

Abstract
Lung epithelium restoration subsequent to injury is of concern in association with the outcomes of diverse inflammatory lung diseases. Previous studies have demonstrated that mesenchymal stem cells (MSCs) may promote epithelial repair subsequent to inflammatory injury, however the mechanism that mediates this effect remains unclear. The current study examined the role of MSCs in alveolar type II epithelial cell (AT-II cell) restoration subsequent to an inflammatory insult. AT-II cells were firstly exposed to inflammatory cytokines including tumor necrosis factor-α, interleukin (IL)-6 and IL-1β, then were co-cultured with MSCs in Transwell for 72 h. Cell proliferation, expression of surfactant protein A (SP-A) and expression of the α1 subunit were evaluated respectively by the Cell Counting Kit-8 assay, western blotting and semiquantitative reverse transcription-polymerase chain reaction. Keratinocyte growth factor (KGF) small interfering RNA (siRNA) was applied to knockdown the main cytoprotective factors in the MSCs. Subsequent to an inflammatory insult, AT-II cells were observed to be impaired, exhibiting the characteristics of injured cell morphology, reduced cell proliferation and reduced expression of SP-A and the α1 subunit. Co-culture with MSCs significantly ameliorated these cell impairments, while these benefits were weakened by the application of KGF siRNA. Simultaneously, expression levels of phosphorylated (p-) protein kinase B (AKT) and p-mammalian target of rapamycin (mTOR) in AT-II cells were upregulated by MSCs, suggesting activation of the phosphoinositide 3-kinase (PI3K) pathway. These data demonstrate that administration of MSCs to the inflammation-insulted AT-II cells may ameliorate the impairments through a KGF-dependent PI3K/AKT/mTOR signaling pathway.
Keywords: MSCs, AT-II cells, SP-A, α1 subunit, PI3K/AKT/mTOR
Introduction
Mesenchymal stem cells (MSCs) have been reported to have therapeutic applications in tissue injury (1,2). In lung tissue, the ability of the lung epithelium to restore itself is of clinical importance, which is correlated with alveolar fluid clearance (AFC). Impaired AFC in patients with acute lung injury and acute respiratory distress syndrome has been demonstrated to be associated with high morbidity and mortality (3,4). AT-II cells, as one of the key types of pulmonary epithelium, are responsible for the secretion of surfactant in addition to active sodium transport from the alveolar surface to the pulmonary interstitium via sodium channels and the Na+-K+-adenosine triphosphatase (ATPase) transporter. AT-II cells have been reported to be important in numerous lung diseases and exert vital functions in the prevention of pulmonary inflammatory formation (5,6). Despite research into MSC administration for lung injury, the effect of MSCs on AT-II cells in an inflammatory microenviroment remains unclear.
Despite initial interest in the multipotency capabilities of MSCs, the differentiation of MSCs into pulmonary epithelium does not appear to serve a key role in lung injury repair. Previous studies have indicated that MSCs may be engrafted to injured pulmonary epithelial cells and express the specific biomarker of the pulmonary epithelium (7–9). However, the engraftment rate of MSCs to alveoli was observed to be too low to replace the damaged cells (10–12), suggesting that direct engraftment and differentiation into pulmonary epithelial cells was unlikely to be the key therapeutic mechanism. At present, it has been suggested that MSCs function in tissue repair in the lungs, and this is predominantly mediated through paracrine factors (13).
In the current study, AT-II cells were exposed to major inflammatory cytokines which led to impairments including damaged cell morphology, and reduced cell proliferation and expression of surfactant protein A (SP-A) and the α1 subunit. In order to study the potential benefits of MSCs on injured AT-II cells and the possible mechanisms underlying this, AT-II cells and MSCs were co-cultured using a Transwell system under inflammatory stimulation. Due to the fact that keratinocyte growth factor (KGF) is a specific epithelial growth factor secreted by MSCs (13,14), KGF was knocked down by small interfering RNA (siRNA) to further investigate whether the therapeutic effects on AT-II cell repair were due to the MSCs (13).
The current study suggested that MSCs ameliorated AT-II cell impairments by increasing cell proliferation and the expression levels of SP-A and the α1 subunit of the Na+-K+-ATPase transporter. In addition, the phosphoinositide 3-kinase (PI3K) signaling pathway was involved in this process. Using KGF siRNA knockdown, it was identified that MSCs increased the expression levels of SP-A and the α1 subunit in injured AT-II cells in part via a KGF-dependent PI3K/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling pathway.
