was defined as P < .05, 2-tailed. Given our a priori hypothesis for a glutamate increase in resistant compared to responsive patients, this contrast is reported uncorrected in relation to other metabolites of interest (Glx, NAA, choline, myo-inositol) while for differences these metabolites, a Bonferroni correction was applied with a threshold of P = .013 (α = .05/4 for the 4 metabolites of interest). For data that were different between groups, metabolite comparisons were assessed with a linear regression where the differential variable was entered as a nuisance covariate. Additional post hoc correction for age was conducted irrespective of group differences as it has been previously shown to be associated with glutamate and glutamine levels.26
To control for potential group differences in tissue composition, we performed tissue segmentation within the anterior cingulate voxel (see supplementary material). Since we only acquired water suppressed spectra referenced to creatine, main contrasts are reported without tissue correction. A brain tissue correction was conduced post hoc. Cohen’s d was used as a measure of effect size. Correlations between metabolite levels and PANSS Positive, Negative, General, and Total scales were conducted using Pearson’s correlation. To control for multiple comparisons, a Bonferroni correction threshold was set to P = .013 (α = .05/4 for the 4 PANSS scales). Residuals were tested for normality using the Shapiro-Wilke Test and Cook’s distance test was used to assess the effect of potential outliers.
An exploratory finite mixture modeling for patient classification was implemented using Matlab 7.9 mixture model functions within the Statistics toolbox (see supplementary material).
Results
Clinical and Demographic Data
21 patients were classified as treatment resistant (14 male and 7 female, mean age 41.2±13.2) and 20 as treatment responsive (12 male and 8 female, mean age 39.2±8.3). There were no differences between the 2 groups in sex, age, smoking, and illness duration (see table 1). The treatment-resistant group had received a greater level of antipsychotic medication over the course of illness compared to the treatment-responsive group (t(35) = 2.50, P = .02). As expected, treatment-resistant patients had higher PANSS scores across all sub-scales (see table 1), which, however, was not statistically different across groups, P > .1
Table 1.
Demographic and Clinical Characteristics of Patients
| Responsive (n = 20) | Resistant (n = 21) | |
|---|---|---|
| Sex (male/female) | 12/8 | 14/7 |
| Age, mean ± SD, y | 39.2±8 | 41.2±13 |
| Smoking (nonsmoker/smoker) | 9/11 | 10/11 |
| Ilness duration,mean ± SD,y | 14.4±6.7 | 14.6±11.3 |
| Medication dose (mg), mean ± SD, (chlorpromazine equivalent) | 315.6±246.1 | 682.5±459.1a |
| PANSS-Positive, mean ± SD | 10±5 | 18±6 |
| PANSS-Negative,mean ± SD | 11±4 | 13±4 |
| PANSS-General,mean ± SD | 22±8 | 33±9 |
| PANSS-Total,mean ± SD | 43±13 | 65±18 |
Note:
PANSS, Positive and Negative Syndrome Scale.aSignificant differences, t-test, 2-tailed, P < .05.
Spectra Quality
Over the course of the study, water-scaled phantom data were acquired and did not provide any evidence of significant scanner drift or other acute change (data not shown). Two patients from each group were excluded due to glutamate CRLB >20% and SNR <8. This reflected movement of these patients in the scanner before 1H-MRS acquisition in relation to their position during the T1 scan that was used for voxel placement. This resulted in LCModel not being able to fit the spectrum adequately due to movement after voxel placement. Hence data from 19 treatment resistant and 18 responders were included in the final analysis. No differences were found between groups in any of the spectra quality measures (see table 2).
Table 2.
1H-MRS Spectra Quality Measures (mean ± SD) in the Anterior Cingulate Cortex in Treatment-Responsive and Treatment-Resistant Patients
| Responsive (n = 18) | Resistant (n = 19) | |
|---|---|---|
| CRLB% | CRLB%a | |
| Glutamate/Cr | 10.1±2.6 | 10.6±2.5 |
| Glx/Cr | 10.8±2.9 | 10.2±2.8 |
| NAA/Cr | 6.5±1.7 | 5.7±2.6 |
| Choline/Cr | 7.2±2.4 | 6.8±3.2 |
| Myo-inositol/Cr | 10.8±4.3 | 10.1±3.3 |
| S/N | 17.1±2.4 | 16.6±3.3 |
| FWHM | 0.053±0.014 | 0.051±0.016 |
Note: CRLB, Cramer-Rao Lower Bounds; Cr, Creatine; Glx, Glutamate plus Glutamine; NAA, N-acetylaspartate; S/N, signal-to-noise ratio; FWHM, full-width-half-maximum.
aNo significant differences across any of the measures
Ps > .1.
