

Abstract
Glucocorticoid hormones (GCs) are important regulators of lipid metabolism, promoting lipolysis with acute treatment but lipogenesis with chronic exposure. Conventional wisdom posits that these disparate outcomes are mediated by the classical glucocorticoid receptor GRα. There is insufficient knowledge of the GC receptors (GRα and GRβ) in metabolic conditions such as obesity and diabetes. We present acute models of GC exposure that induce lipolysis, such as exercise, as well as chronic-excess models that cause obesity and lipid accumulation in the liver, such as hepatic steatosis. Alternative mechanisms are then proposed for the lipogenic actions of GCs, including induction of GC resistance by the GRβ isoform, and promotion of lipogenesis by GC activation of the mineralocorticoid receptor (MR). Finally, the potential involvement of chaperone proteins in the regulation of adipogenesis is considered. This reevaluation may prove useful to future studies on the steroidal basis of adipogenesis and obesity.
Keywords: glucocorticoid receptor, glucocorticoid receptor, glucocorticoid receptor-α, glucocorticoid receptor-β, glucocorticoids, adipose differentiation, adipogenesis, lipolysis
there is much controversy over the role of glucocorticoids (GCs) in either the enhancement or reduction of obesity. Chronic GC treatment results in weight gain as well as increased visceral obesity and hypertension. On the other hand, acute GC treatment typically causes weight loss, especially at adipose tissue. Adipose tissue expansion occurs by increasing either adipocyte number (adipogenesis) or adipocyte size (hypertrophy). During the development of obesity, the adipocyte number can increase, although there is a higher rate of apoptosis (63). Adipocytes are derived from the mesenchyme lineage. Mesenchymal stem cells (MSCs) are committed to preadipocytes with the loss of capacity to differentiate into other cell types (55). The MSCs enter the initiation phase of determination toward mature white adipocyte differentiation and undergo genetic and molecular changes that allow them to accumulate and produce lipids. Typically, a problem arises when there is excessive adipose tissue expansion due to positive energy balance, which results most commonly from overeating or physical inactivity. High-fat diets in particular can lead to weight gain, hepatic lipid accumulation, and insulin insensitivity (7). In obese individuals, adipocyte expansion also leads to increased inflammation as a result of proinflammatory cytokine secretion from adipose tissue and macrophage infiltration of the tissue. This results in a chronic inflammatory state in the obese that leads to significant health risks for other unfavorable conditions, such as type 2 diabetes, cardiovascular diseases, and various types of cancers (57). Typically, GCs exert potent anti-inflammatory actions, which serve as the dominant basis for GC-based therapeutics. Yet chronic GC exposure has been linked to elevated inflammatory states, which has been interpreted as a state of GC resistance (35). However, the underlying mechanisms of GC resistance have yet to be defined.
The process of adipogenesis and adipocyte tissue expansion are complex mechanisms utilizing a number of transcription factors that include the peroxisome proliferator-activated receptor-γ (PPARγ; see appendix a: pparγ promotes adipogenesis), CCAAT-enhancer-binding protein-α (C/EBPα), and purportedly the glucocorticoid receptor-α (GRα). Because GCs are commonly used to induce adipogenesis in tissue culture systems, mediation of this response by GRα has been assumed. Yet under normal physiological conditions, GCs often have the opposite effect, such as the promotion of lipolysis. For this and other reasons, it is reasonable to speculate whether GRα is the actual mediator of GC-induced adipogenesis, and the potential roles of other corticosteroid receptors should be considered. For example, the contributions of other GR isoforms, such as GRβ and the translational isoforms of GRα, to adiposity are largely unknown, but recent evidence suggests that GRβ at least can be regulated by metabolic signals. Moreover, the closely related mineralocorticoid receptor (MR) is now emerging as an important regulator of GC effects on adipose biology. Herein, we will discuss various physiological aspects of obesity and adiposity, with a particular emphasis on GR and MR receptors, and also consider how phosphorylation mechanisms may affect the activities of each receptor.
CORTICOSTEROID RECEPTORS AND OBESITY
Although it is established that extended exposure to GCs causes abdominal obesity and Cushing's syndrome (see appendix b: hpa axis and production of glucocorticoids), the underlying mechanism is complex. Many studies have clearly shown that acute GC exposure is lipolytic at adipose tissue (16, 71). Yet in the long term, pharmacological GC treatment appears to promote lipogenesis at this very same tissue. Clues to understanding this duality may be found in the divergent properties of the corticosteroid receptors. There are two primary nuclear receptors that can bind GCs: 1) GR, nuclear receptor subfamily 3, group C, member 1 (NR3C1), which is located on chromosome 5 (5q31); and 2) MR (NR3C2), located on chromosome 4 (4q31). MR is highly expressed in the white and brown adipose tissue as well as in the brain, heart, liver, and kidney of mice, and almost at similar levels in humans (Table 1). GR expression does not vary as much as MR, but the highest expression was found in T cells, which may be for the regulation of the immune system. However, the specific GR isoform is not reported, which has been a complication of measuring “total” GR expression without regard to the particular isoform. MR is typically activated by aldosterone, a corticosteroid that increases the reabsorption of ions and water in the kidneys as part of the renin-angiotensin-aldosterone system. Thus, MR is involved in the regulation of blood pressure and electrolyte balance, a function that is not directly controlled by GR. However, GCs like cortisol can affect blood pressure because MR has the same affinity for cortisol as it does for aldosterone (2). Indeed, the binding affinity of MR for cortisol is greater than that of GR (2). Thus, it is possible that many GC-mediated responses attributed to GR activation may be under the direct control of MR.
Table 1.
Expression profiles of GR and MR in mouse and human
| Tissue | NR3C1 (GR) | NR3C2 (MR) |
|---|---|---|
| Metabolic | ||
| Human | ||
| Adipose (white) | 1.0 | 0.9 |
| Skeletal muscle | 1.1 | 1.2 |
| Liver | 1.4 | 1.3 |
| Kidney | 0.8 | 0.8 |
| Mouse | ||
| Adipose (brown) | 1.1 | 4.2 |
| Adipose (white) | 0.8 | 2.5 |
| Skeletal muscle | 1.7 | 5.7 |
| Liver | 0.6 | 4.1 |
| Kidney | 0.5 | 4.2 |
| Immune | ||
| Human | ||
| T cells CD4+ | 1.4 | 1.4 |
| T cells CD8+ | 3.2 | 2.3 |
| Mouse | ||
| T cells CD4+ | 3.0 | 1.0 |
| T cells CD8+ | 2.2 | 0.8 |
| HPA axis | ||
| Human | ||
| Adrenal gland | 0.9 | 0.8 |
| Pituitary | 1.1 | 2.2 |
| Hypothalamus | 1.0 | 1.2 |
| Mouse | ||
| Adrenal gland | 0.5 | 4.4 |
| Pituitary | 1.1 | 30.2 |
| Hypothalamus | 0.9 | 9.8 |
| Other brain | ||
| Human | ||
| Cerebellum | 0.8 | 1.0 |
| Cerebellum cortex | 1.1 | 1.1 |
| Thalamus | 1.0 | 1.0 |
| Mouse | ||
| Cerebellum | 0.7 | 5.9 |
| Cerebral cortex | 1.1 | 11.2 |
| Hippocampus | 0.8 | 102.9 |
| Random tissues | ||
| Human | ||
| Lung | 1.1 | 1.1 |
| Heart | 1.3 | 1.3 |
| Bone marrow | 1.0 | 1.0 |
| Prostate | 1.1 | 1.1 |
| Mouse | ||
| Lung | 1.2 | 4.2 |
| Heart | 1.3 | 9.3 |
| Bone | 0.9 | 0.8 |
| Prostate | 0.4 | 9.3 |
GR, glucocorticoid receptor; MR, mineralocorticoid receptor. The high-throughput gene expression data was downloaded for normal tissues and organs from http://biogps.org (69, 70). For normalization, the average value was divided by the graph median.