Materials and methods
Primary culture of AT-II cells
All animal procedures were approved in advance by the Animal Care Committee of Chinese People's Liberation Army General Hospital (Beijing, China). A total of 50 male 8-week old Sprague-Dawley rats (average weight, 175 g; Beijing HFK Bioscience Co., Ltd., Beijing, China) were housed in conditions of 40% humidity and 23°C with a 12 h light/dark cycle and ad libitum access to food and water. Rats were anesthetized by 2% pentobarbital (50 mg/kg; Cascade Biologics; Thermo Fisher Scientific, Inc., Portland, OR, USA), anticoagulated with heparin sodium (ToYongBio, Shanghai, China), disinfected with 75% alcohol and plated on a Superclean bench (Shanghai Boxun Industry & Commerce Co., Ltd., Shanghai, China). The thorax of the rats was opened and the pulmonary microcirculation was flushed through the right ventricle to remove remaining blood subsequent to sacrifice of the rats by exsanguination. The lungs were removed and lavaged with phosphate-buffered saline (PBS). The distal airspaces were then lavaged 10 times and intubated with 20 ml trypsase (0.25%; Beijing Solarbio Science & Technology Co., Ltd., Beijing, China). The lobes were ground in the presence of fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and then digested with DNase (500 µg/ml; Beijing Solarbio Science & Technology Co., Ltd.) at 37°C for 60 min. The cell-rich fraction was filtered through a 200 meshstrainer (Beijing Solarbio Science & Technology Co., Ltd.). The filtrate was centrifuged at 400 × g for 20 min at 4°C, and the supernatant was removed. The deposit was resuspended with PBS and red blood cell lysis buffer (Beijing Solarbio Science & Technology Co., Ltd.) was added into suspension for 5 min subsequent to mixing. The suspension was centrifuged at 400 × g for 5 min at 4°C subsequent to completely dissolving the red blood cells and removing the supernatant. Cells were resuspended, counted and added into culture dishes coated with rat polyclonal IgG antibody (1:500; SP5-10; Beijing Solarbio Science & Technology Co., Ltd.) in an incubator (37°C and 5% CO2) for one hour. The unattached remaining cells were transferred to a centrifuge tube and centrifuged at 400 × g for 10 min at 4°C. The deposit was resuspended and cultured in a dish with (Dulbecco's modified Eagle's medium (DMEM)/F12 containing 2% FBS, 100 U/ml penicillin and 100 U/ml streptomycin (Gibco; Thermo Fisher Scientific, Inc.) for the experiments. AT-II cells were identified using rabbit polyclonal alveolar SP-A (1:100; sc-13977; Santa Cruz Biotechnology, Inc., Heidelberg, Germany) and monoclonal fluorescien isothiocyanate labeled goat anti-rabbit secondary antibody (1:500; A0562; Beyotime Institute of Biotechnology), which exhibited green fluorescence under confocal fluorescence microscopy (Leica TCS SP5; Leica Microsystems, Wetzlar, Germany).
MSC culture and identification
Tibiaes and femurs were excised from rats following anaesthesia. MSCs were flushed with DMEM/F12 and isolated from the tibiae and femur marrow of 8-week old male SD rats (15). bone marrow-derived MSCs were cultured with DMEM/F12 containing 1% glutamine, 2% FBS, 100 U/ml penicillin and 100 U/ml streptomycin in incubator (37°C and 5% CO2). As cells reached 80–90% confluence, MSCs were passaged every 3–4 days by trypsinization (Beijing Solarbio Science & Technology Co., Ltd.) and cells from the 3rd to 8th passage were used for experiments. Cells (5×105) in a plate were cultured with adipogenic or osteogenic induction media (Cyagen Biosciences, Guangzhou, China) every 3 days. After 2 weeks, cells reached 90% confluence and were stained with oil red O or alizarin red (Cyagen Biosciences) in a culture plate. MSCs exhibited osteogenic and adipogenic differentiation. Biological cell surface markers of MSCs, including CD29, CD44 (both allophycocyanin-labeled), CD90, CD45 and CD34 (all phycoerythrin-labeled), were detected by flow cytometry (BD FACSCalibur; BD Biosciences, San Jose, CA, USA).
Impairment assay of AT-II cells subsequent to inflammatory exposure
To injure the cells, primary cultures of AT-II cells were exposed to inflammatory cytokines containing 1.7 ng/ml tumor necrosis factor (TNF)-α, 87.6 ng/ml IL-6 and 4.4 ng/ml IL-1β (PeproTech, Inc., Rocky Hill, NJ, USA), which were determined according to a previous study (16). Cell morphology was observed and cell proliferation were analyzed with the Cell Counting Kit-8 (CCK-8) kit (Beyotime Institute of Biotechnology, Jiangsu, China) in a 96-well plate at 72 h. The medium was replaced by 90 µl fresh DMEM/F12 mixed with 10 µl CCK-8 solution at a final volume of 0.1 ml. Subsequently, cells were incubated (37°C, 5% CO2) for 2 h. The optical density in each well was measured with a microplate reader (Spectra MR; Dynex Technologies, Inc., Chantilly, VA, USA). Protein expression levels of SP-A and the α1 subunit of Na+-K+-ATPase were evaluated by western blotting.