Anterior Cingulate Metabolite Levels in Treatment-Resistant and Treatment-Responsive Patients The Glu/Cr ratio was significantly higher by 11% in the treatment-resistant group compared to the treatment-responsive group t(35) = 2.34, P = .025, 2-tailed, with a large effect size of d = 0.76, (see figure 1). There was no difference between the 2 groups in other quantifiable metabolites of interest (see table 3). The difference between resistant and responsive patients remained significant after accounting for effects of medication dose, T(34) = 2.21, P < .05, brain tissue composition T(34) = 2.29, and age, T(34) = 2.26, P < .05. For completeness, we also correct Glu/Cr with a Bonferroni correction of P = .025 (a = 0.05/2 for Glu/Cr and Glx/Cr ratios). This remains significant in respect to Glx/Cr, for P = .025.
Fig. 1.

Placement of anterior cingulate cortex voxel (right panel) and representative sample 1H-MRS spectra (left panel).
Table 3.
1H-MRS Metabolite Level Ratios (mean ± SD) in the Anterior Cingulate Cortex in Treatment-Responsive and Treatment-Resistant Patients
| Responsive (n = 18) | Resistant (n = 19) | Statistic | |
|---|---|---|---|
| Glutamate/Cr | 1.38±0.23 | 1.57±0.24 | T(35) = −2.34, P = .02 |
| Glx/Cr | 2.01±0.53 | 2.23±0.51 | T(35) = −1.27, P = .21 |
| NAA/Cr | 1.08±0.19 | 1.15±0.11 | T(35) = −1.37, P = .19 |
| Choline/Cr | 0.23±0.03 | 0.25±0.03 | T(35) = −1.89, P = .07 |
| Myo-inositol/Cr | 0.70±0.13 | 0.75±0.11 | T(35) = −1.12, P = .26 |
Note: Cr, Creatine; Glx, Glutamate plus Glutamine; NAA, N-acetylaspartate.
Metabolite Relationship to Symptoms and Medication Dose
We did not find any significant relationship between any of the metabolite levels and any of the scales of PANSS, either when collapsing across both groups or for each group separately. Furthermore, there was no significant correlation between the metabolite and antipsychotic dose (all correlations P > .1).
Patient Classification
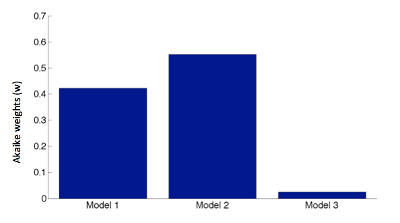
First the distribution was fitted with Model 1 with a mean of 1.47 (ie, the mean of the whole distribution). Importantly, Model 2 produced values of 1.50 and 1.35 for each population, corresponding closely to the actual Glu/Cr means for resistant and responsive patients (1.57 and 1.38, respectively), also reflecting the same difference between the 2 groups (11% in the actual data and 10% for Model 2). Note that these values were produced from the raw data set of all patients (both resistant and responsive), without the inclusion of any priors for each group regarding mean, SD or distribution of the data. However, this model had an inclusion rate of 80% for the high glutamate group and 20% for the low glutamate group, hence it overestimated the high Glu/Cr population. Thus, accurate classification was not possible, probably due to a relatively small sample size not allowing more accurate classification. Model 3 (dummy model) produced means of 1.36, 1.33, and 1.64 for each population. Akaike weight for Model 1 was 0.44, for Model 2 it was 0.55, and for Model 3 was 0.02 (see figure 2, supplementary figure 1). Akaike weights ratio for Model 2 to Model 1 was 1.25. This is a moderate evidence ratio, smaller than the recommended 2.7 when comparing 2 models to conclusively decide the best fit.40
Fig. 2.

