

Abstract
Pulmonary hypertension (PH) complicating chronic parenchymal lung disease, such as idiopathic pulmonary fibrosis, results in significant morbidity and mortality. Since the hypoxia-inducible factor (HIF) signaling pathway is important for development of pulmonary hypertension in chronic hypoxia, we investigated whether HIF signaling in vascular endothelium regulates development of PH related to pulmonary fibrosis. We generated a transgenic model in which HIF is deleted within vascular endothelial cells and then exposed these mice to chronic intraperitoneal bleomycin to induce PH associated with lung fibrosis. Although no differences in the degree of fibrotic remodeling were observed, we found that endothelial HIF-deficient mice were protected against development of PH, including right ventricle and pulmonary vessel remodeling. Similarly, endothelial HIF-deficient mice were protected from PH after a 4-wk exposure to normobaric hypoxia. In vitro studies of pulmonary vascular endothelial cells isolated from the HIF-targeted mice and controls revealed that endothelial HIF signaling increases endothelial cell expression of connective tissue growth factor, enhances vascular permeability, and promotes pulmonary artery smooth muscle cell proliferation and wound healing ability, all of which have the potential to impact the development of PH in vivo. Taken together, these studies demonstrate that vascular endothelial cell HIF signaling is necessary for development of hypoxia and pulmonary fibrosis associated PH. As such, HIF and HIF-regulated targets represent a therapeutic target in these conditions.
Keywords: idiopathic pulmonary fibrosis, hypoxia-inducible factor, connective tissue growth factor, pulmonary hypertension, pulmonary vascular disease
idiopathic pulmonary fibrosis (IPF) is a progressive lung disease with a median survival of 2 to 3 yr. Pulmonary hypertension (PH), defined as a mean pulmonary artery pressure (mPAP) of greater than 25 mmHg, is a frequent comorbidity in patients with IPF (WHO Group III PH, or PH associated with chronic lung disease and hypoxia), affecting 20 to 80% of patients depending on disease severity (32, 39, 53). Although lung volume and extent of fibrosis on imaging do not correlate with development of PH in patients with IPF (52), the presence of PH is associated with a decrease in exercise capacity, independence, quality-of-life, and the 6-min walk distance (7). Furthermore, the relative risk of death in patients with IPF and PH 5 yr after diagnosis is 2.2- to 4.85-fold higher compared with IPF patients without PH (22).
Pulmonary arterial hypertension (PAH, or WHO Group I PH)-focused drugs have not proven beneficial in Group III PH patients, and they may in some cases be harmful. Drugs studied include nearly all classes of PAH-approved therapies, including inhaled vasodilators, such as prostacyclin (40), endothelin-receptor antagonists (45), and phosphodiesterase type 5 inhibitors (28). Supplemental oxygen, however, decreases pulmonary vascular resistance and improves quality of life in these patients (65).
Hypoxia has long been known to contribute to pulmonary vascular remodeling in animal models of PH (49). One of the primary mediators of the cellular response to hypoxia is the protein hypoxia-inducible factor (HIF), a transcription factor that exists as a heterodimer consisting of an ubiquitous β-subunit, and an α-subunit. Under conditions of normoxia, the α-subunit is continually degraded, but in response to low oxygen levels, HIF-α translocates to the nucleus with the β-subunit, where the complex binds to the hypoxia response element, facilitating transcription of various target genes (51). HIF exists as HIF1-α and a homologous HIF2-α protein, both of which have been shown to be integral in the development of PH in a chronic hypoxia model (8, 69). In a recent study, PH in the chronic hypoxia model was shown to be, in part, mediated by HIF activation specifically within vascular smooth muscle cells (SMCs) of the lung (3). A proposed mechanism for this finding is that HIF stabilization in SMCs leads to decreased potassium channel expression, resulting in increased cytosolic calcium concentration and tonic vasoconstriction with attendant expected changes in vascular remodeling leading to PH (50, 63).
Although the role of HIF in development of PH related to chronic lung disease is unknown, experimental data link vascular endothelial cell dysfunction with development of PAH (WHO Group I PH) (9), as well as vascular remodeling associated with chronic obstructive pulmonary disease (COPD) (31) and pulmonary fibrosis (15). To explore the contribution of HIF signaling in endothelial cells to pulmonary vascular remodeling, we developed a model with selective HIF deletion in vascular endothelium and found that these mice were protected from PH in response to pulmonary fibrosis and chronic hypoxia.
METHODS
Transgenic mice.