Co-culture system development and KGF detection
To detect KGF secretion by MSCs, a co-culture system was developed using a 6-well Transwell plate (0.4 µm pore size insert; Corning Incorporated, Corning, NY, USA). MSCs (5×105) were exposed to the inflammatory cytokines or co-cultured with AT-II cells (5×105) in Transwell for 48 h (n=3 per group). The concentration of KGF was detected under following conditions: i) MSCs alone; ii) cytokine-exposed MSCs; iii) cytokine-exposed MSCs + AT-II cells in Transwell; and iv) cytokine-exposed AT-II cells. In advance, MSCs were starved for 24 h using serum-free medium (Gibco; Thermo Fisher Scientific, Inc.) prior to inflammatory exposure. The supernatant of the culture medium was obtained at 48 h and the KGF concentration was measured with the rat specific Enzyme-Linked Immunosorbent Assay (ELISA) kit (R&D Systems, Inc., Minneapolis, MN, USA) according to the manufacturer's protocol.
MSC transfection and efficiency assay
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) was used to transfect MSCs (5×105) with siRNA to knockdown KGF secretion in a dish (60 mm × 15 mm). Lipofectamine 2000 (5 µl) and siRNA (5 µl) were supplemented into 125 µl Opti-MEM (Gibco; Thermo Fisher Scientific, Inc.), then after five min, the two liquids were mixed and placed at room temperature for 20 min. Subsequent to washing of MSCs with PBS, the 260 µl mixture and Opti-MEM were respectively added to the MSC culture plate at final volume of 2 ml. Cells were then cultured in an incubator (37°C and 5%) for 6 h and the transfection liquid was replaced with fresh DMEM/F12 containing 2% FBS, 1% glutamine, 100 U/ml penicillin and 100 U/ml streptomycin. The siRNA against rat KGF [3′-dTdTCGCUGUGUGCUCUUCAAUA-5′ (siG1312692455)] was provided by Guangzhou RiboBio Co., Ltd. (Guangzhou, China). MSCs were transfected with the negative siRNA (Leica TCS SP8; Leica Microsystems) or transfected only with Lipofectamine 2000 (MSCs-Lipof). To test the transfection efficiency, MSCs were transfected with cy5-labeled siRNA (Guangzhou RiboBio Co., Ltd.), which was imaged by confocal microscopy. The knocking down efficiency in each group was evaluated by RT-PCR and ELISA assays at 48 h subsequent to transfection.
Proliferation assay of AT-II cells by CCK-8
Cell proliferation was measured using a CCK-8 assay kit in a 6-well transwell plate. AT-II cells were grown on the upper compartment (5×105), while MSCs were plated in the bottom compartment (5×105). To injure cells, AT-II cells were exposed to the inflammatory cytokines. The cell proliferation assay was conducted under the following conditions at 0, 12, 24, 36, 48 and 72 h: i) Cytokine-exposed AT-II cells + PBS; ii) cytokine-exposed AT-II cells + MSCs; iii) cytokine-exposed AT-II cells + MSCs-KGF siRNA; and iv) cytokine-exposed AT-II cells + MSCs-Lipof.
Total protein isolation and western blot analysis
To further investigate the effects of MSCs on AT-II cells in vitro, the protein levels of the α1 subunit and SP-A were evaluated under the same experimental conditions. In advance, MSC transfection was conducted in the lower Transwell chamber. Subsequent to exposure of AT-II cells to the inflammatory cytokines for 4 h, AT-II cells were co-cultured with MSCs according to the following experimental conditions: i) Cytokine-exposed AT-II cells + PBS (control group); ii) cytokine-exposed AT-II cells + MSCs; iii) cytokine-exposed AT-II cells + MSCs-KGF siRNA; and iv) cytokine-exposed AT-II cells + MSCs-Lipof. Subsequent to culture for 72 h at 37°C, total protein was extracted from the AT-II cells using 0.2 ml radioimmunoprecipitation assay lysate (Applygen Technologies, Inc., Beijing, China) per well. Samples mixed with 10% sodium dodecyl sulfate-polyacrylimide gel electrophoresis (SDS-PAGE) were denatured at 95°C for 10 min. Protein levels were measured with a bicinchoninic acid protein assay kit (Applygen Technologies, Inc.). Western blotting was conducted according to the following protocol. The proteins were transferred onto a polyvinylidene difluoride membrane (PVDF; Shanghai Jiang Lai Biotechnology Co., Ltd., Shanghai, China) and blocked with 5% dried skimmed milk in PBS with Tween-20 at a density of 0.1% for 1.5 h. The PVDF membrane was then exposed to the primary antibody overnight at 4°C. The following three primary antibodies were used: Monoclonal mouse anti-Na+/K+-ATPase α1 (1:200; sc-21712; Santa Cruz Biotechnology, Inc.), monoclonal mouse anti-β-actin antibody (1:800; TA-09; OriGene Technologies, Inc., Beijing, China) and polyclonal rabbit anti-SP-A (1:200; sc-13977; Santa Cruz Biotechnology, Inc.). The PVDF membrane was incubated for 2 h with monoclonal goat anti-mouse (1:5,000; ZB2305) or goat anti-rabbit secondary antibodies (1:5,000; ZB2301) (both purchased from ZSGB-BIO, Beijing, China). The protein blots on the PVDF membranes were visualized by enhanced chemiluminescence reagent detection reagents (Applygen Technologies, Inc.) and semi-quantified with Image J2x software (version 2.1.4.7; Rawak Software, Inc., Germany). Expression of SP-A and the α1 subunit was normalized to β-actin expression. Protein expression levels were detected using monoclonal rabbit anti-mouse AKT (1:1,000; 4685), monoclonal rabbit anti-mouse phosphorylated AKT (p-AKT) (1:1,000; 4058), polyclonal rabbit anti-mouse mTOR (1:1,000; 2972) and polyclonal rabbit anti-mouse p-mTOR (1:1,000; 2974) (all purchased from Cell Signaling Technologies, Inc., Danvers, MA USA) in the PI3K/AKT/mTOR signalling pathway were also analyzed.
