

Abstract
Ischemic injury induces breakdown of the intestinal barrier. Recent studies in porcine postischemic tissues indicate that inhibition of NHE2 results in enhanced recovery of barrier function in vitro via a process involving interepithelial tight junctions. To further study this process, recovery of barrier function was assessed in wild-type (NHE2+/+) and NHE2−/− mice in vivo and wild-type mice in vitro. Mice were subjected to complete mesenteric ischemia in vivo, after which barrier function was measured by blood-to-lumen mannitol clearance over a 3-h recovery period or measurement of transepithelial electrical resistance (TER) in Ussing chambers immediately following ischemia. Tissues were assessed for expression of select junctional proteins. Compared with NHE2+/+ mice, NHE2−/− mice had greater intestinal permeability during the postischemic recovery process. In contrast to prior porcine studies, pharmacological inhibition of NHE2 in postischemic tissues from wild-type mice also resulted in significant reductions in TER. Mucosa from NHE2−/− mice displayed a shift of occludin and claudin-1 expression to the Triton-X-soluble membrane fractions and showed disruption of occludin and claudin-1 localization patterns following injury. This was qualitatively and quantitatively recovered in NHE2+/+ mice compared with NHE2−/− mice by the end of the 3-h recovery period. Serine phosphorylation of occludin and claudin-1 was downregulated in NHE2−/− postischemia compared with wild-type mice. These data indicate an important role for NHE2 in recovery of barrier function in mice via a mechanism involving tight junctions.
Keywords: tight junction, intestinal permeability, ischemia, Na+/H+ exchange
the gastrointestinal tract is lined by a single layer of epithelial cells connected by a series of interepithelial junctions. Collectively, the epithelial monolayer can selectively transport solutes but also prevent the entrance of noxious luminal contents. The latter function is termed gut barrier function. The barrier properties of the epithelium are regulated by the apical tight junctions (TJs), which consist of an array of membrane-spanning proteins (e.g., occludin and claudins) linked by cytoplasmic plaque proteins such as zonula occludens-1 to the cytoskeleton (25, 28). Recent studies have shown that TJs are not static structures but can be regulated to alter solute absorption and barrier properties (26, 28, 30). For example, NHE3 appears to be involved in a signaling cascade that results in decreased transepithelial resistance during Na+-glucose cotransport as a result of TJ alteration (27).
Intestinal ischemic injury is a leading cause of death in critically ill patients because of the onset of sepsis as the damaged intestinal barrier becomes permeable to bacterial toxins (8, 23). Injured mucosa is initially repaired by restitution, during which viable epithelial cells adjacent to the wound migrate to cover denuded basement membrane (17). However, loss of TJ architecture must also be addressed to fully restore barrier function (4, 5, 11). Although epithelial restitution results in recovery of a continuous epithelial monolayer spanning the wound, barrier function is predominantly restored by restoration of TJs in postischemic studies (15, 16). This was associated with recruitment of junctional proteins such as occludin to the apical intercellular space. Pharmacological approaches indicated that a large component of the recovery of intestinal barrier function following ischemic injury was attributable to NHE function. In particular, NHE2, rather than NHE3, played a major role in recovery of barrier function (16). In these porcine experiments, pharmacological NHE2, but not NHE3, enhanced recovered of barrier function as measured in ischemic-injured tissues within Ussing chambers (16).
Currently, the exact role of NHE2 in gastrointestinal function remains obscure. Most of the electroneutral Na+ absorption that occurs between meals has been shown to be mediated by NHE3 whereas NHE2 is thought to have a minor contribution (9). Investigators have demonstrated that NHE3−/− mice develop diarrhea and electrolyte abnormalities, indicative of the importance of this exchanger in intestinal absorption. Alternatively, NHE2−/− mice do not exhibit any noticeable clinical or electrophysiological abnormalities (9, 22). In double knockout experiments, mice with targeted deletion of both NHE2 and NHE3 did not exhibit differences in Na+ malabsorption or severity of diarrheal disease compared with NHE3−/− mice, suggesting that NHE2 does not have a compensatory role in these mice (14). However, there is increasing evidence that NHE2 may play an important role in the large intestine. In addition, there are considerable species differences with regard to the contribution of NHE2 to electroneutral Na+/H+ exchange (2, 12). The objective of the present experiments was to investigate the role of NHE2 in mucosal repair in ischemic intestine utilizing NHE2 knockout mice.
METHODS
Experimental animals.
All studies were approved by the North Carolina State University Institutional Animal Care and Use Committee. NHE2 null (NHE2−/−), heterozygous (NHE2+/−), and wild-type (NHE2+/+) mice 2–4 mo of age were used for this study. All mice were maintained on a standard laboratory diet. The progeny mice were genotyped by PCR using primers specific for amplification of either intact or disrupted murine NHE2 alleles, as described previously (14). For amplification of wild-type and disrupted alleles, two discriminating forward primers (wild-type NHE2; 5′- CATCTCTATCACAAGTTGCCCACAATCGTG-3′, disrupted NHE2; 5′- GACAATAGCAGCCATGCTGG-3′) and a conversed reverse primer (5′-GTGACTTCGTTGAGCAGAGACTCG-3′) were used simultaneously in PCR to yield a wild-type NHE2 product of 450 bp and disrupted NHE2 product of 220 bp. PCR was carried out using a Bio-Rad iCycler in a 50-μl reaction volume of PCR buffer II (Perkin-Elmer; Foster City, CA) containing 2.5 mM MgCl2, 2.5 U AmpliTaq Gold DNA polymerase, 100 pmol of each forward primer, 200 pmol reverse primer, 20 μM each deoxynucleotide diphosphate, and 5 μl DNA template. PCR was performed at the following temperature profiles: initial denaturation at 94°C for 5 min; denaturation at 94°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s. Total number of cycles was 40, followed by a final extension for 5 min at 72°C. PCR products were separated by agarose gal electrophoresis and visualized under UV light after staining with ethidium bromide.