Glutamate levels scaled to creatine in the anterior cingulate cortex for treatment-resistant and treatment-responsive patients. * = Resistant patients had significantly higher levels, t(35) = 2.34, P = .025, 2-tailed. Error bars represent SE of the mean.
Discussion
Glutamate Elevation in Treatment-Resistant Patients
Our results show, for the first time, that chronically ill patients with treatment-resistant schizophrenia show significantly elevated anterior cingulate cortex glutamate levels when directly compared to treatment-responsive schizophrenia patients. A previous study reported resistant patients, but not responsive, showed elevated anterior cingulate glutamate levels compared to controls, but no difference was found when directly comparing resistant and responsive patients as that study was underpowered.27 Other studies28 have found that nonremitted first-episode patients (but not meeting resistance criteria) show elevated anterior cingulate cortex glutamate levels compared to first-episode patients that show remission. Our study, extending the above, suggests that glutamatergic dysfunction plays a key role in antipsychotic treatment resistance, from early to chronic stages of the illness. In our sample, both patient groups had illness duration of approximately 14 years. Importantly, the resistant patients had never been treated with clozapine, which is the only known effective medication to address antipsychotic treatment resistance. In the UK clozapine is initiated approximately after 5 years since treatment resistance has been established,41,42 with similar timeframes reported in other health systems.43–45 Thus, elevated anterior cingulate glutamate appears to be a stable neurobiological trait in treatment-resistant patients, that is preserved after a number of treatment courses with different nonclozapine medications. Attenuation of glutamatergic dysfunction can be related to the effectiveness of clozapine in treatment-resistant patients. Animal studies have reported that clozapine shows specificity over haloperidol in modulating partial NMDA agonists glycine and serine,46,47 which have also been found to be deficient in treatment-resistant patients.48
This is consistent with the glutamate model of schizophrenia, which suggests that NMDA type glutamate receptor hypofunction is a key component of the pathophysiology of the disorder.49–52 Although initially counterintuitive, elevated cortical glutamate seems to reflect compensation for N-methyl-d-aspartate (NMDA)-receptor hypofunction due to increased GABA disinhibition.53 It is unclear, however, what is the exact nature of glutamatergic dysfunction in relation to antipsychotic treatment resistance. Previous studies have shown that unmedicated first-episode patients before treatment show increased striatal glutamate compared to controls, and this is normalized after antipsychotic treatment54,55 (though no such data exist with anterior cingulate measurements). The patients in these studies were treated with risperidone, which has been shown to affect glutamatergic function56 In our sample the patients received a number of different antipsychotic medications (supplementary table 1). This raises the interesting question of whether nonclozapine antipsychotic medications affect glutamate differentially. Animal research57 suggests that all antipsychotic medications affect glutamatergic function similarly, but the exact effects are still unknown.
As it pertains to treatment resistance, there are 2 main possibilities: (1) at first-episode stage, potentially resistant patients have higher glutamate levels than patients who will become responsive and, thus, antipsychotic treatment alone is not sufficiently effective to normalize their glutamate levels; (2) both resistant and responsive patients initially have similar glutamate levels, but antipsychotic treatment does not have an effect on glutamatergic function on the resistant. Glutamate-dopamine interactions appear to be complex with no clear resolution on the primacy of each system to the other in the development of symptomatology (for review see58). If treatment-resistant patients have normal dopaminergic function, it is possible that their symptoms develop through a nondopaminergic and predominantly glutamatergic pathway.59 Such interactions cannot be elucidated by the present study and warrant further investigations. In this context though, our findings are significant as, for the first time, we show that antipsychotic treatment resistance in chronically ill patients is specifically associated with elevated cortical glutamate.
Relationship of Glutamate Levels With Clinical Symptoms
One of the aims of the study was to investigate whether elevated glutamate levels in treatment resistance were associated with positive symptoms. However, we did not find any correlations with any of the scales in PANSS. It has to be noted that such correlations are scarce in 1H-MRS literature. Indeed such associations are not very common in 1H-MRS studies (for comprehensive reviews see60,61). The relatively narrow range of symptom scores in our groups, which will have reduced the power to detect correlations, indicates that we cannot exclude a Type II error accounting for the lack of significant associations between MRS measures and symptom scores. The use of more sensitive clinical scales and the inclusion of patients with a greater range of symptom scores in future studies will address this issue.