Regulation of blood pressure aside, GR and MR appear to have opposing roles in other aspects of physiology. As mentioned already, acute GC targeting of GR stimulates lipolysis in adipose tissue. Yet there is now emerging evidence that adipose tissue also expresses MR possibly to a higher extent than GR (Table 1). MR has been shown to respond to cortisol by promoting adipogenesis and lipid storage (see below for further discussion of this topic) With respect to inflammation, GR and MR are also oppositional, with significant implications for acute vs. chronic GC exposure. GCs are potent anti-inflammatory compounds that are widely used to treat inflammatory diseases such as rheumatoid arthritis and asthma. GCs exert their anti-inflammatory effects through GR by increasing transcription of anti-inflammatory genes (e.g., IκBα) while inhibiting numerous proinflammatory genes (e.g., TNFα). Although this is a common characteristic of GCs, there are many instances where GCs fail to function in this capacity. It is tempting to speculate that this failure may be due to activation of MR, which we now know promotes the expression of inflammatory cytokines in adipose tissue in response to both aldosterone and GCs like cortisol (44). Therefore, this mechanism may underlie several heretofore incongruous reports. For example, in an obese/diabetic mouse model, GCs were found to augment, rather than repress, the immune response and promote neuroinflammation, whereas inhibition of GCs decreased proinflammatory cytokines (IL-1β/TNFα) in the brain (15), where MR is highly expressed (Table 1). In humans, chronic oral GC treatment leads to weight gain and adipocyte expansion compared with acute treatment (6). The latter observation serves to highlight another potentially significant feature of the GR/MR antagonism in adipose tissue. The binding affinity of oral GCs like prednisone (metabolized to prednisolone) has been reported to be higher for MR compared with GR (64). However, studies by Lan et al. (40) in rat kidney found the competitive inhibition (C50) of cortisol to be 115.2 nM for aldosterone and 124 nM for dexamethasone and the C50 for prednisolone to be 65.2 nM for aldosterone and 55 nM for dexamethasone, which suggests that the affinities of cortisol and prednisolone for the two receptors are nearly the same. Contradictory to this investigation, Reul et al. (51) found that competition of steroids for MR and GR in the dog hippocampus was 0.19 nM for cortisol and 0.42 nM for dexamethasone for MR and 5.00 nM for cortisol and 7.75 nM dexamethasone for GR. Thus, low-dose chronic treatment may preferentially activate the prolipogenic actions of adipose MR, whereas high-dose acute treatment is needed to activate GR-mediated lipolysis.
An alternative form of GR may assist the actions of MR in adipose, which is also proinflammatory. The GR (NR3C1) gene is a complex arrangement of 9 exons (Fig. 1) that are alternatively spliced to create multiple isoforms, such as GRα, GRβ, GRγ, GR-A, and GR-P (47). Two of these isoforms, GRα and GRβ, have been implicated in GC resistance. In both mice and humans, the GR protein is composed of exons 2–8, and 50 amino acids are added from exon 9α to produce GRα. Alternatively, 15 amino acids from either 9β for humans or intron 8 for mice are added to exon 8 to produce GRβ. GRα is the classical receptor that is bound by GCs to regulate gene expression. Thus, a decrease in GRα proteins may also lead to a reduced response to GCs and resistance. In contrast, GRβ lacks helix 12 of the ligand-binding domain and does not bind GCs, yet it appears to have intrinsic gene regulatory activity (36). However, most investigations into GRβ actions suggest that its primary function is to serve as an inhibitor of GRα (25, 62). Previously, GRβ expression was thought to be specific to humans. However, GRβ was discovered in the mouse (25) and then later in the rat (18) and most recently in the pig (52). At present, it is not known whether GRβ specifically inhibits the lipolytic actions of GRα. But several reports exist showing that GRβ is regulated metabolically (18, 25, 62). In cell-based studies, elevated expression of GRβ mRNA and protein was observed in response to insulin stimulation. More importantly, increased expression of GRβ was found in the livers of rats following insulin injection and in mouse livers during the refeeding phase after fasting. In all reports, GRα expression was not affected by insulin or refeeding. As is the case of Cushing's syndrome, chronic exposure of humans to GCs leads to increased total GR protein but, more importantly, decreased affinity for GCs (3, 32). Potentially, this suggests a state of GC resistance, which may be due to increasing GRβ expression and negative regulation of GRα (12). This speculation is further supported by the fact that treatment of cells with the GC agonist dexamethasone not only caused GRβ to increase but also reduced levels of GRα. Thus, GRβ may be a method by which to compensate for cortisol excess. The effect of increased GRβ on GRα or MR activity in adipose tissue has yet to be determined. However, it is clear that GRβ is expressed in the adult human white adipose tissue since there is an association of GR polymorphism A3669G in exon 9β and reduced central obesity in women (65). Moreover, in an adipogenesis model, it was found that GRβ increased during differentiation, whereas GRα remained constant (28). Taken together, these observations serve as strong circumstantial evidence that increased GRβ or decreased GRα may be a component of GC resistance in obesity. Critical tests of this hypothesis will be a determination of whether GRβ is essential to adipogenesis in cell-based models and whether it contributes to animal models of diet-induced obesity. A tissue-specific GR isoform overexpression or knockout would reveal their roles in each organ.
Fig. 1.
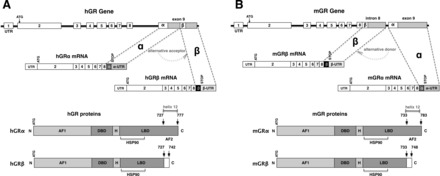
Alternative splicing of the human (hGR) and mouse glucocorticoid (GC) receptor (hGR) genes. Alternative splicing of the hGR (A) and mGR genes (B) constructs the GRα and GRβ isoforms. The mGRβ isoform exhibits a functional domain structure that is nearly identical to hGRβ, and both have 15 amino acids as a result of an alternative acceptor for human and alternative donor for mouse from the β-exon. Compared with the GRα protein, the β-isoforms of both species have reduced and distinct COOH-terminal regions that have a truncated ligand binding AF-2 domain (helix 12). These features account for their lack of ability to bind hormone. AF, activation function; DBD, DNA-binding domain; H, hinge region; LBD, ligand-binding domain; ATG, ATG start site; N, NH2 terminus; UTR, untranslated region. Heat shock protein 90 (HSP90) binding regions for human and mouse GRα and GRβ are shown.