In vivo intestinal ischemia.
Mice had unrestricted access to water but not feed 6 h prior to the experimental surgery. The mice were subjected to general anesthesia with isoflurane (4% for induction and 1.5% for maintenance) and were prepared for surgery by aseptic technique. The midjejunum was approached via a midline laparotomy incision. Intestinal loops 2 cm in length were delineated with Doyen intestinal forceps, and complete ischemia was induced by clamping the mesenteric blood supply with Johns Hopkins Bulldog clamps. Following 45 min of ischemia, clamps were removed and the abdomen was closed with 4-0 silk for the midline and staples for the skin. Toward the end of the surgery, mice were administered 0.01 mg/kg buprenorphine SQ to provide preemptive analgesia. The mice were allowed to recover for 3 h in heated cages.
Blood-to-lumen [3H]mannitol clearance.
Following a 1.5- or 3-h recovery period, mice were reanesthetized by the same technique as before, after which the abdomen was opened and ischemic and control intestinal segments were ligated with 4-0 silk. Control and ischemic loops were injected with 0.25 ml of buffered saline (pH 7.4). Subsequently, 0.2 ml of buffered saline containing [3H]mannitol (10 μCi/ml) was injected into the tail vein. Time zero blood samples (0.3 ml) were collected via retroorbital bleeding into heparinized vials. Throughout the experimental procedures body temperature was maintained by retaining abdominal contents within the body wall and by using water-circulated heating pads and solar blankets. The body temperature was monitored and maintained at 36.5–37.5°C. Mice were euthanized, intestinal segments were weighed, and intestinal perfusates from each loop were emptied into microcentrifuge tubes. The volume of intestinal perfusate was recorded and the perfusates were saved in scintillation fluid. The blood samples were centrifuged at 14,000 rpm for 4 min and 25 μl of plasma was saved in scintillation fluid. The plasma and intestinal perfusate samples were assessed on a scintillation beta counter to measure 3H activity, and the [3H]mannitol clearance was calculated as the percentage recovery of 3H in the lumen relative to the initial plasma 3H activity.
Ussing chamber experiments.
Intestinal ischemia was induced as described above, after which ileal tissues were harvested immediately, cut longitudinally along the antimesenteric surface as for prior porcine experiments (16), and placed on 0.12-cm2-aperture Ussing chambers. Tissues were bathed on the serosal and mucosal sides with Ringer solution. The serosal bathing solution contained 10 mM glucose, which was osmotically balanced on the mucosal side with 10 mM mannitol. Bathing solutions were oxygenated (95% O2-5% CO2) and circulated in water-jacketed reservoirs maintained at 37°C. The spontaneous potential difference (PD) was measured using Ringer-agar bridges connected to calomel electrodes, and the PD was short circuited through Ag-AgCl electrodes using a voltage clamp that corrected for fluid resistance. Transepithelial electrical resistance (TER; Ω·cm2) was calculated from the spontaneous PD and short-circuit current. The NHE2 inhibitor HOE694 (kindly donated by Aventis Pharma, Frankfurt, Germany) was applied to the mucosal surface of select tissues at a final concentration of 25 μM.
Histological examination.
Tissues from control and ischemic-injured intestinal loops were collected in 10% neutral buffered formalin for histological evaluation. Tissues were sectioned (5 μm) and stained with hematoxylin and eosin. For each tissue, three sections were evaluated. Three well-oriented villi and crypts were identified in each of three serial sections. Villus length, villus width, and crypt depth were obtained by using a micrometer in the eyepiece of a light microscope. The surface area of the villus was calculated by using the formula for the surface area of a cylinder. The formula was modified by subtracting the area of the base of the villus and multiplying by a factor accounting for the variable position at which each villus was cross sectioned (1). The percentage of the villous surface area that remained denuded was calculated and the percent denuded villous surface area was used as an index of epithelial restitution.
Cell fraction analyses.