Finite Mixture Model Analysis
We conducted an exploratory analysis to investigate the potential of using glutamatergic indices to classify resistant and responsive patients. Using finite mixture modeling on the whole distribution of anterior cingulate cortex Glu/Cr values, a model that assumed 2 populations fitted the data 25% better than a model that assumed a single population, though this reflected a trend and was not significant.
Importantly, we found that the 2-population model produced values for the high and low Glu/Cr populations that were remarkably similar to the actual Glu/Cr means of the resistant and responsive groups. Computationally, this shows that maximum likelihood estimates best converge for 2 distributions around values almost identical to the actual means of the populations, even without any priors on means, sample distribution and SD. This suggests that significant information can be extracted from glutamate values as measured by 1H-MRS that could contribute to stratifying patients in future work. For instance, 1H-MRS glutamate measures can be used to inform machine learning classification models that could be later applied to first-episode patients to predict treatment response. The finite mixture model in our sample overestimated the inclusion of patients in the high Glu/Cr group and the weight ratio was modest, indicating that further work with larger sample sizes is necessary before the classification is sufficiently accurate for clinical use.
Limitations
One limitation of the present study is the possibility of glutamine contamination of the Glu/Cr was not able to provide accurate quanitification of glutamine. For spectra with short TEs <40ms Glu and Gln peaks overlap by <30% in the 2.25–2.55 ppm range over which they are most distinct, and contribute around 90% and 10%, respectively, to the combined Glx signal.62 Based on this, the contamination of Glu by Gln in the present data is estimated as less than 10%. In our data, Glx/Cr ratio was not estimated for 29 out of 37 participants. This failure to quantify Gln is reflected as added noise in Glx/Cr signal. Although numerically the group difference was the same as the Glu/Cr ratio (10%), it did not reach statistical significance due to larger SD compared to Glu/Cr. Nevertheless, as glutamine increases have been reported in schizophrenia16,19,63,64 future studies in treatment resistance would benefit from studies at higher field strengths that allow accurate quantification of both glutamate and glutamine.
Furthermore, we used the ratio of glutamate concentration to total creatine as calculated by LCModel (ie, creatine plus phosphocreatine). It is a well-validated method implemented in LCModel that has shown to be resilient to signal-to-noise differences and increased sensitivity.65 This can be problematic if the groups have different creatine levels, and since we did not have absolute metabolite quantification we could not directly test that. In our dataset, all metabolite values were increased in the resistant group with differences ranging from 4%–7% which could potentially reflect significantly lower creatine concentrations in treatment-resistant patients. However, similar elevations in other metabolites apart from glutamate were reported for resistant patients in Demjaha et al27 even though those values were referenced to water, while no creatine differences were found between resistant, responsive, and healthy control groups. Egerton et al27 also reports similar metabolite elevations in nonremitted patients (both when referenced to creatine or water with no creatine differences between groups). In our data, only Glu/Cr ratio was found significant with a difference of 11%, almost twice larger than any other metabolite contrast. Although in the present study, we cannot completely exclude creatine differences, it is unlikely that the significant glutamate elevation in the treatment-resistant group can be attributed solely to creatine group differences.
Another potential caveat was that antipsychotic doses were higher in the resistant patients, as is typically the case.41 However, we did not find any relationship between antipsychotic dose and glutamate levels and the Glu/Cr difference between the 2 groups remained significant when using antipsychotic dose as a nuisance variable. Also, while the groups were well matched on demographic variables, it remains possible that there are residual confounders, such as socioeconomic, IQ, or education differences, which we did not measure. We also did not have a healthy control group, thus we cannot elucidate on glutamatergic function of the patient populations. However, the main objective of the present study was to investigate differences directly between resistant and responsive patients in glutamate as a potential marker of antipsychotic treatment resistance in chronically ill patients. No previous study has addressed this before with adequate group numbers to find a significant glutamate difference when comparing resistant and responsive patients. Further studies could include controls to test the differential effects in glutamate compared to treatment-resistant and treatment-responsive patients. Such information about the glutamate distribution in each population will elucidate the neurobiology of treatment resistance and could potentially be used to inform future efforts for patient stratification. Finally, by nature of the cross-sectional design used in the present study, we cannot determine the causal relationship between glutamate levels and treatment resistance. This question can be potentially resolved with a prospective design, measuring glutamate in medication-naïve patients and repeating it when treatment resistance has been determined.