Total RNA extraction and semiquantitative reverse transcription-polymerase chain reaction (RT-PCR) analysis
RT-PCR analysis of SP-A and α1 subunit mRNA levels was conducted. Total RNA of the α1 subunit and SP-A in AT-II cells was extracted from AT-II cells under the following conditions: i) Cytokine-exposed AT-II cells + PBS; ii) cytokine-exposed AT-II cells + MSCs; iii) cytokine-exposed AT-II cells + MSCs-KGF siRNA; and iv) cytokine-exposed AT-II cells + MSCs-Lipof. Cells were extracted using TRIzol reagent (Life Technologies; Thermo Fisher Scienctific, Inc.) according to manufacturer's instruction and RNA quality was assessed by the 260/280 ratio. RT-PCR was conducted following the two step manufacturer's protocol of the PrimeScript™ RT-PCR kit (Takara Bio, Inc., Otsu, Japan) using the C1000 Thermal cycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA). First, the cDNA was prepared using the C1000 Thermal cycler, and the PCR reaction system was prepared in tubes with 1 µl dNTP mixture (25 mM), 1 µl Oligo (dt) primers (2.5 µM), 1 µg template RNA (or positive control RNA) and DEPC-treated water to a final volume of 10 µl. Tubes were placed into the C1000 Thermal Cycler at 65°C for 5 min for denaturation and annealing. Subsequently, reverse transcription was conducted by preparing the following reagent mixture including 10 µl reaction mixture from denaturation and annealing, 4 µl 5X PrimeScript buffer, 0.5 µl RNase inhititor, 0.5 µl PrimeScript RTase and 5 µl RNase DEPC-treated water to a final volume of 20 µl reaction mixture. Tubes were placed into the Thermal Cycler at 42°C for 30 min and at 4°C for 10 min. Subsequently, a 50 µl reaction consisting of 5 µl 10X PCR buffer II, 2 µl dNTP Mixture (10 mM), 0.5 µl upstream primer (0.2 µM), 0.5 µl downstream primer (0.2 µM), Takara Ex TaqHS (5 u/µl), 5 µl reverse transcriptant and DEPC-treated water to a final volume of 50 µl was conducted. Subsequent to mixing, all tubes were placed in the thermal cycler at 94°C for 30 sec, 60°C for 30 sec and 72°C for 1 min. This program was run for 30 cycles. Primer sequences for the α1 subunit, SP-A and β-actin were provided by Sangon Biotech Co., Ltd. (Shanghai, China) (Table I).
Table I.
Primer sequences used in experiment.
| Gene | Sequence |
|---|---|
| ATP-α1 F | 5′-CTCCTTCTGCCTGACGAACA-3′ |
| ATP-α1 R | 5′-ATCAAGCTCAACCGAGTGCT-3′ |
| SP-A F | 5′-ATCAAGCTCAACCGAGTGCT-3′ |
| SP-A R | 5′-TGGACAGGTAGGACGTTTGG-3′ |
| KGF F | 5′-AGCGATCAACTCAAGGTCCA-3′ |
| KGF R | 5′-TATGGTGCCCACAAGACAGA′-3′ |
| Actin F | 5′-CTAAGGCCAACCGTGAAAAGA-3′ |
| Actin R | 5′-CCAGAGGCATACAGGGACAAC-3′ |
ATP, adenosine triphosphate; F, forward; R, reverse; SP-A, surfactant protein A; KGF, keratinocyte growth factor.