Intestinal mucosal scrapings from control and ischemic-injured mucosa were snap frozen and stored at −70°C before SDS-PAGE. Triton X-soluble and -insoluble fractions were extracted by using respective buffers (Triton X-soluble extraction buffer: 0.5% Triton X-100, 50 mM Tris·HCl, 140 mM EGTA, 30 mM sodium pyrophosphate, 50 mM sodium fluoride, 100 μM sodium orthovanadate, Complete Protease Inhibitor cocktail tablet from Roche; Triton X-insoluble extraction buffer: 1% SDS, 50 mM Tris·HCl, 140 mM EGTA, 30 mM sodium pyrophosphate, 50 mM sodium fluoride, 100 μM sodium orthovanadate, Complete Protease Inhibitor cocktail tablet from Roche). Protein analysis of extract aliquots was performed (DC protein assay; Bio-Rad, Hercules, CA). Tissue extracts (amounts equalized by protein concentration) were mixed with an equal volume of 2× SDS-PAGE sample buffer and boiled for 4 min. Lysates were loaded on a 10% SDS polyacrylamide gel, and electrophoresis was carried out according to standard protocols. Proteins were transferred to a polyvinylidene difluoride membrane (Immobilon, Millipore, Billerica, MA) by using an electroblotting minitransfer apparatus. Membranes were blocked at room temperature for 60 min in Tris-buffered saline plus 0.05% Tween 20 (TBST) and 5% dry powdered milk and then incubated for 60 min in primary antibody (rabbit polyclonal occludin, claudin-1; Zymed, San Francisco, CA). After washings in TBST, membranes were incubated with horseradish peroxidase-conjugated secondary antibody and developed for visualization of protein with chemiluminescence enhancer solution (Pierce, Rockford, IL).
Immunolocalization of TJ proteins.
Intestinal tissues were embedded in OCT media (Tissue Tek, Sakura, Torrance, CA), frozen, sectioned at 5 μm, and stored at −80°C. Sectioned tissues were fixed in cold acetone, and blocked with normal goat serum. The sections were incubated with rabbit polyclonal occludin, or claudin-1 (Zymed, San Francisco, CA) antibodies diluted in 5% normal goat serum for 90 min. Following washes in PBS, the sections were incubated for 60 min with goat anti-rabbit IgG conjugated with FITC or Cy3 (Zymed) diluted in 5% normal goat serum. The slides were washed in PBS, mounted in media with DAPI (Vectashield, Vector Laboratories, CA), and examined with a microscope capable of detecting epifluorescence. Duplicate sections stained similarly but with omission of primary antibodies served as controls.
Tissue immunoprecipitation experiments.
Tissue aliquots were thawed at 4°C and added to 3 ml of chilled lysis buffer, including protease inhibitors (0.5 mM Pefabloc, 0.1 mM 4-nitrophenyl phosphate, 0.04 mM β-glycerophosphate, 0.1 mM Na3VO4, 40 μg/ml bestatin, 2 μg/ml aprotinin, 0.54 μg/ml leupeptin, and 0.7 μg/ml pepstatin A) at 4°C. This mixture was homogenized on ice and then centrifuged at 4°C, and the supernatant was saved. Extracted proteins (1 mg) were solubilized in RIPA buffer [50 mM Tris·HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.1% (wt/vol) SDS, 1% Triton X-100 with protease inhibitors], precleared with protein A/G-agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) and incubated with either NHE2 or NHE3 anti-goat polyclonal antibodies or normal goat preimmune serum (Santa Cruz Biotechnology) for 1 h at 37°C. The antibody-protein complexes were adsorbed from solution with protein A/G-agarose beads. Protein samples were washed three times in ice cold RIPA buffer and centrifuged at 2,500 rpm for 2 min at 4°C. Protein pellets were solubilized in 2× SDS-PAGE sample buffer, and electrophoresis and transfer to membrane were carried out according to standard procedures described previously. The blots were probed for respective proteins, then treated with stripping buffer, washed in TBS, and probed with anti-phosphoserine antibodies (Chemicon, Billerica, MA). In further coimmunoprecipitation experiments, immunoprecipitated NHE2 or NHE3 was subjected to SDS-PAGE and probed by use of a rabbit polyclonal occludin or claudin-1 (Zymed, San Francisco, CA) antibody. Immunoprecipitates obtained by using normal preimmune serum served as negative control. The experiments were repeated more than three times and the blot images were analyzed by densitometry using SigmaScan Pro 5.0 (Sigmastat, Systat Software, San Jose, CA).
Statistics.
Data were reported as means ± SE. All data were analyzed by using an ANOVA for repeated measures or a standard one-way ANOVA (Sigmastat). A Tukey's test was used to determine differences between treatments following ANOVA (P < 0.05).
RESULTS
Recovery of mucosal barrier function is impaired in NHE2−/− mice.
Mucosal clearances of [3H]mannitol were greater in tissues subjected to ischemia compared with control tissues across all time periods regardless of genotype (P < 0.05). However, there was evidence of mucosal recovery following ischemic injury, based on progressive reductions in blood-to-lumen mannitol clearance (Fig. 1). Nonetheless, clearances from ischemic-injured tissues from NHE2−/− mice were significantly greater at the 1.5-h and 3-h time periods compared with injured ileum from wild-type mice. Similar permeability values for control and ischemic ileal loops were observed in NHE2 heterozygote (NHE2+/−) ileum (data not shown). There were no apparent histological differences when examining intestinal tissues from wild-type and NHE2−/− mice. In both genotypes, 45 min of intestinal ischemia resulted in sloughing of apical villous enterocytes and contraction of villi (Fig. 2). After 1.5-h of postischemic recovery, the villi were contracted and the epithelium was near-fully restituted in wild-type and NHE2−/− mice. There was no evidence of ischemia-reperfusion injury. Following a 3-h recovery period, the epithelium was completely restituted in wild-type and NHE2−/− mice. The villus height, villus width, and crypt depth measurements were comparable in control and ischemic-injured tissues from wild-type and NHE2−/− mice (Table 1). Our histological findings led us to conclude that differences in mucosal permeability between genotypes of mice were independent of epithelial restitution and more likely attributable to changes in paracellular permeability.