All transgenic mice generated for this study were on the C57BL/6J background, were greater than 8 wk of age at the study onset, included both males and females, and ranged in weight from 20 to 30 g. Transgenic mice expressing Cre-recombinase under control of the mouse VE-cadherin promoter (VECad.Cre) (2) were crossed with mice in which HIF1-α and HIF2-α are each flanked by two loxP sites (HIF1-αfl/fl and HIF2-αfl/fl, respectively) to create Cre-mediated constitutive gene deletion of HIF1-α and HIF2-α within vascular endothelium (26). These mice were then crossed with a ROSA26.Stop.lacZ transgenic mouse (55) to create irreversible activation of lacZ within vascular endothelium (VECad.Cre.HIF1-alphafl/fl.HIF2-alphafl/fl.ROSA.Stop.lacZ) for cell fate lineage confirmation of the model. Breedings were set up, such that HIF constructs and the lacZ constructs were maintained in homozygous state, while VECad.Cre was in the heterozygous state, yielding Cre+ mice with endothelial deletion of HIF1-α and HIF2-α, while Cre- mice served as littermate controls. To control for potential Cre+ effects, in some experiments, VECad.Cre.ROSA.Stop.lacZ mice were used as additional controls.
Mice were housed in the central animal care facility at Vanderbilt University Medical Center (Nashville, TN) and were given food and water ad libitum. The experimental protocol was reviewed and approved by the Institutional Animal Care and Use Committee at Vanderbilt University.
Bleomycin model.
Mice underwent intraperitoneal injection with 0.035 U/g bleomycin (Bedford Laboratories, Bedford, OH) or vehicle twice weekly for 4 wk (4). One week after the last injection, mice were then harvested for histology and hemodynamic measurements. In some mice, 3 h prior to lung harvest, pimonidazole (60 mg/kg) was administered by intraperitoneal injection. Mouse tail pulse oximetry was determined using a Starr MouseOx device per manufacturer's recommendations (MouseOx; Starr Life Sciences, Oakmont, PA).
Chronic hypoxic exposure.
Mice exposed to chronic hypoxia were placed in a normobaric chamber where the oxygen concentration is controlled through flow of nitrogen to provide the desired FiO2 (ProO2 monitor/controller and chamber; Biospherix, Lacona, NY) with continuous monitoring of oxygen and carbon dioxide concentration. Ventilation is maintained, such that carbon dioxide levels remain less than 1,000 parts per million (0.1%) (ProCO2 Monitor, Biospherix). Mice were housed in the same room under normoxia (room air, FiO2 21%) or hypoxia (FiO2 10%) for a period of 4 wk. At the completion of the chronic hypoxia protocol, mice underwent harvest for histology and hemodynamic measurement.
Human samples.
Explanted lung tissue was obtained from subjects undergoing lung transplant for IPF and from lungs rejected for transplant from normal controls per the National Institutes of Health Lung Tissue Research Consortium (protocol no. 14-99-0011). n = 10 per group, except in IPF with PH, where n = 4. The protocol for collection of lung tissue samples, and subsequent studies, were approved by the institutional review board at Vanderbilt University and the University of Florida.
Hemodynamic measurements: right ventricular systolic pressure and right ventricle remodeling.
Invasive hemodynamic measurement was conducted, as described in previous studies (66). In brief, mice were given 0.75 mg/g of 2.5% Avertin (a mixture of tert-amyl alcohol and 2,2,2-tribromoethanol; Sigma-Aldrich, St. Louis, MO) to induce anesthesia. Mice were then placed on a heating pad, and systemic blood pressure and pulse were measured via a tail cuff transducer. The internal jugular vein was dissected and catheterized with a 1.4-French pressure catheter (SPR-839; Millar Instruments, Houston, TX), which was advanced into the right ventricle (RV). The right ventricular pressure tracing was then recorded using LabChart Pro v7 software (ADInstruments, Colorado Springs, CO). After completion of the measurements, blood was collected. The heart was then excised with removal of the atria, and the RV and left ventricle (LV) plus septum were isolated for measurement of the Fulton index (RV:LV+S) as previously described (24).
Antibodies.
Antibodies used were as follows: α-smooth muscle actin (α-SMA) rabbit polyclonal (Abcam, Cambridge, MA); β-actin rabbit polyclonal (Abcam); HIF1-α rabbit polyclonal (GeneTex, Irvine, CA); HIF2-α rabbit polyclonal (GeneTex); CTGF rabbit polyclonal (Novus Biologicals, Littleton, CO); von Willebrand Factor (vWF) rabbit polyclonal (Abcam); pimonidazole rabbit polyclonal (Hypoxyprobe, Burlington, MA); β-galactosidase (β-gal) chicken polyclonal (Abcam); CD34 rat monoclonal (BioLegend, San Diego, CA). Secondary fluorescent antibodies were from Jackson Immunoresearch (West Grove, PA).
Histology, Western blot, semiquantitative scoring, and collagen content.