Statistical analysis
All experiments were conducted three times for each group. Results are expressed as the mean ± standard deviation. Comparisons between two groups were made using the unpaired two-tailed t-test. Multiple comparisons between more than two groups were made using one-way analysis of variance using SPSS software, version 13.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Characterization of MSCs and AT-II cells
Rat MSCs are adherent and spindle-like cells, which were observed to differentiate into the predominant mesenchymal lineages, adipocytes and osteocytes. Rat MSCs were identified to positively express the cell surface markers CD29, CD44, CD90 and negatively express CD34 and CD45 by flow cytometry (Fig. 1). The primary culture of AT-II cells was observed to exhibit adherent and round cells in the culture plate, which were identified by the fact that SP-A was bound to the fluorescein isothiocyanate-labeled secondary antibody. Green fluorescence from AT-II cells was detected in the cell membrane and cytoplasm by confocal fluorescence microscopy (Fig. 2A and B).
Figure 1.

Characterization of MSCs. (A) Undifferentiated passage three MSCs are adherent and spindle-like. (B) Osteogenesis was detected by alizarin red. (C) Adipogenesis was detected by oil red O. (Scale bars in A–C, 100 µm). (D) Flow cytometry indicated that MSCs uniformly expressed CD29, CD44 and CD90, and negatively expressed CD34 and CD45 surface markers. MSCs, mesenchymal stem cells; SSC, side scatter; FSC, forward scatter; PE, phycoerythrin; APC, allophycocyanin.
Figure 2.

AT-II cell impairments by inflammatory cytokines. (A and B) Primary culture of AT-II cells were stained with green fluorescence, measured using immunofluorescence. Exposure to cytokines (tumor necrosis factor α and interleukins 6 and 1β) resulted in (C) damaged cell morphology and (D) reduced AT-II cell proliferation measured by the Cell Counting Kit-8 assay. *P<0.05 vs. AT-II cells (normal medium). (E) Western blotting indicated downregulated protein levels of SP-A and the α1-subunit following inflammatory insult. (Scale bars in A–C, 50 µm). AT-II cell, alveolar type II epithelial cell; SP-A, surfactant protein A; OD, optical density.
Impairments of AT-II cells caused by inflammatory cytokines
To injure AT-II cells, AT-II cells were exposed to inflammatory cytokines (TNF-α, IL-6 and IL-1β) for 72 h, which resulted in impaired cell morphology, delayed cell proliferation after 12 h (Fig. 2C and D) and a downregulation in protein expression of SP-A and the α1 subunit compared with AT-II cells cultured in normal medium (Fig. 2E).
KGF secretion by MSCs
KGF secretion reached 884.17 pg/ml in MSCs that were cultured for 48 h subsequent to exposure to inflammatory cytokines in comparison with 437.65 pg/ml in MSCs cultured in normal medium (P<0.01). However, the KGF concentration was detected to be 382.37 pg/ml in the co-culture system of AT-II cells and MSCs under the condition of inflammation. The results indicated that AT-II cells produced significantly less KGF under inflammatory condition vs. MSCs under the same conditions (Fig. 3A and B).
Figure 3.

KGF secretion and knockdown efficiency of KGF siRNA. (A) A co-culture system was developed with AT-II cells in the upper compartment, and MSCs in the bottom compartment of a Transwell chamber. (B) KGF secretion was detected by ELISA under different conditions. Experiments were run three times and data were expressed as the mean ± standard deviation (n=3/group, **P<0.01 vs. MSCs; ##P<0.01 vs. MSCs (cytokines); ††P<0.01 vs. MSCs (cytokines). KGF knockdown efficiency was evaluated by (C) ELISA and (D) semiquantitative reverse transcription-polymerase chain reaction (n=3 per group). The mRNA level was normalized to β-actin expression. Data were expressed as the mean ± standard deviation. (**P<0.01 vs. all other groups). KGF, keratinocyte growth factor; siRNA, small interfering RNA; AT-II cell, alveolar type II epithelial cell; MSCs, mesenchymal stem cells; ELISA, enzyme-linked immunosorbent assay; Lipof, lipofectamine.
Knockdown efficiency of KGF siRNA
The knockdown efficiency was confirmed by RT-PCR and ELISA at 48 h after transfection of MSCs with KGF siRNA. According to the results, the concentration of KGF in MSC culture medium was significantly reduced 48 h subsequent to transfection with KGF siRNA in comparison to MSCs without transfection. In addition, it was identified that KGF expression at the mRNA level was significantly downregulated by KGF siRNA. Protein or mRNA expression levels of mock-transfected MSCs were unchanged compared with those transfected with negative-siRNA (Fig. 3C and D).