Fig. 1.
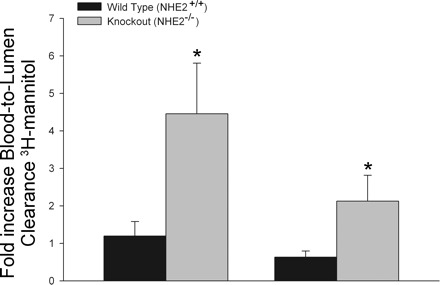
In vivo [3H]mannitol permeability in ischemic murine ileum. Data are presented as the increase in blood-lumen [3H]mannitol clearance relative to control (nonischemic) intestinal segments. Ischemia induced elevations in [3H]mannitol clearance in both NHE+/+ and NHE2−/− measured at 1.5 and 3 h postischemia. However, in NHE2−/− mice, the ischemic loops had greater [3H]mannitol clearance compared with NHE2+/+ mice (*P < 0.05).
Fig. 2.
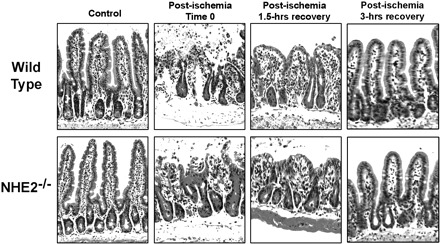
Histological examination of jejunal tissues from wild-type and NHE2−/− mice subjected to ischemia and recovery. Ischemia results in epithelial sloughing at the tips of villi and contraction of villi. After 1.5 and 3 h of recovery, epithelial restitution in NHE2−/− mice was comparable to wild-type mice with gradual increases in villus height. Hematoxylin and eosin stain, ×100.
Table 1.
Morphometric assessment of ischemic jejunal epithelium of wild type and NHE2−/− mice
| Genotype | Villus Height, μm |
Villus Width, μm |
Crypt Depth, μm |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Control | Ischemic | Control | Ischemic | Control | Ischemic | ||||
| NHE2+/+ | 162.9±1.6 | 122.1±1.9 | 40.83±2.3 | 44.72±2.7 | 32.4±7.3 | 28.4±2.7 | |||
| NHE2−/− | 172.2±5.4 | 131.8±5.9 | 40.31±1.1 | 41.8±0.4 | 34.7±4.2 | 29.1±3.9 | |||
Values are means ± SE; n = 6. Uninjured and ischemic intestinal segments from NHE2+/+ and NHE2-deficient mice were fixed in 10% buffered formalin and processed for histological examination according to standard protocol. There was no significant difference in villus height, villus width, and crypt depth measurements of ischemic loops of wild-type and NHE2−/− mice.
Pharmacological blockade of NHE2 is deleterious to mucosal barrier integrity in ischemic murine jejunum.
Previous experiments using porcine ileum subjected to 45 min of complete ischemia and mounted on Ussing chambers demonstrated that blockade of NHE2 with the selective NHE2 inhibitor HOE694 stimulated elevations in TER. This is in contrast to the in vivo findings in the present study in which NHE2−/− mice had reduced recovery of mucosal barrier function following ischemic injury. To better understand the discrepancies between previous pharmacological studies in porcine ileum and that of the present experiments, we conducted Ussing chamber experiments with ischemic-injured murine ileum. Blockade of NHE2 with mucosal application of HOE694 resulted in reductions in TER in ischemic-injured tissues compared with nontreated ischemic-injured tissues (Fig. 3). However, TER continued to decline during the experimental period, whereas in the pig tissues had increasing levels of TER in the Ussing chamber. This data suggests that species differences may exist with regard to the role of NHE2 in recovery of the mucosal barrier.
Fig. 3.
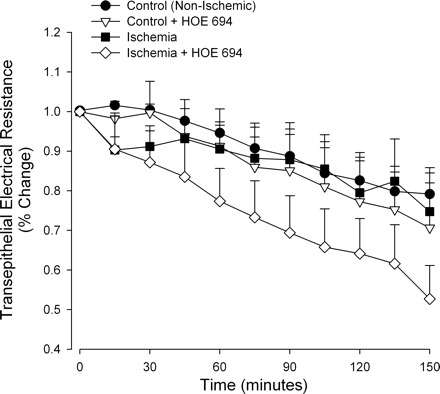
Percent change in transepithelial electrical resistance (TER) in murine ileum mounted on Ussing chambers. Data are presented as the % change in TER relative to initial starting TER values. Ileal tissues were subjected to 45 min of intestinal ischemia in vivo and then mounted on Ussing chambers for monitoring of TER over a 150-min period. The selective NHE2 blocker HOE694 (25 μM) was applied to the mucosal side of select tissues. Ischemic tissues treated with HOE694 exhibited significant reduction in TER compared with other treatments (2-way ANOVA on repeated measures, P < 0.05).