Upon harvest, the left lobe of the lung was inflated and placed in either 10% formalin or 4% PFA followed by sucrose for histological processing, as previously described (12, 57), and the right lobes were snap frozen in liquid nitrogen for RNA and protein processing. Sections were prepared and processed for immunohistochemistry, immunofluorescence staining, and Xgal staining, as previously described (58). Immunostaining was performed for α-SMA to identify muscularized pulmonary vessels, which were then identified and counted per high-powered field, as previously described (30). Western blots were performed on tissue and cell lysates, as previously described (56). Semiquantitative lung fibrosis scoring (34) and hydroxyproline microplate assay were performed, as previously described (33).
Quantitative RT-PCR.
Total RNA was isolated from frozen whole lung tissue and human cell lysate using a RNeasy kit (Qiagen, Valencia, CA) per manufacturer's recommendations, DNase treated and prepared for quantitative RT-PCR. Specific transcript levels for target genes were then determined by normalization to 18s. Values are presented as mean normalized transcript level using the comparative Ct method (2−ΔΔCt).
Cell isolation and culture.
Primary isolates of PMVECs were obtained from transgenic mice and littermate controls, as previously described (67). In brief, cells were prepared from uninjured mice using collagenase type 2 and red blood cell lysis buffer. Endothelium was then isolated by fluorescence-activated cells sorting based on CD31-PECAM-1 expression. To induce endothelial differentiation, sorted cells were plated on gelatin-coated plastic and were cultured in endothelial growth medium (Lonza, Walkersville, MD), until cells were confluent. Cells were then incubated with Alexa Fluor 488-labeled AcDiLDL (Life Technologies, Grand Island, NY), as previously described (29), and positively stained cells were enriched by flow cytometry (35). Cells were then expanded and phenotyped as shown. Human lung PMVECs obtained from explanted lungs rejected for transplant were used as well.
In experiments with hypoxic exposure (O2 1%), cells were placed in a hypoxia cabinet (Coy Lab Products, Grass Lake, MI) for either 6 or 24 h. Transendothelial electrical resistance (TEER) was determined, as per previously described protocol (48). In brief, cells were plated to confluence in transwell chambers, with resistance measured across the endothelial cell monolayers with an epithelial voltohmmeter and STX-2 electrodes (EVOM, World Precision Instruments, Sarasota, FL). A decrease in TEER represents disruption of endothelial cell-cell junctions and increased permeability, and an increased TEER denotes increased monolayer integrity. Relative intracellular calcium concentration was determined via fura-2 AM assay (Molecular Devices, Sunnyvale, CA), as previously described (13). Calcium content of media EGM-2 media was 1.598 mM, or 0.235 g/l of CaCl2·(H2O)2 (verbal communication per Lonza scientific support representative). Cells were transfected with hypoxia response element (HRE)-luciferase construct (Promega, Madison, WI), and HRE activity was determined as described in the literature (27).
Scratch assay, proliferation detection, ELISA, and siRNA transfection.
Primary isolates of PMVECs from both the control and transgenic mice were exposed to 24 h of either normoxia (O2 21%) or hypoxia (O2 1%), with collection of conditioned media (CM). Pulmonary artery smooth muscle cells (PASMCs) were then cultured with 50% CM and 50% smooth muscle cell growth media (Lonza) for 24 h. A scratch assay was performed on 80–90% of confluent cells, as previously described (57), and wound closure was documented at 0, 2, 4, 8, 12, 18, and 24 h. Similarly, PASMCs were plated at 60–70% confluence and exposed to 50% CM to assess for cell proliferation by BrdU assay (Cell Signaling, Danvers, MA), per manufacturer's instructions. CTGF was quantified in CM by ELISA (Kamiya, Seattle, WA).
Negative control siRNA (Life Technologies) and siRNAs targeting HIF1-α, endothelial PAS domain-containing protein (EPAS1; HIF2-α), and CTGF (Life Technologies) were used according to manufacturer's instructions in generation of knockdown CM from PMVECs exposed to 24 h of normoxia (O2 21%) and hypoxia (O2 1%). In brief, control PMVECs were plated at 60–70% confluence and were transfected with siRNAs by Lipofectamine RNAiMAX reagent (Life Technologies). Twenty-four hours posttransfection, cells were cultured in oxygen conditions prior to collecting for CM, RNA, and protein. Cell proliferation was detected as per above using BrdU proliferation assay.
Vascular permeability.
Mice underwent near-infrared spectroscopic imaging to determine degree of vascular permeability in the tissue bed of interest (the lung), according to a previously described protocol (23). In brief, mice were prepared for imaging by having the chest and abdomen shaved 1 day prior to imaging, whereupon they also had injection of 2 nmol/100 μl of AngioSense 750 EX per mouse tail vein (Perkin Elmer, Waltham, MA). The following day, lung fluorescent signal was captured and quantified with normalization to the mouse bladder infrared signal intensity.