MSCs ameliorated AT-II cell impairment
The CCK-8 assay demonstrated that AT-II cell proliferation was delayed subsequent to the exposure of cells to inflammatory cytokines. The reduced AT-II cell proliferation was increased with co-culture with MSCs, in particular between 24 h and 72 h. In addition, AT-II cell proliferation was reduced in AT-II cells cultured with MSCs-KGF siRNA when compared with AT-II cells cultured with MSCs (Fig. 4). However, proliferation in the MSCs-KGF siRNA group was still enhanced compared with the proliferation of cytokine-exposed AT-II cells. Results of SP-A and α1 subunit expression by western blot analysis indicated that co-culture with MSCs increased the protein expression levels, which were reduced by MSCs-KGF siRNA. However, the protein levels of the α1 subunit and SP-A were observed to be unchanged between the AT-II cells treated with MSCs-Lipof and those treated with MSCs alone (Fig. 5A). Therapeutic benefits were further investigated by RT-PCR. Co-culture with MSCs was observed to increase the mRNA expression levels of the α1 subunit and SP-A in AT-II cells. However, co-culture with MSCs-KGF siRNA attenuated the increased expression of α1 subunit and SP-A. It was observed that there was no significant difference in the mRNA levels of SP-A and the α1 subunit between AT-II cells treated with MSCs-Lipof and MSCs alone (Fig. 5B).
Figure 4.
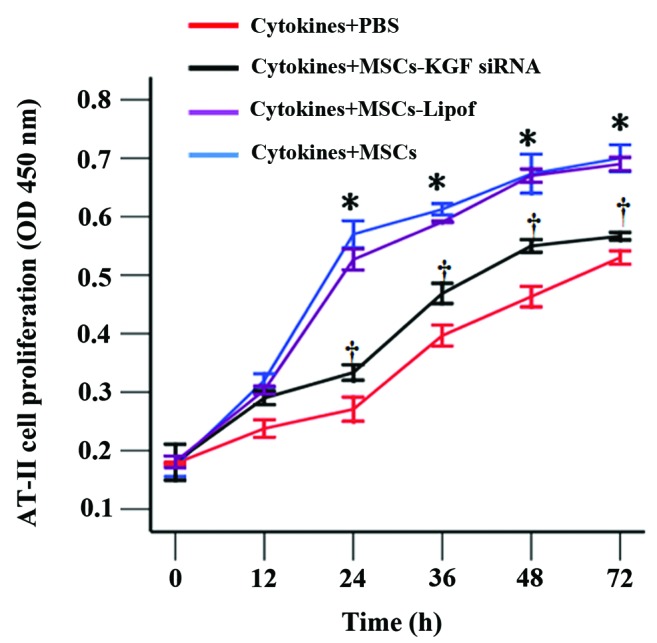
MSCs increased AT-II cell proliferation. AT-II cell proliferation was evaluated by the Cell Counting Kit-8 assay under four different conditions. MSCs were observed to improve AT-II cell proliferation, and this effect was reduced by MSCs-KGF siRNA. *P<0.05 vs. AT-II cells (cytokines + PBS); †P<0.05 vs. AT-II cells (cytokines + PBS) or AT-II cells (cytokines + MSCs). MSCs, mesenchymal stem cells; AT-II cell, alveolar type II epithelial cell; KGF, keratinocyte growth factor; siRNA, small interfering RNA; PBS, phosphate-buffered saline; OD, optical density; Lipof, lipofectamine.
Figure 5.

Co-culture with MSCs increased expression of the α1 subunit and SP-A. Expression levels of the α1-subunit and SP-A by (A) western blotting and (B) semiquantitative reverse transcription-polymerase chain reaction were analyzed in order to observe MSC effects on AT-II cells. Experiments were run three times and the protein levels were normalized to β-actin expression. Data are presented as the mean ± standard deviation. **P<0.01 vs. AT-II cells (phosphate-buffered saline); ††P<0.01 vs. AT-II cells (MSCs). MSCs, mesenchymal stem cells; SP-A, surfactant protein A; AT-II cell, alveolar type II epithelial cell; KGF, keratinocyte growth factor; siRNA, small interfering RNA; Lipof, lipofectamine.
MSCs activated PI3K signaling pathway by KGF secretion
To further investigate the underlying mechanisms, the proteins levels of AKT, p-AKT, mTOR and p-mTOR in the PI3K/AKT/mTOR pathway were measured by western blotting. Treatment with MSCs significantly increased the protein expression levels of p-AKT and p-mTOR at 72 h. MSCs-KGF siRNA significantly attenuated the protein increase of p-AKT and p-mTOR. Protein levels of AKT and mTOR were not markedly altered apparently under the same experimental conditions (Fig. 6).
Figure 6.