TJ protein expression in tissue fractions from NHE2+/+ and NHE2−/− mice.
To determine whether impaired mucosal barrier recovery in NHE2−/− mice was associated with changes in TJ composition, we performed Western blot analyses for transmembrane TJ proteins occludin and claudin-1. Immunoblots revealed low expression of occludin and claudin-1 in Triton-X-soluble fractions of NHE2+/+ mice after 3 h of postischemic recovery, with expression of these proteins concentrated in the Triton-X-insoluble fraction (Fig. 4A). Alternatively, Triton-X-soluble fractions from NHE2−/− mice had increased expression of occludin and claudin-1, whereas the expression of these proteins was reduced in Triton-X-insoluble fractions. Thus there was a shift of occludin and claudin-1 from the Triton-X-insoluble fractions toward the Triton-X-soluble fraction in NHE2−/− mice compared with NHE2+/+ mice. The total expression of occludin and claudin-1 was comparable in control and postischemic tissues following a 3-h recovery period in wild-type and NHE2−/− mice, as revealed by Western analyses of respective whole tissue lysates, and verified with the loading control β-actin (Fig. 4B).
Fig. 4.
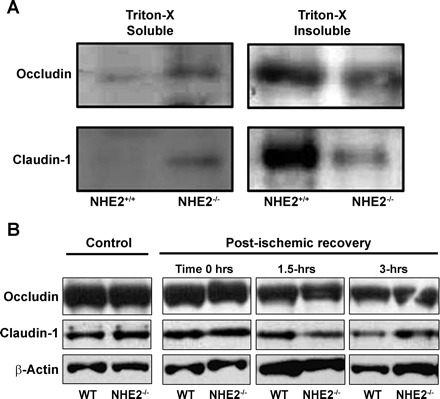
Western blot analysis of cell fractions for tight junction proteins. A: following the postischemic recovery period of 3 h, protein extracts from NHE2+/+ and NHE2−/− mice were homogenized in a buffer containing Triton X-100 (Triton X-100 soluble) or a more stringent buffer (Triton X-100 insoluble, see methods). Supernatants were subsequently analyzed by using anti-occludin and anti-claudin-1 antibodies. There was increased expression of both occludin and claudin-1 in the Triton X-100-soluble fraction in NHE2-deficient mice following ischemia, suggesting movement of tight junction proteins to the cytosol. B: whole tissue lysates from control and ischemic tissues of wild-type (WT) and NHE2−/− mice revealed comparable expression of occludin and claudin-1.
TJ structure is disrupted in NHE2−/− mice.
We further investigated TJ structure by performing immunofluorescence (IF) labeling for occludin and claudin-1 in wild-type and NHE2−/− ischemic mucosa. IF analyses revealed uniform, punctate fluorescence for occludin that was confined to the region of the apical TJ in NHE2+/+ recovering ischemic-injured mucosa. Likewise for claudin-1, the normal chicken wire pattern was seen in cross sections from NHE2+/+ ileal tissues (Fig. 5). In contrast, IF analyses for occludin and claudin-1 from ischemic tissues from NHE2−/− mice showed a disrupted and diffuse staining pattern.
Fig. 5.
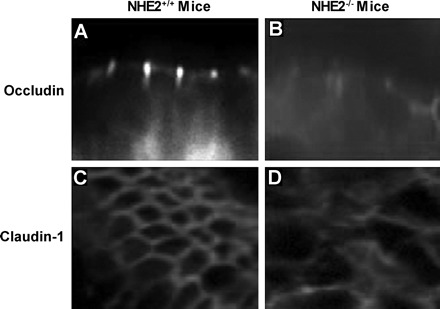
Localization of the tight junction proteins occludin and claudin-1 in control and ischemic-injured jejunal mucosa of wild-type and NHE2−/− mice. After 3-h postischemic recovery intestinal samples were collected and processed for immunofluorescence by use of rabbit polyclonal occludin and claudin-1 antibodies, followed by goat anti-rabbit IgG conjugated with FITC and Cy3, respectively. A: punctate localization of occludin to the region of the tight junction was noted in NHE2+/+ mice compared with diffuse localization of occludin in NHE2−/− mice (B). C: an en face view of the immunofluorescence pattern of claudin-1, showing a “chicken-wire” configuration in NHE2+/+ mice, compared with diffuse staining in NHE2−/− mice (D).
TJ proteins have reduced phosphorylation in NHE2−/− mice.
To further assess the role of NHE2-mediated barrier function recovery, we studied the phosphorylation status of TJ protein in mouse tissues. Western blot analyses of immunoprecipitated occludin and claudin-1 showed significantly increased phosphoserine expression of both junctional proteins in ischemic intestine of wild-type mice, but not in NHE−/− mice (Fig. 6). These data suggest that phosphorylation of NHE2 is an important component of the signaling mechanism that induces recovery of injured TJs.
Fig. 6.