Statistics.
Statistical analyses were performed using GraphPad Prism (GraphPad, La Jolla, CA). Differences among groups were assessed using one-way ANOVA or Kruskal-Wallis rank ANOVA. Differences between pairs were assessed using Mann-Whitney U-test. Results are presented as means ± SE. P < 0.05 was considered significant.
RESULTS
HIF1-α and HIF2-α are expressed in the vascular endothelium of patients with IPF and PH and in the lung of bleomycin-treated wild-type mice.
Immunostaining for HIF1-α and HIF2-α was performed in lung sections from control subjects, patients with IPF, and patients with IPF-associated PH. In control lungs, staining for HIF1-α and HIF2-α was absent. In IPF lungs, there was scattered HIF expression throughout the lung parenchyma; however, staining for both proteins was observed within the vascular endothelium in lungs from subjects with IPF and PH (Fig. 1A). In isolated control and IPF PMVECs exposed to hypoxia (O2 1% for 6 h), IPF PMVECs demonstrated an increase in HIF1-α and HIF2-α expression compared with controls (Fig. 1, B and C). Furthermore, using a HRE-luciferase assay, isolated IPF PMVECs also demonstrated an exaggerated response under hypoxic conditions compared with control cells (Fig. 1D).
Fig. 1.

Lungs of patients with idiopathic pulmonary fibrosis (IPF) and pulmonary hypertension (PH) have increased expression of hypoxia-inducible factor 1-α (HIF1-α) and HIF2-α within the vascular endothelium. A: representative images of HIF1-α and HIF2-α immunohistochemical staining in a normal patient, in a patient with IPF, and in a patient with IPF and known PH. Scale bars: 50 μm at ×60 magnification. Arrows point to immunostain positive cells. n = 10 per group, except in IPF with PH where n = 4. Quantitative PCR of whole cell lysates of pulmonary microvascular endothelial cells (PMVECs) from normal subjects and patients with IPF and PH exposed to hypoxia (O2 1%) for 6 h, demonstrating increased mRNA expression of HIF1-alpha (B) and HIF2-α (C). D: normal and IPF PMVECs were transiently transfected with an hypoxia response element (HRE)-luciferase reporter and cotransfected with renilla construct and exposed to hypoxia (FiO2 1%) for 24 h. With hypoxia exposure, HRE-luciferase activity was greater in IPF PMVECs compared with normal PMVECs. n = 4 per group. *P < 0.05.
We next analyzed HIF expression in wild-type C57BL/6J mice after treatment with intraperitoneal bleomycin. Compared with vehicle-treated controls, bleomycin-treated mice had upregulation of HIF1-α and HIF2-α in the vascular endothelium at 33 days (Fig. 2, A and B). HIF expression was low to absent in an endothelial distribution (CD34), in vehicle-exposed mice. To determine whether increased HIF expression correlated with local tissue hypoxia in this model, we injected mice with pimonidazole 3 h prior to harvest, a reagent used as a surrogate to detect cellular hypoxia as it is reduced to reactive intermediates at low oxygen tension (Po2 < 10 mmHg) (61). Pimonidazole immunostaining was detected in the lungs from 1 wk postbleomycin until the 33-day time point, including within the vascular endothelium (Fig. 2C). Together, these studies show increased HIF protein expression and local tissue hypoxia in pulmonary vascular endothelial cells in humans and mice with PH associated with lung fibrosis.
Fig. 2.

Lungs of mice exposed to intraperitoneal bleomycin have increased expression of HIF1-α and HIF2-α within the vascular endothelium. A: representative images of immunohistochemical staining for HIF1-α and HIF2-α in wild-type mice given vehicle (PBS) vs. intraperitoneal bleomycin. Scale bars: 50 μm at ×60 magnification. Arrows point to immunostain positive cells. B: confocal IF images from lung sections of bleomycin-treated mice for HIF1-α or HIF2-α (green), endothelial cell marker CD34 (red), and DAPI (blue). Magnification ×100. C: In vehicle (PBS)-treated lungs, immunostaining for pimonidazole was not detected, while after bleomycin was administered, immunostaining for pimonidazole was noted in multiple cell populations in the lung parenchyma, including alveolar epithelium (arrow in panel at 1 wk) and vascular endothelium (arrow in panel at 33 days). Scale bars: 50 μm at × 60 magnification. Counterstains with methyl green.
Deletion of HIF1-α and HIF2-α in the vascular endothelium is protective against development of PH and RV remodeling.