Conclusion
Here we show that glutamatergic dysfunction is associated with chronic antipsychotic treatment resistance as a neurobiological trait of treatment-resistant patients. This suggests that medications that specifically target the glutamatergic system may be particularly effective in such patients. Also, given that 1H-MRS is a quick, safe, and relatively cost-effective examination, it has potential as a clinical tool for early detection of treatment resistance so patients could be fast tracked to more efficient treatments such as clozapine or novel glutamatergic compounds. However, before this could be tested further prospective studies are deemed necessary to elucidate the nature of this glutamatergic dysfunction both before antipsychotic treatment and in early stages of the disease after exposure to medication. This is a crucial next step to establish whether glutamatergic indices have the potential to be a biomarker.
Supplementary Material
Supplementary material is available at http://schizophreniabulletin.oxfordjournals.org.
Funding
This work was supported by a Medical Research Council (UK) grant to Dr Howes (grant number: MC-A656-5QD30) and a Maudsley Charity to Dr Howes (grant number 666). Additional support was received from the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. Dr Howes has received investigator-initiated research funding from and/or participated in advisory/speaker meetings organized by Astra-Zeneca, BMS, Eli Lilly, Jansenn, Lundbeck, Lyden-Delta, Servier, and Roche. Neither Dr Howes or his family have been employed by or have holdings/a financial stake in any biomedical company. Dr Egerton has received consultant fees from “Heptares Therapeutics Ltd”. Dr Stone has received honoraria from Janssen, Hoffman la Roche, and Sunovion.
Supplementary Material
Acknowledgments
We would like to thank the patients that took part in the study, the radiographers and staff at the scanner site and Dr David Lythgoe for his useful comments. The authors have declared that there are no conflicts of interest in relation to the subject of this study.
References
- 1. Howes OD, Kambeitz J, Kim E, et al. The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch Gen Psychiatry. 2012;69:776–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Uchida H, Takeuchi H, Graff-Guerrero A, Suzuki T, Watanabe K, Mamo DC. Dopamine D2 receptor occupancy and clinical effects: a systematic review and pooled analysis. J Clin Psychopharmacol. 2011;31:497–502. [DOI] [PubMed] [Google Scholar]
- 3. Lindenmayer JP. Treatment refractory schizophrenia. Psychiatr Q. 2000;71:373–384. [DOI] [PubMed] [Google Scholar]
- 4. Kapur S, Zipursky R, Jones C, Remington G, Houle S. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157:514–520. [DOI] [PubMed] [Google Scholar]
- 5. Pilowsky LS, Costa DC, Ell PJ, Murray RM, Verhoeff NP, Kerwin RW. Antipsychotic medication, D2 dopamine receptor blockade and clinical response: a 123I IBZM SPET (single photon emission tomography) study. Psychol Med. 1993;23:791–797. [DOI] [PubMed] [Google Scholar]
- 6. Kane JM. Addressing nonresponse in schizophrenia. J Clin Psychiatry. 2012;73. [DOI] [PubMed] [Google Scholar]
- 7.Lehman AF, Lieberman JA, Dixon LB, et al. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161:1–56. [PubMed] [Google Scholar]
- 8. Howes OD, Kapur S. A neurobiological hypothesis for the classification of schizophrenia: type A (hyperdopaminergic) and type B (normodopaminergic). Br J Psychiatry. 2014;205:1–3. [DOI] [PubMed] [Google Scholar]
- 9. Demjaha A, Murray RM, McGuire PK, Kapur S, Howes OD. Dopamine synthesis capacity in patients with treatment-resistant schizophrenia. Am J Psychiatry. 2012;169:1203–1210. [DOI] [PubMed] [Google Scholar]
- 10. Bartlett EJ, Brodie JD, Simkowitz P, et al. Effect of a haloperidol challenge on regional brain metabolism in neuroleptic-responsive and nonresponsive schizophrenic patients. Am J Psychiatry. 1998;155:337–343. [DOI] [PubMed] [Google Scholar]