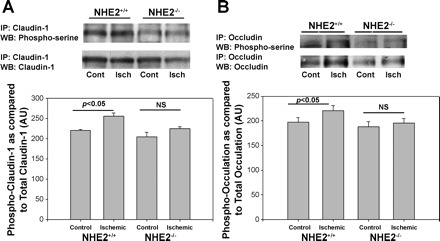
Phosphorylation of tight junction proteins during postischemic recovery. A: expression of phosphorylated claudin-1 immunoprecipitates (IP) appears greater in postischemic 3-h recovered tissues from NHE2+/+ mice compared with similar tissues from NHE2−/− mice on representative Western blots (WB). Bottom: densitometry analysis, which accounted for total claudin-1 expression against phosphorylated claudin-1 in individual lanes, revealed ∼15% increase in claudin-1 phosphoserine expression in ischemic intestine (Isch) over control intestine (Cont) in NHE2+/+ mice; n = 4, *P < 0.05; NS, not significant. B: expression of serine phosphorylated occludin appears greater in postischemic 3-h recovered tissues from NHE2+/+ mice compared with similar tissues from NHE2−/− mice on a representative Western blot. Bottom: densitometry analysis that also considered total occludin expression in individual lanes revealed ∼14% increase in occludin phosphoserine expression in ischemic intestine over control intestine in NHE2+/+ mice; n = 4, *P < 0.05. The expression of neither serine phosphorylated claudin-1 or occludin changed significantly between control and ischemic intestine in NHE2−/− mice.
DISCUSSION
The initial striking finding in the present studies is that there is no evidence of ischemia-reperfusion injury. This may relate to the degree to which the mesenteric arterial supply is occluded. For example, in studies on ischemia-reperfusion injury, rodents have typically been subjected to low-flow ischemia that lessens the loss of viable cells and primes oxidant enzyme systems such as xanthine oxidase. In the present series of experiments, the level of injury following 45 min was severe, and therefore any exacerbation during reperfusion may have been difficult to detect. This has been documented in prior studies (20). Instead, the tissue begins a rapid reparative phase as has been described for large animals such as the pig (5).
The present studies demonstrate an important role for NHE2 in intestinal injury and repair of mucosal barrier function in ischemic-injured mouse ileum. Histological examination of ischemic tissues and the ensuing recovery period revealed that epithelial restitution was complete by the 1.5-h postischemic period, indicating that increased [3H]mannitol permeability in NHE2−/− mice was attributable to increased paracellular permeability. The link between NHE2 and epithelial barrier function has been reported. However, in contrast to the present work, studies in the literature describe a beneficial role of NHE2 inhibition. For example, Nowak et al. (19) showed that pharmacological inhibition of NHE2 in Caco2 cell monolayers stimulated elevations in transepithelial resistance. Furthermore, the authors showed that NHE2 inhibition prevented PMA-induced disruption of TJ proteins and barrier properties suggested a protective role of NHE2. We have previously demonstrated in ischemic-injured porcine ileum that inhibition of NHE2 enhances recovery of barrier function. In contrast, pharmacological blockade of NHE2 in ischemic-injured murine ileum resulted in significant reductions in TER, suggesting that both pharmacological blockade of NHE2 and genetic deletion of NHE2 in knockout mice are detrimental to recovery of intestinal barrier function in injured mucosa. This suggests that NHE2 plays a different role in murine mucosa compared with porcine mucosa in response to barrier injury.
Impaired barrier recovery in ischemic NHE2−/− mouse ileum was associated with a marked disruption of occludin and claudin-1 localization and a shift in expression of these proteins to the cytosol, suggesting that loss of NHE2 impairs the ability of the TJ to repair. The signaling events associated with impaired TJ recovery in NHE2−/− mouse ileum are not known. However, it is known that TJs are regulated by phosphorylation events during repair or biogenesis (3, 21, 29). For example, recovery of barrier function and occludin localization to the TJs in Madin-Darby canine kidney cells coincided with the phosphorylation status of occludin (29). In the present study, occludin and claudin-1 phosphorylation was increased in recovering ischemic-injured ileum of wild-type mice but not NHE2−/− mouse ileum, thus suggesting that signaling events that result in phosphorylation of TJs are disrupted in the absence of NHE2. The mechanisms coordinating these events remain to be elucidated.
In the present study, coimmunoprecipitation experiments revealed that the TJ proteins occludin and claudin-1 were expressed in NHE2 and 3 immunoprecipitates, indicating their close proximity. Intracellular trafficking of NHE2 has not been studied in detail. However, compared with NHE3 that has greater membrane turnover (6, 32), NHE2 is considered to be a resident protein of the plasma membrane. For example, recent work has demonstrated rapid recruitment of NHE2 to the plasma membrane in response to lower cytosolic pH (10). Such recruitment to the membrane may provide an opportunity for NHE2-junctional protein complexes to form.
Several studies indicate that apical NHEs are regulated by phosphorylation, and this phosphorylation is in large part facilitated by accessory proteins such as EBP50 (31, 33). The linkage between EBP50 and NHE isoforms appears to occur via PDZ domains and allows access of kinases such as PKA to the cytosolic tail of NHE (32). Although advances have been made in understanding the regulation of NHE3 through interaction with Na+/H+ exchange regulatory factors such as EBP50 (24), which in turn links NHE3 to the cytoskeleton via ezrin (32), little is known about regulation of NHE2. Nonetheless, recent studies have shown that NHE2 is coexpressed with EBP50 in porcine small intestinal tissues (16). In addition, α-spectrin, a cytoskeletal protein, has been shown to interact with a COOH-terminal proline-rich area of NHE2 (7). In the present studies, we showed that EBP50 was expressed in both NHE2 and NHE3 immunoprecipitates, but there was greater expression of EBP50 in NHE2 immunoprecipitates during recovery from ischemia. Members of the NHERF family have been shown to be involved in cytoskeleton remodeling (13), but the mechanisms of NHE-EBP50 interactions and their potential role in TJ repair need to be investigated further.