To eliminate HIF signaling, we created a model of conditional deletion of both HIF1-α and HIF2-α in the vascular endothelium using VE-cadherin promoter-driven Cre. Transgenic mice (VECad.Cre, HIF1-alphafl/fl, HIF2-alphafl/fl, and Rosa.Stop.lacZ) were crossed to generate the desired model (VECad.Cre.HIF1-alphafl/fl.HIF2-alphafl/fl.ROSA.Stop.lacZ) (Fig. 3A). Our breeding colony strategy is such that the Cre construct is maintained in the heterozygous state, while the HIF1-α, HIF2-α, and Rosa.Stop.lacZ constructs are homozygous. Thus, 50% of the pups in a litter are gene targeted and the other 50% serve as littermate controls. Mice with endothelial targeted HIF1-α and HIF2-α deletion [endothelial HIF1/2 KO mice (HIF1/2 KO) and littermates (Control)] that were the wild type for HIF1-α and HIF2-α (without VECad.Cre) were born in expected ratios (50% each) and grew normally into adulthood with no evidence of aberrant effects on growth or reproductive capacity. To verify endothelial cell recombination we evaluated β-gal expression as a marker of Cre-recombination in lung sections and in isolated vascular endothelial cells from transgenic mice. In lung sections, Xgal staining was prominent in endothelial cells from the large blood vessels down to the capillaries (Fig. 3B). In addition, PMVECS isolated from the endothelial HIF-targeted mice demonstrated strong dual immunofluorescence for the endothelial cell marker vWF and the β-gal reporter, a pattern not seen with the littermate controls (Fig. 3C). Effective HIF1/2 deletion was corroborated by immunohistochemical staining in mice undergoing intraperitoneal bleomycin, showing absence of HIF1-α and HIF2-α staining in endothelial HIF1/2 KO samples (Fig. 3D). Furthermore, in primary isolated PMVECs exposed to 6 h of hypoxia (O2 1%), control cells demonstrated an increase in both HIF1-α and HIF2-α expression, with minimal expression within the HIF1/2 KO cells (Fig. 3, E and F), indicative of efficient gene deletion.
Fig. 3.

Development of transgenic model for deficiency of HIF1-α and HIF2-α within vascular endothelium. A: schematic illustrating the combination of four individual transgenic mice resulting in the desired model. B: Xgal staining in a lung section from an adult VECad.Cre.HIF1-αfl/fl.HIF2-αfl/fl.ROSA.Stop.lacZ (HIF1/2 KO) mouse. Endothelial cells from the large vessels down to the capillaries (lung parenchyma) stain blue. ×20 magnification. C: confocal microscopic images on isolated pulmonary microvascular endothelial cells (PMVECs) from the gene-targeted mice [VECad.Cre positive (HIF1/2 KO)] demonstrated dual immunofluorescence for the endothelial cell marker vWF and the reporter system-based β-galactosidase (β-gal), a pattern not seen in PMVECs from the littermate controls [VECad.Cre negative (Control)]. D: immunohistochemical staining for HIF1-α and HIF2-α in vehicle (PBS) and bleomycin-exposed mice. Vascular endothelial cells were immunostained positive for HIF1-α and HIF2-α at 3 wk after bleomycin administration in control mice (arrowheads), but not in HIF1/2 KO mice. Scale bars 50 μm at ×60 magnification. Counterstain with methyl green. E: quantitative PCR of PMVEC lysates for HIF1-α and HIF2-α, under hypoxic conditions (1% O2 for 6 h), demonstrating a marked reduction in expression in the HIF-targeted mice. n = 3 per group, *P < 0.05. F: Western blot for HIF1-α from these same PMVECs demonstrated 40% increased blotting intensity of HIF1-α in hypoxia in control cells compared with HIF1/2 KO cells. Immunoblot for HIF1-α is shown with β-actin for loading control.
Endothelial HIF1/2 KO mice and controls, whether Cre- littermates of the endothelial targeted mice or VECad.Cre.ROSA.Stop.lacZ, had normal lung architecture after treatment with vehicle (PBS) and a similar degree of fibrosis after bleomycin treatment, as assessed by lung microscopy with semiquantitative fibrosis scoring of the lung sections (Fig. 4, A and B). Similarly, hydroxyproline measurements revealed no significant difference between groups (control 205 ± 23 μg and HIF1/2 KO 247 ± 16 μg, n = 18 per group) (Fig. 4C). Upon assessment of pulmonary hemodynamics, control mice treated with bleomycin were found to have an elevated right ventricular systolic pressure (RVSP) (26.9 ± 1.2 mmHg). In contrast, RVSP was normalized in the HIF1/2 KO group (22.2 ± 1.0 mmHg, P < 0.05, n = 18 per group) (Fig. 5A). In addition, RV remodeling was not observed in bleomycin-treated HIF1/2 KO mice, as assessed by the Fulton Index (RV:LV+S), with control mice displaying increased RV mass by percentage (29.0 ± 6.5%) compared with HIF1/2 KO (14.5 ± 1.6%) (P < 0.05, n = 18 per group) (Fig. 5B). After staining of lung sections for α-SMA to assess for vascular remodeling, and confirming normal vascular architecture within the vehicle-exposed mice, muscularized pulmonary vessel counts revealed a dramatic decrease in small and medium completely muscularized vessels between lungs of bleomycin-treated HIF1/2 KO mice compared with bleomycin-treated controls (*P < 0.05, n = 18 per group) (Fig. 5, C–E). There was no significant difference in systemic oxygen saturation by tail pulse oximetry in either control or transgenic mice exposed to bleomycin (average SpO2 of 92%, for both groups) or vehicle (average SpO2 of 99% for both groups; data not shown).