CORTICOSTEROID RECEPTORS AND ADIPOGENESIS
Unlike the accumulation of fat in adipocytes, the process of lipolysis is the breakdown and release of lipids from hydrolysis of triglycerides to glycerol and free fatty acids. In acute treatment, GCs function to promote lipolysis (16), which occurs by GRα sensitizing catecholamine induction of protein kinase A (PKA) activity by downregulating the transcription of phosphodiesterase 3B (PDE3B) (72) (Fig. 2). Suppression of PDE3B results in increased cyclic AMP (cAMP) levels that activate PKA, leading to enhanced phosphorylation of hormone-sensitive lipase (HSL) and hydrolysis of intracellular lipids (38, 72). Also, GCs induce the expression of angiopoietin-like 4 (ANGPTL4) protein, which promotes lipolysis by further stimulating cAMP signaling (23). GCs also directly upregulate the expression of adipose triglyceride lipase (ATGL), which in combination with HSL are the two critical genes that promote hydrolysis of triglycerides in adipose tissue (59, 72). A second target of PKA is perilipin, a protective protein that surrounds lipid droplets. Phosphorylation causes the release of perilipin from the droplets, allowing lipase access to triglycerides (11). In summary, by regulating lipases and perilipin through the PKA pathway, GCs can robustly promote catalysis of triglycerides and subsequent release of free fatty acids (FFA) and glycerol into the bloodstream (11).
Fig. 2.
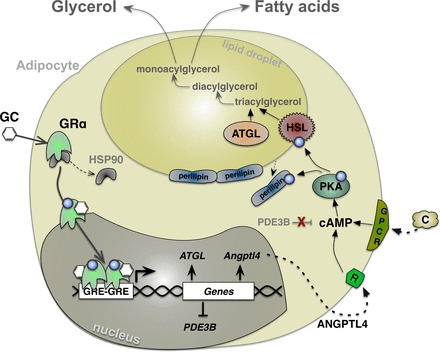
GCs induce lipolysis through enhancement of lipase genes. GCs bind to GRα, causing phosphorylation, dissociation of HSP90, translocation to the nucleus, and binding to GC response elements (GREs) in promoters of genes. In adipocytes, GRα enhances genes involved in lipolysis [adipose triglyceride lipase (ATGL) and angiopoietin-like 4 (Angptl4)] and suppresses genes that are lipogenic [phosphodiesterase 3B (PDE3B)]. Catecholamines (C) and ANGPTL4 bind to their receptors and increase cellular cAMP levels, which activate PKA, causing phosphorylation of hormone-sensitive lipase (HSL) and perilipins. Phosphorylation causes the release of perilipins from the lipid droplet and exposure of HSL to triacylglycerol, resulting in breakdown and release of glycerol and fatty acids in the blood. GPCR, G protein-coupled receptor.
As Fig. 2 explains the cellular basis for GRα stimulation of lipolysis, rodent models confirm that administration of GCs increases FFAs throughout circulation, indicating a systemic stimulation of lipolysis (72). However, the effects of GCs on lipolysis are fat depot specific. Xiao et al. (71) showed that rats treated for 12 wk with prednisolone had reduced body weight and differential effects in visceral (VAT) and subcutaneous (SAT) adipose tissues. While prednisolone increased adipocyte number in VAT (∼100 adipocytes/slide) and SAT (∼200 adipocytes/slide), adipocyte diameter was reduced in SAT by 50% compared with VAT (71). Furthermore, following prednisolone treatment, SAT adipocyte diameter was accompanied by increased ATGL and HSL expression in contrast to VAT (71). An investigation by Goedecke et al. (19) supported these findings in an ethnic-based study that found that African-American women had decreased GRα expression in their SAT, which was associated with increased abdominal SAT, inflammatory markers, and reduced insulin sensitivity.
There is substantial literature supporting the fact that GCs promote adipogenesis by inducing adipose progenitors (e.g., C/EBPβ) during the early stages of differentiation (41). However, the conventional wisdom that this effect is mediated solely by GRα is being called into question by the recent discovery that MR also promotes adipogenesis. Aldosterone has been shown to increase adipocyte expansion more effectively than cortisol. Moreover, aldosterone has a much higher binding affinity for MR compared with GR. Thus, aldosterone promotion of adipogenesis is most likely through activation of MR (10) and not GRα. Conversely, as stated above, the binding affinity of MR for cortisol is greater than that of GR, suggesting that the effects of GCs in the early stages of adipogenesis might be mediated by MR. Evidence to support this hypothesis has been reported recently. During adipose differentiation, MR mRNA and protein are upregulated, and knockdown of MR inhibited adipogenesis (10). Hoppmann et al. (30) demonstrated that selectively stimulating MR by aldosterone treatment resulted in increased expression of proinflammatory adipokines, whereas GR stimulation with dexamethasone suppressed proinflammatory adipokine expression. Blockade of MR by gene knockout or with antagonists substantially decreased the differentiation potential of primary mouse adipocytes (10, 30), whereas adipocytes obtained from GR-knockout mice displayed no significant alteration in adipogenesis (30). Several studies have demonstrated that MR expressed in brown adipose tissue can be activated by aldosterone, and in response they sustain brown adipogenesis (49, 73).
The synthetic corticosteroid dexamethasone is a common component of most adipocyte differentiation cocktails, and this property has been widely viewed as evidence that GRα activation is needed for the early stages of differentiation, at least in 3T3-L1 preadipocytes. However, dexamethasone can be eliminated from some adipogenesis assays, resulting in cells that still undergo differentiation to the fully competent lipid-bearing state (28). In the studies by Hoppmann et al. (30), GR-knockout preadipocytes showed no deficiency of differentiation, whereas MR-knockout cells completely failed to store lipid, suggesting that an alternative non-GRα pathway exists for the promotion of adipogenesis. Importantly, the adipogenic cocktail used to obtain those results contained dexamethasone, which suggests that MR may possibly be the primary mediator. Although this might seem incongruous to some, it is an overlooked fact that dexamethasone can selectively bind MR, albeit with lower affinity compared with GR in adipose, and activate MR-mediated gene expression (2, 54). In summary, it appears that a comprehensive reevaluation of the relative contributions of GR and MR to adipogenesis is needed. This evaluation, along with more definitive experimental results, will have important implications for the development of the next generation of drugs targeting the steroidal basis of obesity and metabolic conditions linked to excessive adiposity.