Our results suggest a signal transduction pathway involving NHE2 and reorganization of junctional proteins following ischemic injury. PKA regulation of NHE2 involves interaction with its COOH terminus (18). This may in turn signal the recruitment of junctional proteins to the apical epithelial membrane. The strong role of PKA in phosphorylating NHE may explain in part the reparative effects of prostaglandins, which signal through the PKA pathway. Alternatively, the NHE2-deficient mice used in this study lack a complete COOH terminus, although a truncated NH2 terminus is produced (personal communication, L. Gawenis, Shull Laboratory, University of Cincinnati). Nonetheless, it is not known whether this truncated NH2 terminus is correctly targeted to the apical membrane and it would likely not be phosphorylated by PKA because of the missing COOH terminus. Overall, our results suggest that phosphorylation of the COOH terminus plays a role in facilitating junction reassembly. Since tissue permeability of NHE2−/− mice is equivalent to wild-type mice in the control state, it appears that this mechanism is only required for reassembly rather than maintenance of the epithelial barrier. In addition, this mechanism may be species specific, as highlighted by differences in the present study from prior porcine studies.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Argenzio RA, Lecce J, Powell DW. Prostanoids inhibit intestinal NaCl absorption in experimental porcine cryptosporidiosis. Gastroenterology 104: 440–447, 1993. [DOI] [PubMed] [Google Scholar]
- 2.Bachmann O, Riederer B, Rossmann H, Groos S, Schultheis PJ, Shull GE, Gregor M, Manns MP, Seidler U. The Na+/H+ exchanger isoform 2 is the predominant NHE isoform in murine colonic crypts and its lack causes NHE3 upregulation. Am J Physiol Gastrointest Liver Physiol 287: G125–G133, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Balda MS, Gonzalez-Mariscal L, Matter K, Cereijido M, Anderson JM. Assembly of the tight junction: the role of diacylglycerol. J Cell Biol 123: 293–302, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blikslager AT, Roberts MC, Argenzio RA. Prostaglandin-induced recovery of barrier function in porcine ileum is triggered by chloride secretion. Am J Physiol Gastrointest Liver Physiol 276: G28–G36, 1999. [DOI] [PubMed] [Google Scholar]
- 5.Blikslager AT, Roberts MC, Rhoads JM, Argenzio RA. Prostaglandins I2 and E2 have a synergistic role in rescuing epithelial barrier function in porcine ileum. J Clin Invest 100: 1928–1933, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cavet ME, Akhter S, Murtazina R, Sanchez de Medina F, Tse CM, Donowitz M. Half-lives of plasma membrane Na+/H+ exchangers NHE1–3: plasma membrane NHE2 has a rapid rate of degradation. Am J Physiol Cell Physiol 281: C2039–C2048, 2001. [DOI] [PubMed] [Google Scholar]
- 7.Chow CW. Regulation and intracellular localization of the epithelial isoforms of the Na+/H+ exchangers NHE2 and NHE3. Clin Invest Med 22: 195–206, 1999. [PubMed] [Google Scholar]
- 8.Deitch EA, Rutan R, Waymack JP. Trauma, shock, and gut translocation. New Horiz 4: 289–299, 1996. [PubMed] [Google Scholar]
- 9.Gawenis LR, Stien X, Shull GE, Schultheis PJ, Woo AL, Walker NM, Clarke LL. Intestinal NaCl transport in NHE2 and NHE3 knockout mice. Am J Physiol Gastrointest Liver Physiol 282: G776–G784, 2002. [DOI] [PubMed] [Google Scholar]
- 10.Gens JS, Du H, Tackett L, Kong SS, Chu S, Montrose MH. Different ionic conditions prompt NHE2 and NHE3 translocation to the plasma membrane. Biochim Biophys Acta 1768: 1023–1035, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gookin JL, Galanko JA, Blikslager AT, Argenzio RA. PG-mediated closure of paracellular pathway and not restitution is the primary determinant of barrier recovery in acutely injured porcine ileum. Am J Physiol Gastrointest Liver Physiol 285: G967–G979, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guan Y, Dong J, Tackett L, Meyer JW, Shull GE, Montrose MH. NHE2 is the main apical NHE in mouse colonic crypts but an alternative Na+-dependent acid extrusion mechanism is upregulated in NHE2-null mice. Am J Physiol Gastrointest Liver Physiol 291: G689–G699, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Lamprecht G, Weinman EJ, Yun CH. The role of NHERF and E3KARP in the cAMP-mediated inhibition of NHE3. J Biol Chem 273: 29972–29978, 1998. [DOI] [PubMed] [Google Scholar]
- 14.Ledoussal C, Woo AL, Miller ML, Shull GE. Loss of the NHE2 Na+/H+ exchanger has no apparent effect on diarrheal state of NHE3-deficient mice. Am J Physiol Gastrointest Liver Physiol 281: G1385–G1396, 2001. [DOI] [PubMed] [Google Scholar]