Fig. 4.

Mice with deletion of HIF1-α and HIF2-α in vascular endothelium are not protected against the development of bleomycin-associated pulmonary fibrosis. A: representative images of Masson trichrome-stained lung sections, demonstrating similar degrees of bleomycin-induced pulmonary fibrosis between controls and HIF1/2 KO mice. With the intraperitoneal bleomycin model, lung fibrosis is predominantly peripheral and subpleural. Scale bars: 100 μm at ×20 magnification. B: quantification of degree of fibrosis by scoring on lung sections, with no difference between control and HIF1/2KO mice after bleomycin administration. C: collagen content quantification as determined by microplate hydroxyproline assay of the right lower lobe, showing no difference between control and HIF1/2 KO bleomycin-exposed mice. n = 18 per group.
Fig. 5.

Deletion of HIF1-α and HIF2-α within vascular endothelium is protective against the development of pulmonary hypertension and cardiac remodeling, with evidence of decreased pulmonary vessel remodeling in a model of pulmonary fibrosis. A: right ventricular systolic pressure (RVSP) increased following bleomycin exposure in littermate controls, while RVSP was similar to baseline measures in bleomcyin-treated endothelial HIF1/2 KO mice. B: right-ventricular remodeling as determined by Fulton Index (RV:LV+S, %) was prominent following bleomycin exposure in littermate controls, while such remodeling was prevented in bleomycin-treated endothelial HIF1/2 KO mice. C: representative images of lung sections with α-smooth muscle actin (α-SMA; representative vessel, highlighted by arrowhead) immunostaining for muscularized pulmonary vessel counting. Vessels shown are in the peripheral alveolar parenchyma. Scale bars: 50 μm at ×60 magnification. Counterstain with hematoxylin. D: Small and medium-sized muscularized pulmonary vessels were equivalent in vehicle-treated mice, suggesting normal vascular morphometric development in transgenic mice vs. controls. E: however, small and medium muscularized vessels were increased after bleomcyin administration in the lungs of control littermates compared with endothelial HIF1/2 KO mice. n= 18 per group. *P < 0.05.
Separately, we exposed endothelial HIF1/2 KO and control mice to 4 wk of chronic hypoxia (FiO2 10%). Similar to the bleomycin model, the hypoxia-induced increase in RVSP was blunted in endothelial HIF1/2 KO mice compared with controls (27.0 ± 2.6 mmHg vs. 32.5 ± 3.4 mmHg, respectively; n = 18 per group, P < 0.05) (Fig. 6A). HIF1/2 KO mice had a marked reduction in RV remodeling following exposure to hypoxia, as determined by the Fulton Index (28.6 ± 3.0% in controls compared with 18.3 ± 2.1% in HIF1/2 KO mice) (n = 18 per group, P < 0.05) (Fig. 6B). As in the bleomycin model, mice without HIF expression displayed a decrease in muscularized vessel count, primarily in small and completely muscularized vessels (Fig. 6C). Together, these studies indicate that HIF signaling in endothelial cells is a critical pathway for regulating pulmonary vascular and RV remodeling.
Fig. 6.

Deletion of HIF1-α and HIF2-α within vascular endothelium is protective against the development of pulmonary hypertension and cardiac remodeling in a model of chronic hypoxia. A: RVSP increased to a greater degree with hypoxia exposure in littermate controls compared with endothelial HIF1/2 KO mice. B: right-ventricular remodeling was more prominent with hypoxia exposure in littermate controls compared with endothelial HIF1/2 KO mice, which had an index similar to baseline values. C: consistent with this finding, small pulmonary vessels displayed increased evidence of complete muscularization in control animals exposed to hypoxia, vs. HIF1/2 KO mice. n = 18 per group, *P < 0.05.
In vitro studies on isolated pulmonary microvascular endothelial cells suggest multiple pathways by which endothelial cell HIF expression regulates development of PH.