EFFECT OF EXERCISE ON CORTICOSTEROID RECEPTOR AND 11β-HYDROXYSTEROID DEHYDROGENASE TYPE 1 EXPRESSION
Exercise is known to reduce adiposity and increase lean muscle mass. This process includes utilization of fat stores in adipose by the release of lipids through lipolysis and the breakdown and rebuilding of muscle. Ultimately, exercise causes an enlargement of muscle tissue and a reduction in fat stores. Exercise is a metabolic stress that elevates GCs in circulation (42). Exercise promotes the production of proopiomelanocortin, which is a necessary precursor of ACTH in the anterior pituitary gland, and stimulates cortisol production from the adrenals (50). Although exercising mice have body weights comparable with control mice, they have lower abdominal fat but elevated muscle mass, along with heavier adrenal glands (17). There are two interesting components that correlate with and are even necessary for a proper GC response during exercise. The two factors are 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) and GRα, whose expression levels may directly affect GC availability and utilization. In response to exercise, 11β-HSD1 increases significantly in visceral adipose stores (9, 14). 11β-HSD1 converts inactive cortisone to cortisol, and studies have confirmed that tissue-specific 11β-HSD1 overexpression elevates intracellular levels of active GCs (45). In contrast to adipose, exercise causes 11β-HSD1 expression and activity to decrease in skeletal muscle (14). Because GCs promote myopathy in muscle but lipolysis in adipose, the inverse phenomenon of 11β-HSD1 expression may be a contributor to the increased muscle mass and reduced adipose mass observed in response to exercise. As a direct consequence of increased GCs and elevated lipolysis, decreased adipose tissue mass has been found. This relationship between GC levels in adipose and lipolysis was further confirmed using a GR antagonist, which blocked GC action and decreased lipolysis (8).
Not only does exercise increase active GC content in adipose tissue, it also upregulates GR protein expression, a necessary component of GC signaling to induce lipolysis (Fig. 3) (8). Thus, it is reasonable to conclude that elevated GR expression in adipose during exercise is also a contributing factor to reduced adipose tissue mass. Conversely, since GCs break down muscle (24), it is also reasonable to speculate that decreased GRα expression would increase muscle mass during exercise. Although this has not been tested directly, muscle-specific knockout of GR in mice has been shown to increase muscle mass and, curiously, reduce adipose stores. The latter effect in the muscle-specific GR-KO mouse appears to be mediated by increased expression of the fibroblast growth factor 21 endocrine factor in the liver to promote increased fat utilization via a muscle-liver-adipose axis (58).
Fig. 3.
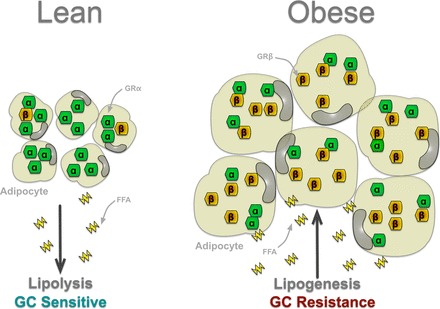
Speculation that obesity is a state of GC resistance and the relationship of GRα and GRβ. The participation of the GR isoforms in obesity and adipocyte hypertrophy is unknown. However, the role of GCs and GRα in lipolysis has been demonstrated. Therefore, lean patients may have GC sensitivity that enhances GRα activity in adipocytes, which elevates lipolysis and the release of free fatty acids (FFA), whereas obese patients may have GC resistance by increasing levels of GRβ, resulting in adipocyte hypertrophy, lipogenesis, and FFA uptake.
Because GRβ antagonizes GRα action, it is interesting to speculate that upregulation of its expression could simultaneously inhibit lipolysis at adipose (Fig. 3) while increasing muscle mass. However, liberated fatty acids are needed as an energy source during exercise, making it unlikely that upregulation of GRβ in adipose occurs under these conditions. But mechanisms may exist for selective upregulation of GRβ in muscle in response to exercise. To date, there have been no studies to compare GRα and GRβ during exercise or in response to obesity or high-fat diets. A potential role for MR in exercise also exists. As discussed already, it is now clear that MR promotes adipogenesis and lipid storage in response to cortisol. Studies have shown that MR levels in the hippocampus decrease in response to exercise (17). Although not yet demonstrated in adipose, a reduction of MR in response to exercise would be an important observation. Such a result would imply that fatty acid mobilization during exercise results from the synergistic inhibition of MR-mediated lipid storage and stimulation of GRα-mediated lipolysis. The above observations suggest the intriguing possibility that the metabolic response to exercise may result from the coordinated interplay of GRα, GRβ, and MR at multiple organs, especially in adipose and muscle. An adipose or muscle-specific overexpression of the GR isoforms is warranted to uncover their roles in lean and fat mass.
GLUCOCORTICOID RECEPTOR AND FATTY LIVER
Adipose tissue expansion increases fat storage and also causes elevated lipids in the serum. The lipids are taken up by the liver and may promote the development of a fatty liver (hepatic steatosis), which can impair insulin turnover and cause insulin resistance. During the time of feeding, the pancreas releases insulin, and the liver regulates the amount in circulation by clearing it from the hepatic portal vein. A fatty liver reduces insulin clearance, inducing peripheral tissue insulin resistance, possibly due to the increased duration of circulating insulin, which may cause visceral adiposity, type 2 diabetes, and the metabolic syndrome. Normally, the liver responds to energy deprivation by producing glucose by the process of gluconeogenesis. The liver is under homeostatic control by various hormones, including GCs. The GC-GR interaction is essential for appropriate liver function and has been implicated in several liver dysfunctions, including hepatic steatosis (fatty liver). Jenson et al. (34) reported that the fatty livers of obese rats displayed reduced GR expression compared with lean rats. In a study by Mueller et al. (46), mice with liver-specific deletion of GR maintained on a regular diet developed fatty liver. Similarly, mice with reduced hepatic GRα activity due to deletion of the GR chaperone protein FKBP52 were found to be more susceptible to diet-induced steatosis (68). These reports implicate decreased GR expression and activity as a contributory factor to fatty liver disease. In both prior works, reduced hepatic GR activity also resulted in hypercortisolism due to compensation by the hypothalamic-pituitary-adrenal (HPA) axis in response to peripheral glucocorticoid insensitivity. Liver-specific GR-knockout mice also display increased corticosteroid-binding globulin, which keeps the circulating GCs in an inactive form, thereby further inducing compensatory activation of the HPA axis (46). In Cushing's disease, hypercortisolism commonly leads to hepatic steatosis (53). These facts suggest that GC overstimulation may also contribute to steatosis. Indeed, Mueller et al. (46) demonstrated that hypercortisolism due to GR deletion from hepatocytes caused increased GR activation in adipocytes, which consequently led to increased lipolysis via upregulation of ATGL and HSL and reduction of perilipins. This phenomenon ultimately results in the overall diminution of adipose storage and the subsequent elevation of circulating FFAs, which accumulate in the liver (46). Moreover, due to impairment of β-oxidation in fatty liver disease, which was possibly due to the decreased GR expression, the FFAs are inversely used for the synthesis of triglycerides (5, 50). Another key mechanism underlying hepatic steatosis is insulin resistance and hypercortisolism, which increases the production of glucose and insulin, eventually leading to a state of insulin resistance and hepatic steatosis (43, 48). The effect of GCs on lipid accumulation in the liver may be mediated by overstimulation of lipolysis from adipose with long-term GC treatment, which may enhance the development of a fatty liver and insulin resistance.