- 11. Remington G, Kapur S, Foussias G, et al. Tetrabenazine augmentation in treatment-resistant schizophrenia: a 12-week, double-blind, placebo-controlled trial. J Clin Psychopharmacol. 2012;32:95–99. [DOI] [PubMed] [Google Scholar]
- 12. Wolkin A, Duncan E, Sanfilipo M, Wieland S, Cooper TB, Rotrosen J. Persistent psychosis after reduction in pre- and post-synaptic dopaminergic function. J Neural Transm Gen Sect. 1994;95:49–61. [DOI] [PubMed] [Google Scholar]
- 13. Abi-Dargham A, Rodenhiser J, Printz D, et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci USA. 2000;97:8104–8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bartha R, Williamson PC, Drost DJ, et al. Measurement of glutamate and glutamine in the medial prefrontal cortex of never-treated schizophrenic patients and healthy controls by proton magnetic resonance spectroscopy. Arch Gen Psychiatry. 1997;54:959–965. [DOI] [PubMed] [Google Scholar]
- 15. Kegeles LS, Mao X, Stanford AD, et al. Elevated prefrontal cortex γ-aminobutyric acid and glutamate-glutamine levels in schizophrenia measured in vivo with proton magnetic resonance spectroscopy. Arch Gen Psychiatry. 2012;69:449–459. [DOI] [PubMed] [Google Scholar]
- 16. Théberge J, Bartha R, Drost DJ, et al. Glutamate and glutamine measured with 4.0 T proton MRS in never-treated patients with schizophrenia and healthy volunteers. Am J Psychiatry. 2002;159:1944–1946. [DOI] [PubMed] [Google Scholar]
- 17. Théberge J, Williamson KE, Aoyama N, et al. Longitudinal grey-matter and glutamatergic losses in first-episode schizophrenia. Br J Psychiatry. 2007;191:325–334. [DOI] [PubMed] [Google Scholar]
- 18. Stone JM, Day F, Tsagaraki H, et al. ; OASIS. Glutamate dysfunction in people with prodromal symptoms of psychosis: relationship to gray matter volume. Biol Psychiatry. 2009;66:533–539. [DOI] [PubMed] [Google Scholar]
- 19. Bustillo JR, Rowland LM, Mullins P, et al. 1H-MRS at 4 tesla in minimally treated early schizophrenia. Mol Psychiatry. 2010;15:629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bustillo JR, Chen H, Gasparovic C, et al. Glutamate as a marker of cognitive function in schizophrenia: a proton spectroscopic imaging study at 4 Tesla. Biol Psychiatry. 2011;69:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kraguljac NV, Reid MA, White DM, den Hollander J, Lahti AC. Regional decoupling of N-acetyl-aspartate and glutamate in schizophrenia. Neuropsychopharmacol. 2012;37:2635–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reid MA, Stoeckel LE, White DM, et al. Assessments of function and biochemistry of the anterior cingulate cortex in schizophrenia. Biol Psychiatry. 2010;68:625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wood SJ, Yücel M, Wellard RM, et al. Evidence for neuronal dysfunction in the anterior cingulate of patients with schizophrenia: a proton magnetic resonance spectroscopy study at 3 T. Schizophr Res. 2007;94:328–331. [DOI] [PubMed] [Google Scholar]
- 24. Ongür D, Jensen JE, Prescot AP, et al. Abnormal glutamatergic neurotransmission and neuronal-glial interactions in acute mania. Biol Psychiatry. 2008;64:718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aoyama N, Théberge J, Drost DJ, et al. Grey matter and social functioning correlates of glutamatergic metabolite loss in schizophrenia. Br J Psychiatry. 2011;198:448–456. [DOI] [PubMed] [Google Scholar]
- 26. Marsman A, van den Heuvel MP, Klomp DW, Kahn RS, Luijten PR, Hulshoff Pol HE. Glutamate in schizophrenia: a focused review and meta-analysis of ¹H-MRS studies. Schizophr Bull. 2013;39:120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Demjaha A, Egerton A, Murray RM, et al. Antipsychotic treatment resistance in schizophrenia associated with elevated glutamate levels but normal dopamine function. Biol Psychiatry. 2014;75:11–13. [DOI] [PubMed] [Google Scholar]