MSCs increased protein expression levels of p-AKT and p-mTOR in AT-II cells. Protein expression of (A) p-AKT and AKT, and (B) p-mTOR and mTOR were analyzed by western blotting subsequent to treatment of AT-II cells with phosphate-buffered saline, MSCs, MSC-KGF siRNA or MSC-Lipof. Experiments were run three times and protein levels were normalized to β-actin expression. Data are presented as the mean ± standard deviation. **P<0.01 vs. the PBS group; ††P<0.01 vs. the MSC group. MSCs, mesenchymal stem cells; p-, phosphorylated; AKT, protein kinase B; mTOR, mechanistic target of rapamycin; AT-II cell, alveolar type II epithelial cell; KGF, keratinocyte growth factor; siRNA, small interfering RNA; Lipof, lipofectamine.
Discussion
The key observations of the current study are summarized as following: i) To the best of our current knowledge, this is the first study investigating AT-II cell impairments characterized by impaired cell morphology accompanied by reductions in cell proliferation and the expression of SP-A and the α1 subunit following an inflammatory insult in vitro. ii) Co-culture with MSCs was observed to increase AT-II cell proliferation and expression levels of SP-A and the α1 subunit of Na+-K+-ATPase significantly via activation of the KGF-dependent PI3K/AKT/mTOR pathway.
As has been previously demonstrated, AT-II cell impairment is involved in multiple inflammation-associated lung diseases (6). To imitate inflammation, the AT-II cells were exposed to the key inflammatory cytokines including TNF-α, IL-6 and IL-1β at concentrations of 1.7, 87.6 and 4.4 ng/ml, respectively (16). The results of the current study demonstrated that exposure to these inflammatory cytokines led to impairment of cell morphology, and reductions in cell proliferation and the expression levels of SP-A and the α1 subunit, suggesting that the impairments of AT-II cells by inflammation had been reproduced in vitro.
As hypothesized, co-culture with MSCs in Transwell enhanced AT-II cell proliferation subsequent to exposure to inflammation, which suggested that certain paracrine factors may serve a role in improving AT-II cell growth. Among the previously reported paracrine factors produced by MSCs, KGF has been regarded as the key epithelial promoter (17,18). The results of the current study indicated that the KGF concentration in MSC culture medium was higher under inflammatory conditions than that of normal medium, thus suggesting that MSCs may produce increased quantities of KGF in response to an inflammatory stimulus. However, KGF concentration was observed to be reduced in the MSC and AT-II cell co-culture medium, which indicated that MSC-secreted KGF may be consumed in the crosstalk between MSCs and AT-II cells.
Notably, AT-II cells alone produced significantly less KGF in comparison with MSCs, which suggested that MSCs are likely to be the primary source of KGF in the co-culture system. Therefore, KGF was knocked down in MSCs using siRNA, in order to confirm whether the therapeutic effect was due to MSCs and dependent on its secretion of KGF. The results demonstrated that the increase in SP-A and α1 subunit expression was reduced by the addition of MSCs pretreated with KGF siRNA. Thus, this suggested that MSCs-secreted KGF may contribute to this therapeutic effect in addition to cell proliferation.
In order to investigate the underlying mechanisms, the critical proteins of the PI3K pathway that are associated with cell prolifieration, p-AKT and p-mTOR, were detected (19–21). It was identified that co-culture with MSCs increased the protein expression levels of p-AKT and p-mTOR under inflammatory conditions, while these effects were reduced by KGF siRNA pretreatment. This also implied that the PI3K/AKT/mTOR signaling pathway may be activated by MSC-secreted KGF. Notably, it was identified that the increases in expression of the α1 subunit and SP-A were in line with alterations of p-AKT and p-mTOR under the same experimental conditions. This indicated that MSCs may enhance the expression of the α1 subunit and SP-A via a KGF-dependent PI3K/AKT/mTOR signaling pathway.
In conclusion, MSCs were identified to be able to ameliorate AT-II cell impairment by increasing cell proliferation and the expression levels of SP-A and the α1 subunit. Furthermore, KGF secretion may account in part for the protective benefits of MSCs by activating the PI3K/AKT/mTOR signaling pathway. The current study explored the mechanism of MSCs in reducing the impairment of AT-II cells caused by inflammatory cytokines. These observations provide a novel insight into MSC-based cell therapy for treating acute lung injury and acute respiratory distress syndrome.
Acknowledgments
The present study was supported partly by the National Nature Science Foundation of China (grant nos. 81121004, 81230041 and 81372066) and the National Basic Science and Development Program (973 Program; grant no. 2012CB518105).