CTGF is a known downstream target of HIF pathway activation (10) that is integral in ECM remodeling and has been implicated in the development of perivascular remodeling in other forms of vascular disease (43). Therefore, we hypothesized that CTGF would be decreased in our animal model, and increased in human samples of lung disease. In PMVECs isolated from endothelial HIF1/2 KO mice, CTGF expression was markedly diminished with exposure to hypoxia compared with controls (Fig. 7, A and B). Furthermore, in PMVECs isolated from control and IPF lung and exposed to hypoxia (O2 1% for 6 h), IPF PMVECs demonstrated an increase in CTGF expression compared with control cells (Fig. 7, C and D). In addition, increased CTGF was detected by immunohistochemical staining in lung sections from patients with IPF with PH compared with control lungs from normal subjects and those with IPF without PH (Fig. 7E). These results suggest that PMVEC HIF activation and CTGF expression may play a role in vascular remodeling in IPF with PH.
Fig. 7.

Connective tissue growth factor (CTGF) expression is decreased in pulmonary microvascular endothelial cells (PMVECs) isolated from transgenic HIF1/2 KO mice. Conversely, CTGF expression is increased in primary human PMVECs isolated from a patient with IPF exposed to hypoxia. A: by quantitative PCR on cell lysates, CTGF mRNA expression increased dramatically in control PMVECs with exposure to hypoxia (O2 1% for 24 h) compared with PMVECs from endothelium-targeted mice, confirmed at the protein level by immunoblot on whole cell lysates for CTGF (B), demonstrating a reduction in CTGF in HIF1/2 KO compared with the hypoxic control. β-actin shown as loading control. C: quantitative PCR of whole cell lysates of PMVECs from normal subjects and patients with IPF and PH exposed to hypoxia (O2 1%) for 6 h, demonstrating increased mRNA expression of CTGF in IPF PMVECs compared with normal control. D: by immunoblot of whole cell lysates collected after exposure to hypoxia, CTGF expression was increased in IPF PVMECs compared with normal PMVECs. β-actin shown for loading control. E: representative images from lung sections with immunohistochemical staining for CTGF from normal subjects with no immunostain observed, diffuse parenchymal staining in samples from patients with IPF alone, and in patients with IPF and PH, demonstrating immunostain-positive cells in the lung, including the vascular endothelium (arrowheads points to representative cell). Scale bars: 50 μm at ×60 magnification. n = 4 per group for in vitro experiments. *P < 0.05. n = 10 per group for immunohistochemistry, except in IPF + PH where n = 4.
PASMCs are a key cell type of interest in the pathogenesis of PH (60), and given our in vivo findings above, we wanted to explore whether endothelial cell HIF signaling might have an impact on PASMCs. For these studies, we exposed wild-type murine PASMCs to CM from either HIF1/2 KO or control PMVECs cultured for 24 h in normoxia or hypoxia (O2 1%). PASMCs exposed to hypoxic CM from HIF1/2 KO PMVECs had a decrease in scratch closure by 18 and 24 h, compared with control PMVEC hypoxic CM (Fig. 8A), as well as a corresponding decrease in cell proliferation by BrdU assay (Fig. 8B). Interestingly, and building on our observations above, these findings correlated with CTGF levels in the CM, with transgenic HIF1/2 KO PMVECs producing a decreased amount of CTGF (Fig. 8C). To explore these relationships further, we used siRNA to knockdown expression of HIF1-α, HIF2-α, and CTGF (vs. nontargeted siRNA) in primary isolated PMVECs from wild-type mice, collecting CM from these targeted cells after culture for 24 h under both normoxic and hypoxic (O2 1%) conditions (Fig. 8, D and E). PASMCs exposed to hypoxic CM from HIF siRNA-targeted cells had a decrease in scratch assay closure (both HIF1-α and HIF2-α targeted) (Fig. 8F) and a decrease in proliferation (HIF2-α) (Fig. 8G) compared with PASMCs exposed to hypoxic nontarget siRNA control-treated CM. Similar to the HIF1/2 KO PMVECs, there was a corresponding decrease in CTGF concentration within the CM under both normoxic and hypoxic conditions with HIF1-α or HIF2-α siRNA knockdown (Fig. 8H). Using a similar approach with studies of siRNA knockdown of CTGF in PMVECs, PASMCs exposed to CM from CTGF siRNA targeted PMVECs had decreased wound closure and proliferation (Fig. 8, I and J). As expected with siRNA knockdown, CM from CTGF siRNA-targeted cells had a decrease in CTGF concentration, although it was not eliminated entirely (Fig. 8K). Taken together, these in vitro studies demonstrate that endothelial cell HIF signaling may have the potential to impact adjacent PASMCs and that the CTGF axis may be involved in this process as well.
Fig. 8.