- 28. Egerton A, Brugger S, Raffin M, et al. Anterior cingulate glutamate levels related to clinical status following treatment in first-episode schizophrenia. Neuropsychopharmacology. 2012;37:2515–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Szulc A, Konarzewska B, Galinska-Skok B, et al. Proton magnetic resonance spectroscopy measures related to short-term symptomatic outcome in chronic schizophrenia. Neurosci Lett. 2013;547:37–41. [DOI] [PubMed] [Google Scholar]
- 30. Barksdale KA, Lahti AC, Roberts RC. Synaptic proteins in the postmortem anterior cingulate cortex in schizophrenia: relationship to treatment and treatment response. Neuropsychopharmacology. 2014;39:2095–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lahti AC, Holcomb HH, Weiler MA, et al. Clozapine but not haloperidol Re-establishes normal task-activated rCBF patterns in schizophrenia within the anterior cingulate cortex. Neuropsychopharmacology. 2004;29:171–178. [DOI] [PubMed] [Google Scholar]
- 32. Lahti AC, Holcomb HH, Weiler MA, Medoff DR, Tamminga CA. Functional effects of antipsychotic drugs: comparing clozapine with haloperidol. Biol Psychiatry. 2003;53:601–608. [DOI] [PubMed] [Google Scholar]
- 33. Conley RR, Kelly DL. Management of treatment resistance in schizophrenia. Biol Psychiatry. 2001;50:898–911. [DOI] [PubMed] [Google Scholar]
- 34. Gardner DM, Murphy AL, O’Donnell H, Centorrino F, Baldessarini RJ. International consensus study of antipsychotic dosing. Am J Psychiatry. 2010;167:686–693. [DOI] [PubMed] [Google Scholar]
- 35. Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13:261–276. [DOI] [PubMed] [Google Scholar]
- 36. Andreasen NC, Carpenter WT, Jr, Kane JM, Lasser RA, Marder SR, Weinberger DR. Remission in schizophrenia: proposed criteria and rationale for consensus. Am J Psychiatry. 2005;162:441–449. [DOI] [PubMed] [Google Scholar]
- 37. Taylor D, Paton C, Kapur S. The Maudsley Prescribing Guidelines in Psychiatry. 11th ed. London, UK: Wiley-Blackley; 2012. [Google Scholar]
- 38. Stone JM, Pepper F, Fam J, et al. Glutamate, N-acetyl aspartate and psychotic symptoms in chronic ketamine users. Psychopharmacology (Berl). 2014;231:2107–2116. [DOI] [PubMed] [Google Scholar]
- 39. Provencher SW. Automatic quantitation of localized in vivo 1H spectra with LCModel. NMR Biomed. 2001;14:260–264. [DOI] [PubMed] [Google Scholar]
- 40. Wagenmakers EJ, Farrell S. AIC model selection using Akaike weights. Psychon Bull Rev. 2004;11:192–196. [DOI] [PubMed] [Google Scholar]
- 41. Howes OD, Vergunst F, Gee S, McGuire P, Kapur S, Taylor D. Adherence to treatment guidelines in clinical practice: study of antipsychotic treatment prior to clozapine initiation. Br J Psychiatry. 2012;201:481–485. [DOI] [PubMed] [Google Scholar]
- 42. Taylor DM, Young C, Paton C. Prior antipsychotic prescribing in patients currently receiving clozapine: a case note review. J Clin Psychiatry. 2003;64:30–34. [DOI] [PubMed] [Google Scholar]
- 43. Harrison J, Janlöv M, Wheeler AJ. Patterns of clozapine prescribing in a mental health service in New Zealand. Pharm World Sci. 2010;32:503–511. [DOI] [PubMed] [Google Scholar]
- 44. Kelly DL, Kreyenbuhl J, Dixon L, Love RC, Medoff D, Conley RR. Clozapine underutilization and discontinuation in African Americans due to leucopenia. Schizophr Bull. 2007;33:1221–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alessi-Severini S, Le Dorze JA, Nguyen D, Honcharik P, Eleff M. Clozapine prescribing in a Canadian outpatient population. PLoS One. 2013;8:e83539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schwieler L, Engberg G, Erhardt S. Clozapine modulates midbrain dopamine neuron firing via interaction with the NMDA receptor complex. Synapse. 2004;52:114–122. [DOI] [PubMed] [Google Scholar]
- 47. Tanahashi S, Yamamura S, Nakagawa M, Motomura E, Okada M. Clozapine, but not haloperidol, enhances glial D-serine and L-glutamate release in rat frontal cortex and primary cultured astrocytes. Br J Pharmacol. 2012;165:1543–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hons J, Vasatova M, Cermakova E, Doubek P, Libiger J. Different serine and glycine metabolism in patients with schizophrenia receiving clozapine. J Psychiatr Res. 2012;46:811–818. [DOI] [PubMed] [Google Scholar]