GR PHOSPHORYLATION ATTENUATES ADIPOSITY
Adipogenesis is an intricate process involving various nuclear receptors, including PPARγ and GRα. These two receptors can be viewed as physiological antagonists at adipose tissue, with PPARγ promoting adipogenesis and lipid storage and GRα promoting lipolysis and lipid secretion. Of particular note is the recent discovery that the oppositional properties of the receptors are regulated by phosphorylation (28). Upon hormone binding, GRα is hyperphosphorylated at serines 203, 211, and 226, followed by dissociation from heat shock protein 90 (HSP90) and translocation into the nucleus, where it dimerizes on gene promoters for transcriptional activity (Fig. 2) (26, 28). In contrast, ligand binding of PPARγ results in dephosphorylation at serine 112, which increases its gene activity to regulate adipogenesis (see appendix a: pparγ promotes adipogenesis). Through its dephosphorylating activity, protein phosphatase 5 (PP5) governs the critical balance between lipogenesis and lipolysis by binding to HSP90 via its tetratricopeptide repeat (TPR) domains, resulting in the inhibition of GRα and activation of PPARγ (28). The reciprocal regulation of these two nuclear receptors is what allows PP5 to function as a fulcrum in maintaining the balance between lipogenesis and lipolysis. PP5-null mice on a high-fat diet are lean with glucose resistance and insulin intolerance (21, 22). Jacob et al. (33) showed that mice with inactivated PP5 had decreased adipogenesis and adipose tissue, which was due to increased GR phosphorylation. The FK506-binding protein-51 (FKBP51) has also been found to regulate the activities of GRα and PPARγ in lipid metabolism reciprocally. FKBP51 achieves this by being a negative regulator of the Akt/p38 kinase pathway that targets each receptor (60, 61). Stechschulte et al. (60) found that FKBP51-knockout cells were also resistant to lipid accumulation, similar to PP5-deficient cells. Although structurally related to FKBP51, FKBP52 is a TPR protein that serves to promote rather than inhibit GRα activity. Thus, mice with heterozygous loss of FKBP52 exhibit reduced GRα hepatic activity that results in hepatic steatosis and glucose intolerance (68). FKBP51 and FKBP52 have been shown to regulate GRα phosphorylation inversely, and drugs such as timcodar (VX-853) that target FKBP51 increase GRα activity (29). Therefore, timcodar and other drugs that target these TPR proteins may prove useful in the treatment of obesity.
CONCLUDING REMARKS AND FUTURE PERSPECTIVES
Glucocorticoid hormones are essential but paradoxical regulators of lipid metabolism, promoting lipolysis under acute exposure but lipogenesis and GC resistance with chronic treatment. Not surprisingly, the exact mechanisms governing each of these actions are unclear and controversial. In this review, we provide evidence that our lack of understanding may be due to a failure to differentiate between the effects of the classical GC receptor GRα and two emerging antagonistic receptors, MR and GRβ. The latest studies confirm that GRα is lipolytic and not a major contributor to adipogenesis. Conversely, MR appears to be a major stimulator of adipogenesis in response not only to aldosterone but to GC agonists as well. Although the evidence is presently less strong for GRβ, data showing it to be regulated by GCs and insulin in both cellular and animal models suggest that it is also a promising candidate for further studies on GC resistance and lipid metabolism. Finally, a better understanding of the central roles of the TPR proteins PP5, FKBP51, and FKPB52 in adipose metabolism and their ability to regulate not only GRα and PPARγ but potentially GRβ and MR may prove invaluable in the development of new drugs for the treatment of obesity.
GRANTS
The research reported in this publication was supported by the National Heart, Lung, and Blood Institute under award nos. K01-HL-125445 (T. D. Hinds, Jr.) and L32-MD-009154 (T. D. Hinds, Jr.) and the National Institutes of Health (NIH) PRIDE grant (HL-106365; T. D. Hinds, Jr.). The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
DISCLOSURES
The authors have nothing to disclose.
AUTHOR CONTRIBUTIONS
K.J. and T.D.H. conception and design of research; K.J., J.S.M., E.R.S., and T.D.H. drafted manuscript; K.J., J.S.M., E.R.S., and T.D.H. edited and revised manuscript; K.J., J.S.M., E.R.S., and T.D.H. approved final version of manuscript; T.D.H. prepared figures.
Appendix A: PPARγ PROMOTES ADIPOGENESIS
PPARγ is the master regulator of adipocyte differentiation; it promotes adipogenesis and lipid storage. Conversely, PPARγ-null embryonic stem cells fail to undergo adipogenesis (56). Similarly, an adipose-specific knockout of PPARγ in mice results in complete lipoatrophy and loss of white adipose tissue (67). However, a whole body knockout of PPARγ is lethal due to myocardial thinning (4). In a high-fat-fed state, PPARγ activity promotes the uptake of free fatty acids from circulation to be stored in adipose tissue for future utilization. Increased PPARγ activity can lead to excess adipose tissue expansion and obesity (39). Conversely, PPARα is reduced in obesity and a high-fat-fed diet (20, 27). Numerous endogenous and exogenous ligands, including eicosanoids, fatty acids, and some antidiabetic drugs such as thiazolidinediones, can bind PPARγ and activate it (37, 66); its activation then leads to increased adipocyte differentiation and lipid storage. In contrast to many other phosphoproteins, PPARγ activity is inhibited by phosphorylation of its serine (Ser112), which has been shown to be done by the mitogen-activated protein kinase signaling cascade and thereby inhibit adipose differentiation (Fig. 4) (1). The Ser112 site is a key regulatory site on PPARγ that mediates the adipogenic properties and lipid accumulation (31), and a mutation of serine to alanine (Ser112A) prevents phosphorylation and increases PPARγ activity (28). In an adipogenesis assay with a mutated Ser112A, PPARγ resulted in significantly higher lipid accumulation compared with the wild-type PPARγ (28, 31). Hinds et al. (28) recently showed that PP5 specifically dephosphorylated Ser112 on PPARγ, leading to increased activity, and a knockout of PP5 resulted in decreased lipid accumulation (Fig. 4). Another regulatory site on PPARγ is Ser273, which is phosphorylated by cyclin-dependent kinase, which adversely affects glucose uptake and the antidiabetic properties of PPARγ (13). However, it has not yet been shown whether Ser273 has a role in adipogenesis or whether PP5 also targets this site.
Fig. 4.