- 15.Little D, Dean RA, Young KM, McKane SA, Martin LD, Jones SL, Blikslager AT. PI3K signaling is required for prostaglandin-induced mucosal recovery in ischemia-injured porcine ileum. Am J Physiol Gastrointest Liver Physiol 284: G46–G56, 2003. [DOI] [PubMed] [Google Scholar]
- 16.Moeser AJ, Nighot PK, Ryan KA, Wooten JG, Blikslager AT. Prostaglandin-mediated inhibition of Na+/H+ exchanger isoform 2 stimulates recovery of barrier function in ischemia-injured intestine. Am J Physiol Gastrointest Liver Physiol 291: G885–G894, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Moore R, Carlson S, Madara JL. Rapid barrier restitution in an in vitro model of intestinal epithelial injury. Lab Invest 60: 237–244, 1989. [PubMed] [Google Scholar]
- 18.Nath SK, Kambadur R, Yun CH, Donowitz M, Tse CM. NHE2 contains subdomains in the COOH terminus for growth factor and protein kinase regulation. Am J Physiol Cell Physiol 276: C873–C882, 1999. [DOI] [PubMed] [Google Scholar]
- 19.Nowak P, Blaheta R, Schuller A, Cinatl J, Wimmer-Greinecker G, Moritz A, Scholz M. The Na+/H+ exchange inhibitor HOE642 prevents stress-induced epithelial barrier dysfunction. Int J Mol Med 14: 175–178, 2004. [PubMed] [Google Scholar]
- 20.Park PO, Haglund U, Bulkley GB, Falt K. The sequence of development of intestinal tissue injury after strangulation ischemia and reperfusion. Surgery 107: 574–580, 1990. [PubMed] [Google Scholar]
- 21.Sakakibara A, Furuse M, Saitou M, Ando-Akatsuka Y, Tsukita S. Possible involvement of phosphorylation of occludin in tight junction formation. J Cell Biol 137: 1393–1401, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schultheis PJ, Clarke LL, Meneton P, Miller ML, Soleimani M, Gawenis LR, Riddle TM, Duffy JJ, Doetschman T, Wang T, Giebisch G, Aronson PS, Lorenz JN, Shull GE. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat Genet 19: 282–285, 1998. [DOI] [PubMed] [Google Scholar]
- 23.Stechmiller JK, Treloar D, Allen N. Gut dysfunction in critically ill patients: a review of the literature. Am J Crit Care 6: 204–209, 1997. [PubMed] [Google Scholar]
- 24.Sun F, Hug MJ, Lewarchik CM, Yun CH, Bradbury NA, Frizzell RA. E3KARP mediates the association of ezrin and protein kinase A with the cystic fibrosis transmembrane conductance regulator in airway cells. J Biol Chem 275: 29539–29546, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Turner JR. Molecular basis of epithelial barrier regulation: from basic mechanisms to clinical application. Am J Pathol 169: 1901–1909, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turner JR, Angle JM, Black ED, Joyal JL, Sacks DB, Madara JL. PKC-dependent regulation of transepithelial resistance: roles of MLC and MLC kinase. Am J Physiol Cell Physiol 277: C554–C562, 1999. [DOI] [PubMed] [Google Scholar]
- 27.Turner JR, Black ED, Ward J, Tse CM, Uchwat FA, Alli HA, Donowitz M, Madara JL, Angle JM. Transepithelial resistance can be regulated by the intestinal brush-border Na+/H+ exchanger NHE3. Am J Physiol Cell Physiol 279: C1918–C1924, 2000. [DOI] [PubMed] [Google Scholar]
- 28.Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, Madara JL. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol Cell Physiol 273: C1378–C1385, 1997. [DOI] [PubMed] [Google Scholar]
- 29.Wong V. Phosphorylation of occludin correlates with occludin localization and function at the tight junction. Am J Physiol Cell Physiol 273: C1859–C1867, 1997. [DOI] [PubMed] [Google Scholar]
- 30.Yoo J, Nichols A, Mammen J, Calvo I, Song JC, Worrell RT, Matlin K, Matthews JB. Bryostatin-1 enhances barrier function in T84 epithelia through PKC-dependent regulation of tight junction proteins. Am J Physiol Cell Physiol 285: C300–C309, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Yun CH, Oh S, Zizak M, Steplock D, Tsao S, Tse CM, Weinman EJ, Donowitz M. cAMP-mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein. Proc Natl Acad Sci USA 94: 3010–3015, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zachos NC, Tse M, Donowitz M. Molecular physiology of intestinal Na+/H+ exchange. Annu Rev Physiol 67: 411–443, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Zizak M, Lamprecht G, Steplock D, Tariq N, Shenolikar S, Donowitz M, Yun CH, Weinman EJ. cAMP-induced phosphorylation and inhibition of Na+/H+ exchanger 3 (NHE3) are dependent on the presence but not the phosphorylation of NHE regulatory factor. J Biol Chem 274: 24753–24758, 1999. [DOI] [PubMed] [Google Scholar]