- 49. Carlsson A, Waters N, Holm-Waters S, Tedroff J, Nilsson M, Carlsson ML. Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence. Annu Rev Pharmacol Toxicol. 2001;41:237–260. [DOI] [PubMed] [Google Scholar]
- 50. Howes OD, Egerton A, Allan V, McGuire P, Stokes P, Kapur S. Mechanisms underlying psychosis and antipsychotic treatment response in schizophrenia: insights from PET and SPECT imaging. Curr Pharm Des. 2009;15:2550–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Javitt DC. Glutamate and schizophrenia: phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int Rev Neurobiol. 2007;78:69–108. [DOI] [PubMed] [Google Scholar]
- 52. Krystal JH, Perry EB, Jr, Gueorguieva R, et al. Comparative and interactive human psychopharmacologic effects of ketamine and amphetamine: implications for glutamatergic and dopaminergic model psychoses and cognitive function. Arch Gen Psychiatry. 2005;62:985–994. [DOI] [PubMed] [Google Scholar]
- 53. Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. de la Fuente-Sandoval C, León-Ortiz P, Azcárraga M, et al. Glutamate levels in the associative striatum before and after 4 weeks of antipsychotic treatment in first-episode psychosis: a longitudinal proton magnetic resonance spectroscopy study. JAMA psychiatry. 2013;70:1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. de la Fuente-Sandoval C, León-Ortiz P, Favila R, et al. Higher levels of glutamate in the associative-striatum of subjects with prodromal symptoms of schizophrenia and patients with first-episode psychosis. Neuropsychopharmacol. 2011;36:1781–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Choi YK, Gardner MP, Tarazi FI. Effects of risperidone on glutamate receptor subtypes in developing rat brain. Eur Neuropsychopharmacol. 2009;19:77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tarazi FI, Baldessarini RJ, Kula NS, Zhang K. Long-term effects of olanzapine, risperidone, and quetiapine on ionotropic glutamate receptor types: implications for antipsychotic drug treatment. J Pharmacol Exp Ther. 2003;306:1145–1151. [DOI] [PubMed] [Google Scholar]
- 58. Howes O, McCutcheon R, Stone J. Glutamate and dopamine in schizophrenia: an update for the 21st century. J Psychopharmacol. 2015;29:97–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33:523–533. [DOI] [PubMed] [Google Scholar]
- 60. Poels EMP, Kegeles LS, Kantrowitz JT, et al. Imaging glutamate in schizophrenia: review of findings and implications for drug discovery. Mol Psychiatry. 2014;19:20–29. [DOI] [PubMed] [Google Scholar]
- 61. Merritt K, McGuire P, Egerton A. Relationship between glutamate dysfunction and symptoms and cognitive function in psychosis. Front Psychiatry. 2013;4:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Snyder J, Wilman A. Field strength dependence of PRESS timings for simultaneous detection of glutamate and glutamine from 1.5 to 7T. J Magn Reson. 2010;203:66–72. [DOI] [PubMed] [Google Scholar]
- 63. Bustillo JR, Chen H, Jones T, et al. Increased glutamine in patients undergoing long-term treatment for schizophrenia: a proton magnetic resonance spectroscopy study at 3 T. JAMA Psychiatry. 2014;71:265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Théberge J, Al-Semaan Y, Williamson PC, et al. Glutamate and glutamine in the anterior cingulate and thalamus of medicated patients with chronic schizophrenia and healthy comparison subjects measured with 4.0-T proton MRS. Am J Psychiatry. 2003;160:2231–2233. [DOI] [PubMed] [Google Scholar]
- 65. Kanowski M, Kaufmann J, Braun J, Bernarding J, Tempelmann C. Quantitation of simulated short echo time 1H human brain spectra by LCModel and AMARES. Magn Reson Med. 2004;51:904–912. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}