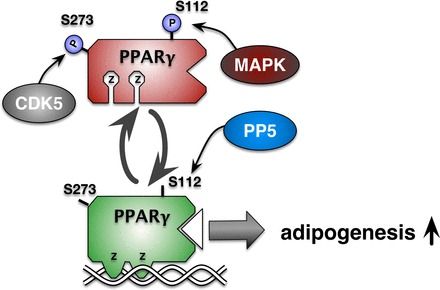
Protein phosphatase (PP5) dephosphorylates peroxisome proliferator-activated receptor-γ (PPARγ) to increase adipogenesis. In adipocytes, PP5 dephosphorylates Ser112 (S112) in PPARγ, increasing adipogenic gene expression. The MAPK pathway causes phosphorylation (P) of S112, resulting in the inhibition of lipid accumulation. Cyclin-dependent kinase-5 (CDK5) targeting of Ser273 (S273) leads to insulin resistance. At present, phosphatase targeting of S273 is unknown. Z, zinc finger DNA-binding domain.
Appendix B: HPA AXIS AND PRODUCTION OF GLUCOCORTICOIDS
GCs are a family of steroid hormones that are made in the adrenal cortex and participate in a wide variety of functions. These range from immune functions such as immunosuppressive and anti-inflammatory roles to the production of glucose from the liver. In a healthy individual, stress induces the release of corticotropin-releasing hormone (CRH), the principal regulator of the hypothalamic-pituitary-adrenal (HPA) axis, from the hypothalamus. CRH enters the hypophyseal portal vasculature and binds to its receptors, stimulating the release of adrenocorticotropic hormone (ACTH) from the anterior pituitary gland. Stress-induced vasopressin release from posterior pituitary can also induce ACTH release. ACTH in turn mediates the synthesis and release of GCs, especially cortisol, from the adrenal cortex. The release of cortisol causes the breakdown of FFAs (lipolysis) in adipose and amino acids from muscle tissues. These nonhexose substrates are shuffled to the liver to be used for glucose production (gluconeogenesis) during the “fight or flight” activation of the sympathetic nervous system. As with other physiological systems that utilize feedback mechanisms to maintain homeostasis, in the HPA axis, cortisol via negative feedback inhibits further release of both CRH and ACTH. Cushing's syndrome is attributed to a systemic overproduction of cortisol and prolonged exposure of the body to high levels of GCs. It is caused by the constant activation of the HPA axis and continuous production of cortisol by the adrenal glands as a result of a tumor either in the pituitary or in the adrenal gland. Disruption of the HPA axis prevents cortisol from exerting its negative feedback effect on the release of CRH and ACTH. In addition to tumors, Cushing's syndrome can also result from the chronic use of corticosteroid medication. This mechanism similarly exposes the body to high GC levels.
REFERENCES
- 1.Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J Biol Chem 272: 5128–5132, 1997. [DOI] [PubMed] [Google Scholar]
- 2.Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, Evans RM. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science 237: 268–275, 1987. [DOI] [PubMed] [Google Scholar]
- 3.Bamberger CM, Bamberger AM, de Castro M, Chrousos GP. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest 95: 2435–2441, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell 4: 585–595, 1999. [DOI] [PubMed] [Google Scholar]
- 5.Berson A, De Beco V, Letteron P, Robin MA, Moreau C, El Kahwaji J, Verthier N, Feldmann G, Fromenty B, Pessayre D. Steatohepatitis-inducing drugs cause mitochondrial dysfunction and lipid peroxidation in rat hepatocytes. Gastroenterology 114: 764–774, 1998. [DOI] [PubMed] [Google Scholar]
- 6.Berthon BS, MacDonald-Wicks LK, Wood LG. A systematic review of the effect of oral glucocorticoids on energy intake, appetite, and body weight in humans. Nutr Res 34: 179–190, 2014. [DOI] [PubMed] [Google Scholar]
- 7.Buettner R, Parhofer KG, Woenckhaus M, Wrede CE, Kunz-Schughart LA, Scholmerich J, Bollheimer LC. Defining high-fat-diet rat models: metabolic and molecular effects of different fat types. J Mol Endocrinol 36: 485–501, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Campbell JE, Fediuc S, Hawke TJ, Riddell MC. Endurance exercise training increases adipose tissue glucocorticoid exposure: adaptations that facilitate lipolysis. Metabolism 58: 651–660, 2009. [DOI] [PubMed] [Google Scholar]
- 9.Campbell JE, Kiraly MA, Atkinson DJ, D'Souza AM, Vranic M, Riddell MC. Regular exercise prevents the development of hyperglucocorticoidemia via adaptations in the brain and adrenal glands in male Zucker diabetic fatty rats. Am J Physiol Regul Integr Comp Physiol 299: R168–R176, 2010. [DOI] [PubMed] [Google Scholar]
- 10.Caprio M, Feve B, Claes A, Viengchareun S, Lombes M, Zennaro MC. Pivotal role of the mineralocorticoid receptor in corticosteroid-induced adipogenesis. FASEB J 21: 2185–2194, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Carmen GY, Victor SM. Signalling mechanisms regulating lipolysis. Cell Signal 18: 401–408, 2006. [DOI] [PubMed] [Google Scholar]
- 12.Chatzopoulou A, Roy U, Meijer AH, Alia A, Spaink HP, Schaaf MJ. Transcriptional and metabolic effects of glucocorticoid receptor alpha and beta signaling in zebrafish. Endocrinology 156: 1757–1769, 2015. [DOI] [PubMed] [Google Scholar]
- 13.Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, Griffin PR, Spiegelman BM. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 466: 451–456, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coutinho AE, Campbell JE, Fediuc S, Riddell MC. Effect of voluntary exercise on peripheral tissue glucocorticoid receptor content and the expression and activity of 11β-HSD1 in the Syrian hamster. J Appl Physiol 100: 1483–1488, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Dey A, Hao S, Erion JR, Wosiski-Kuhn M, Stranahan AM. Glucocorticoid sensitization of microglia in a genetic mouse model of obesity and diabetes. J Neuroimmunol 269: 20–27, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Divertie GD, Jensen MD, Miles JM. Stimulation of lipolysis in humans by physiological hypercortisolemia. Diabetes 40: 1228–1232, 1991. [DOI] [PubMed] [Google Scholar]
- 17.Droste SK, Gesing A, Ulbricht S, Muller MB, Linthorst AC, Reul JM. Effects of long-term voluntary exercise on the mouse hypothalamic-pituitary-adrenocortical axis. Endocrinology 144: 3012–3023, 2003. [DOI] [PubMed] [Google Scholar]
- 18.DuBois DC, Sukumaran S, Jusko WJ, Almon RR. Evidence for a glucocorticoid receptor beta splice variant in the rat and its physiological regulation in liver. Steroids 78: 312–320, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goedecke JH, Chorell E, Livingstone DE, Stimson RH, Hayes P, Adams K, Dave JA, Victor H, Levitt NS, Kahn SE, Seckl JR, Walker BR, Olsson T. Glucocorticoid receptor gene expression in adipose tissue and associated metabolic risk in black and white South African women. Int J Obes 39: 303–311, 2015. [DOI] [PubMed] [Google Scholar]