References
- 1.Matthay MA, Goolaerts A, Howard JP, Lee JW. Mesenchymal stem cells for acute lung injury: Preclinical evidence. Crit Care Med. 2010;38(Suppl 10):S569–S573. doi: 10.1097/CCM.0b013e3181f1ff1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee JW, Gupta N, Serikov V, Matthay MA. Potential application of mesenchymal stem cells in acute lung injury. Expert Opin Biol Ther. 2009;9:1259–1270. doi: 10.1517/14712590903213651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee JW, Fang X, Dolganov G, Fremont RD, Bastarache JA, Ware LB, Matthay MA. Acute lung injury edema fluid decreases net fluid transport across human alveolar epithelial type II cells. J Biol Chem. 2007;282:24109–24119. doi: 10.1074/jbc.M700821200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berthiaume Y, Matthay MA. Alveolar edema fluid clearance and acute lung injury. Respir Physiol Neurobiol. 2007;159:350–359. doi: 10.1016/j.resp.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mutlu GM, Sznajder JI. Mechanisms of pulmonary edema clearance. Am J Physiol Lung Cell Mol Physiol. 2005;289:L685–L695. doi: 10.1152/ajplung.00247.2005. [DOI] [PubMed] [Google Scholar]
- 6.Fehrenbach H. Alveolar epithelial type II cell: Defender of the alveolus revisited. Respir Res. 2001;2:33–46. doi: 10.1186/rr36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kotton DN, Ma BY, Cardoso WV, Sanderson EA, Summer RS, Williams MC, Fine A. Bone marrow-derived cells as progenitors of lung alveolar epithelium. Development. 2001;128:5181–5188. doi: 10.1242/dev.128.24.5181. [DOI] [PubMed] [Google Scholar]
- 8.Krause DS, Theise ND, Collector MI, Henegariu O, Hwang S, Gardner R, Neutzel S, Sharkis SJ. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell. 2001;105:369–377. doi: 10.1016/S0092-8674(01)00328-2. [DOI] [PubMed] [Google Scholar]
- 9.Wang G, Bunnell BA, Painter RG, Quiniones BC, Tom S, Lanson NA, Jr, Spees JL, Bertucci D, Peister A, Weiss DJ, et al. Adult stem cells from bone marrow stroma differentiate into airway epithelial cells: Potential therapy for cystic fibrosis. Proc Natl Acad Sci USA. 2005;102:186–191. doi: 10.1073/pnas.0406266102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mei SH, McCarter SD, Deng Y, Parker CH, Liles WC, Stewart DJ. Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin-1. PLoS Med. 2007;4:e269. doi: 10.1371/journal.pmed.0040269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ortiz LA, Dutreil M, Fattman C, Pandey AC, Torres G, Go K, Phinney DG. Interleukin 1 receptor antagonist mediates the antiinflammatory and antifibrotic effect of mesenchymal stem cells during lung injury. Proc Natl Acad Sci USA. 2007;104:11002–11007. doi: 10.1073/pnas.0704421104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kotton DN, Fabian AJ, Mulligan RC. Failure of bone marrow to reconstitute lung epithelium. Am J Respir Cell Mol Biol. 2005;33:328–334. doi: 10.1165/rcmb.2005-0175RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JW, Fang X, Krasnodembskaya A, Howard JP, Matthay MA. Concise review: Mesenchymal stm cells for acute lung injury: Role of paracrine soluble factors. Stem Cells. 2011;29:913–919. doi: 10.1002/stem.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayes M, Curley G, Laffey JG. Mesenchymal stem cells - a promising therapy for acute respiratory distress syndrome. F1000. Med Rep. 2012;4:2. doi: 10.3410/M4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y, Cheng Y, Lian QQ, Yang L, Qi W, Wu DR, Zheng X, Liu YJ, Li WJ, Jin SW, Smith FG. Contribution of CFTR to alveolar fluid clearance by lipoxin A4 via PI3K/Akt pathway in LPS-induced acute lung injury. Mediators Inflamm. 2013;2013:862628. doi: 10.1155/2013/862628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Semaeva E, Tenstad O, Bletsa A, Gjerde EA, Wiig H. Isolation of rat trachea interstitial fluid and demonstration of local cytokine production in lipopolysaccharide-induced systemic inflammation. J Appl Physiol (1985) 2008;104:809–820. doi: 10.1152/japplphysiol.00846.2007. [DOI] [PubMed] [Google Scholar]
- 17.Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol. 2010;298:L715–L731. doi: 10.1152/ajplung.00361.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panos RJ, Bak PM, Simonet WS, Rubin JS, Smith LJ. Intratracheal instillation of keratinocyte growth factor decreases hyperoxia-induced mortality in rats. J Clin Invest. 1995;96:2026–2033. doi: 10.1172/JCI118250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubbard PA, Moody CL, Murali R. Allosteric modulation of Ras and the PI3K/AKT/mTOR pathway: Emerging therapeutic opportunities. Front Physiol. 2014;5:478. doi: 10.3389/fphys.2014.00478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/Mtor pathway: Effective combinations and clinical considerations. Drug Resist Updat. 2008;11:32–50. doi: 10.1016/j.drup.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang L, Graham PH, Hao J, Ni J, Bucci J, Cozzi PJ, Kearsley JH, Li Y. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013;4:e875. doi: 10.1038/cddis.2013.407. [DOI] [PMC free article] [PubMed] [Google Scholar]