- 20.Goto T, Lee JY, Teraminami A, Kim YI, Hirai S, Uemura T, Inoue H, Takahashi N, Kawada T. Activation of peroxisome proliferator-activated receptor-alpha stimulates both differentiation and fatty acid oxidation in adipocytes. J Lipid Res 52: 873–884, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grankvist N, Amable L, Honkanen RE, Sjoholm A, Ortsater H. Serine/threonine protein phosphatase 5 regulates glucose homeostasis in vivo and apoptosis signalling in mouse pancreatic islets and clonal MIN6 cells. Diabetologia 55: 2005–2015, 2012. [DOI] [PubMed] [Google Scholar]
- 22.Grankvist N, Honkanen RE, Sjoholm A, Ortsater H. Genetic disruption of protein phosphatase 5 in mice prevents high-fat diet feeding-induced weight gain. FEBS Lett 587: 3869–3874, 2013. [DOI] [PubMed] [Google Scholar]
- 23.Gray NE, Lam LN, Yang K, Zhou AY, Koliwad S, Wang JC. Angiopoietin-like 4 (Angptl4) protein is a physiological mediator of intracellular lipolysis in murine adipocytes. J Biol Chem 287: 8444–8456, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansen D, Meeusen R, Mullens A, Dendale P. Effect of acute endurance and resistance exercise on endocrine hormones directly related to lipolysis and skeletal muscle protein synthesis in adult individuals with obesity. Sports Med 42: 415–431, 2012. [DOI] [PubMed] [Google Scholar]
- 25.Hinds TD Jr, Ramakrishnan S, Cash HA, Stechschulte LA, Heinrich G, Najjar SM, Sanchez ER. Discovery of glucocorticoid receptor-beta in mice with a role in metabolism. Mol Endocrinol 24: 1715–1727, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinds TD Jr, Sánchez ER. Protein phosphatase 5. Int J Biochem Cell Biol 40: 2358–2362, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hinds TD Jr, Sodhi K, Meadows C, Fedorova L, Puri N, Kim DH, Peterson SJ, Shapiro J, Abraham NG, Kappas A. Increased HO-1 levels ameliorate fatty liver development through a reduction of heme and recruitment of FGF21. Obesity (Silver Spring) 22: 705–712, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Hinds TD Jr, Stechschulte LA, Cash HA, Whisler D, Banerjee A, Yong W, Khuder SS, Kaw MK, Shou W, Najjar SM, Sanchez ER. Protein phosphatase 5 mediates lipid metabolism through reciprocal control of glucocorticoid receptor and peroxisome proliferator-activated receptor-gamma (PPARgamma). J Biol Chem 286: 42911–42922, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinds TD, Stechschulte LA, Elkhairi F, Sanchez ER. Analysis of FK506, timcodar (VX-853) and FKBP51 and FKBP52 chaperones in control of glucocorticoid receptor activity and phosphorylation. Pharmacol Res Perspect 2: e00076, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoppmann J, Perwitz N, Meier B, Fasshauer M, Hadaschik D, Lehnert H, Klein J. The balance between gluco- and mineralo-corticoid action critically determines inflammatory adipocyte responses. J Endocrinol 204: 153–164, 2010. [DOI] [PubMed] [Google Scholar]
- 31.Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science 274: 2100–2103, 1996. [DOI] [PubMed] [Google Scholar]
- 32.Huizenga NA, de Lange P, Koper JW, de Herder WW, Abs R, Kasteren JH, de Jong FH, Lamberts SW. Five patients with biochemical and/or clinical generalized glucocorticoid resistance without alterations in the glucocorticoid receptor gene. J Clin Endocrinol Metab 85: 2076–2081, 2000. [DOI] [PubMed] [Google Scholar]
- 33.Jacob W, Rosenzweig D, Vázquez-Martin C, Duce SL, Cohen PT. Decreased adipogenesis and adipose tissue in mice with inactivated protein phosphatase 5. Biochem J 466: 163–176, 2015. [DOI] [PubMed] [Google Scholar]
- 34.Jenson M, Kilroy G, York DA, Braymer D. Abnormal regulation of hepatic glucocorticoid receptor mRNA and receptor protein distribution in the obese Zucker rat. Obes Res 4: 133–143, 1996. [DOI] [PubMed] [Google Scholar]
- 35.Johannsson G, Ragnarsson O. Cardiovascular and metabolic impact of glucocorticoid replacement therapy. Front Horm Res 43: 33–44, 2014. [DOI] [PubMed] [Google Scholar]
- 36.Kino T, Manoli I, Kelkar S, Wang Y, Su YA, Chrousos GP. Glucocorticoid receptor (GR) beta has intrinsic, GRalpha-independent transcriptional activity. Biochem Biophys Res Commun 381: 671–675, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell 83: 813–819, 1995. [DOI] [PubMed] [Google Scholar]
- 38.Krintel C, Morgelin M, Logan DT, Holm C. Phosphorylation of hormone-sensitive lipase by protein kinase A in vitro promotes an increase in its hydrophobic surface area. FEBS J 276: 4752–4762, 2009. [DOI] [PubMed] [Google Scholar]
- 39.Kubota N, Terauchi Y, Miki H, Tamemoto H, Yamauchi T, Komeda K, Satoh S, Nakano R, Ishii C, Sugiyama T, Eto K, Tsubamoto Y, Okuno A, Murakami K, Sekihara H, Hasegawa G, Naito M, Toyoshima Y, Tanaka S, Shiota K, Kitamura T, Fujita T, Ezaki O, Aizawa S, Kadowaki T, et al. PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance Mol Cell 4: 597–609, 1999. [DOI] [PubMed] [Google Scholar]
- 40.Lan NC, Graham B, Bartter FC, Baxter JD. Binding of steroids to mineralocorticoid receptors: implications for in vivo occupancy by glucocorticoids. J Clin Endocrinol Metab 54: 332–342, 1982. [DOI] [PubMed] [Google Scholar]
- 41.Lefterova MI, Haakonsson AK, Lazar MA, Mandrup S. PPARgamma and the global map of adipogenesis and beyond. Trends Endocrinol Metab 25: 293–302, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luger A, Deuster PA, Kyle SB, Gallucci WT, Montgomery LC, Gold PW, Loriaux DL, Chrousos GP. Acute hypothalamic-pituitary-adrenal responses to the stress of treadmill exercise. Physiologic adaptations to physical training. N Engl J Med 316: 1309–1315, 1987. [DOI] [PubMed] [Google Scholar]
- 43.Marchesini G, Brizi M, Morselli-Labate AM, Bianchi G, Bugianesi E, McCullough AJ, Forlani G, Melchionda N. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med 107: 450–455, 1999. [DOI] [PubMed] [Google Scholar]
- 44.Marzolla V, Armani A, Feraco A, De Martino MU, Fabbri A, Rosano G, Caprio M. Mineralocorticoid receptor in adipocytes and macrophages: a promising target to fight metabolic syndrome. Steroids 91: 46–53, 2014. [DOI] [PubMed] [Google Scholar]
- 45.Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, Flier JS. A transgenic model of visceral obesity and the metabolic syndrome. Science 294: 2166–2170, 2001. [DOI] [PubMed] [Google Scholar]
- 46.Mueller KM, Kornfeld JW, Friedbichler K, Blaas L, Egger G, Esterbauer H, Hasselblatt P, Schlederer M, Haindl S, Wagner KU, Engblom D, Haemmerle G, Kratky D, Sexl V, Kenner L, Kozlov AV, Terracciano L, Zechner R, Schuetz G, Casanova E, Pospisilik JA, Heim MH, Moriggl R. Impairment of hepatic growth hormone and glucocorticoid receptor signaling causes steatosis and hepatocellular carcinoma in mice. Hepatology 54: 1398–1409, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oakley RH, Cidlowski JA. Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem 286: 3177–3184, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Owen OE, Cahill GF Jr. Metabolic effects of exogenous glucocorticoids in fasted man. J Clin Invest 52: 2596–2605, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Penfornis P, Viengchareun S, Le Menuet D, Cluzeaud F, Zennaro MC, Lombès M. The mineralocorticoid receptor mediates aldosterone-induced differentiation of T37i cells into brown adipocytes. Am J Physiol Endocrinol Metab 279: E386–E394, 2000. [DOI] [PubMed] [Google Scholar]
- 50.Reddy JK, Rao MS. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am J Physiol Gastrointest Liver Physiol 290: G852–G858, 2006. [DOI] [PubMed] [Google Scholar]
- 51.Reul JM, de Kloet ER, van Sluijs FJ, Rijnberk A, Rothuizen J. Binding characteristics of mineralocorticoid and glucocorticoid receptors in dog brain and pituitary. Endocrinology 127: 907–915, 1990. [DOI] [PubMed] [Google Scholar]
- 52.Reyer H, Ponsuksili S, Wimmers K, Murani E. Transcript variants of the porcine glucocorticoid receptor gene (NR3C1). Gen Comp Endocrinol 189: 127–133, 2013. [DOI] [PubMed] [Google Scholar]
- 53.Rockall AG, Sohaib SA, Evans D, Kaltsas G, Isidori AM, Monson JP, Besser GM, Grossman AB, Reznek RH. Hepatic steatosis in Cushing's syndrome: a radiological assessment using computed tomography. Eur J Endocrinol 149: 543–548, 2003. [DOI] [PubMed] [Google Scholar]
- 54.Rogerson FM, Dimopoulos N, Sluka P, Chu S, Curtis AJ, Fuller PJ. Structural determinants of aldosterone binding selectivity in the mineralocorticoid receptor. J Biol Chem 274: 36305–36311, 1999. [DOI] [PubMed] [Google Scholar]
- 55.Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol 7: 885–896, 2006. [DOI] [PubMed] [Google Scholar]
- 56.Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell 4: 611–617, 1999. [DOI] [PubMed] [Google Scholar]
- 57.Roth J, Qiang X, Marbán SL, Redelt H, Lowell BC. The obesity pandemic: where have we been and where are we going? Obes Res 12, Suppl 2: 88S–101S, 2004. [DOI] [PubMed] [Google Scholar]
- 58.Shimizu N, Maruyama T, Yoshikawa N, Matsumiya R, Ma Y, Ito N, Tasaka Y, Kuribara-Souta A, Miyata K, Oike Y, Berger S, Schutz G, Takeda S, Tanaka H. A muscle-liver-fat signalling axis is essential for central control of adaptive adipose remodelling. Nat Commun 6: 6693, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Slavin BG, Ong JM, Kern PA. Hormonal regulation of hormone-sensitive lipase activity and mRNA levels in isolated rat adipocytes. J Lipid Res 35: 1535–1541, 1994. [PubMed] [Google Scholar]
- 60.Stechschulte LA, Hinds TD Jr, Ghanem SS, Shou W, Najjar SM, Sanchez ER. FKBP51 reciprocally regulates GRα and PPARγ activation via the Akt-p38 pathway. Mol Endocrinol 28: 1254–1264, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stechschulte LA, Hinds TD Jr, Khuder SS, Shou W, Najjar SM, Sanchez ER. FKBP51 controls cellular adipogenesis through p38 kinase-mediated phosphorylation of GRα and PPARγ. Mol Endocrinol 28: 1265–1275, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stechschulte LA, Wuescher L, Marino JS, Hill JW, Eng C, Hinds TD Jr. Glucocorticoid receptor β stimulates Akt1 growth pathway by attenuation of PTEN. J Biol Chem 289: 17885–17894, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW 2nd, DeFuria J, Jick Z, Greenberg AS, Obin MS. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 56: 2910–2918, 2007. [DOI] [PubMed] [Google Scholar]
- 64.Sutanto W, de Kloet ER. Mineralocorticoid receptor ligands: biochemical, pharmacological, and clinical aspects. Med Res Rev 11: 617–639, 1991. [DOI] [PubMed] [Google Scholar]
- 65.Syed AA, Irving JA, Redfern CP, Hall AG, Unwin NC, White M, Bhopal RS, Weaver JU. Association of glucocorticoid receptor polymorphism A3669G in exon 9beta with reduced central adiposity in women. Obesity 14: 759–764, 2006. [DOI] [PubMed] [Google Scholar]
- 66.Villacorta L, Schopfer FJ, Zhang J, Freeman BA, Chen YE. PPARgamma and its ligands: therapeutic implications in cardiovascular disease. Clin Sci 116: 205–218, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang F, Mullican SE, DiSpirito JR, Peed LC, Lazar MA. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARgamma. Proc Natl Acad Sci USA 110: 18656–18661, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Warrier M, Hinds TD Jr, Ledford KJ, Cash HA, Patel PR, Bowman TA, Stechschulte LA, Yong W, Shou W, Najjar SM, Sanchez ER. Susceptibility to diet-induced hepatic steatosis and glucocorticoid resistance in FK506-binding protein 52-deficient mice. Endocrinology 151: 3225–3236, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu C, Macleod I, Su AI. BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res 41: D561–D565, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, Hodge CL, Haase J, Janes J, Huss JW 3rd, Su AI. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol 10: R130, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xiao X, Li H, Yang J, Qi X, Zu X, Yang J, Zhong J, Cao R, Liu J, Wen G. Wnt/beta-catenin signaling pathway and lipolysis enzymes participate in methylprednisolone induced fat differential distribution between subcutaneous and visceral adipose tissue. Steroids 84: 30–35, 2014. [DOI] [PubMed] [Google Scholar]
- 72.Xu C, He J, Jiang H, Zu L, Zhai W, Pu S, Xu G. Direct effect of glucocorticoids on lipolysis in adipocytes. Mol Endocrinol 23: 1161–1170, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zennaro MC, Le Menuet D, Viengchareun S, Walker F, Ricquier D, Lombes M. Hibernoma development in transgenic mice identifies brown adipose tissue as a novel target of aldosterone action. J Clin Invest 101: 1254–1260, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]