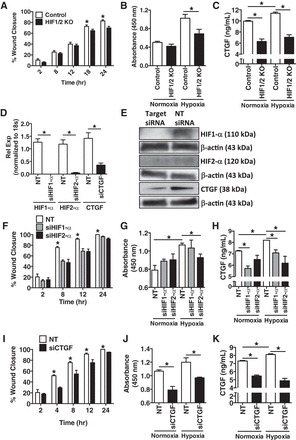
Exposure of pulmonary artery smooth muscle cells (PASMCs) to hypoxic conditioned media (CM) from primary isolated pulmonary microvascular endothelial cells (PMVECs) of control and HIF1/2 KO mice demonstrates a decrease in PASMC wound healing and cellular proliferation, to a similar degree as individual knockdown of HIF1-α, HIF2-α, and connective tissue growth factor (CTGF). A and B: murine PASMC exposure to hypoxia CM (O2 1%, 24 h) from primary isolated murine PMVECs demonstrates decreased wound healing by scratch assay (A) and decreased proliferation by BrdU assay (B) in HIF1/2 KO vs. control conditions. C: CM from HIF1/2 KO PMVECs exposed to hypoxia had a lower concentration of CTGF compared with control cells. D and E: Individually, HIF1-α, HIF2-α, and CTGF were knocked down in wild-type murine PMVECs by siRNA, with a decrease in expression by both real-time PCR (D) and immunoblot (E), compared with nontarget (denoted as NT, in figure) siRNA control. F–H: similar to above, PASMC exposure to CM from PMVECs with siRNA knockdown of either HIF1-α or HIF2-α, vs. nontarget (NT) siRNA, displayed a decrease in PASMC wound closure (both HIF1α and HIF2-α) (F) and PASMC proliferation rate (HIF2-α) (G). H: CTGF concentration in the PMVEC CM was lower with both HIF1-α and HIF2-α siRNA targeting. I–K: PASMC exposure to CM from PMVECs with siRNA knockdown of CTGF vs. nontarget (NT) siRNA, displayed a decrease in PASMC wound closure (I) and PASMC proliferation rate (J). K: CTGF concentration in the PMVEC CM was lower with CTGF siRNA targeting, but was not completely eliminated. n = 4 per group. *P <0.05.
Notably, HIF signaling in PASMCs influences PH development through alteration of transmembrane ion movement (37) via suppression of the potassium channel Kv1.5 (38). Moreover, Kv1.5 protein is known to be integral in the development of oxidative vascular endothelial cell injury (11). Given these facts, along with the results of Kv1.5 expression from our exploratory microarray study, we examined Kv1.5 expression in hypoxia-exposed PMVECs and found an increase in expression in HIF1/2 KO PMVECs exposed to hypoxia (Fig. 9A). Since increased potassium channel expression reduces cell layer permeability by preventing the disruption of tight junctions (5), we examined electrical resistance across a confluent monolayer of PMVECs using a TEER assay. We found that resistance was increased in the HIF1/2 KO cell monolayer (3.5 ± 0.2 ohm·cm2) compared with control (2.2 ± 0.2 ohm·cm2, n = 4 per group, P < 0.05), consistent with tighter cell-cell adhesion and decreased permeability (Fig. 9B). Increases in cell permeability and conversely decreases in TEER have been associated with reduced resting membrane potential and increased sarcolemmal calcium release (20). Therefore, we hypothesized that HIF deletion in PMVECs would result in a decrease in intracellular calcium compared with control cells, accounting for the decreased permeability. Upon exposure to hypoxia (O2 1%) for 24 h and measurement of bound vs. unbound calcium, HIF1/2 KO PMVECs demonstrated a marked reduction in intracellular calcium concentration compared with control PMVECs (Fig. 9C). Providing an in vivo confirmation of the finding of vascular endothelial barrier compromise, a decrease in vascular permeability was seen through near-infrared spectroscopic imaging of endothelial HIF1/2 KO mice compared with controls when analyzed at 2 wk in the intraperitoneal bleomycin protocol (Fig. 9, D and E). These results suggest that endothelial HIF signaling may impact vascular permeability and subsequently be a critical contributor to vascular remodeling in PH.
Fig. 9.

Primary isolated murine pulmonary microvascular endothelial cells (PMVECs) with endothelial HIF deletion have decreased monolayer permeability and increased potassium channel (Kv1.5) expression. A: by quantitative PCR on cell lysates, PMVECs from endothelial HIF-targeted mice had an increase in Kv1.5 channel mRNA expression in response to hypoxia compared with controls. B: PMVECs from endothelial HIF-targeted mice had an increased transcellular resistance as measured by TEER after exposure to hypoxia (O2 1%) for 24 h, compared with control cells under normoxic condition. C: relative intracellular calcium concentration, as measured by fura-2 AM, was decreased in HIF1/2 KO PMVECs compared with control, upon exposure to 24 h of hypoxia (O2 1%). For all experiments; n = 3 per group. D: representative vascular leak images, as measured by Angiosense tissue permeability, of control and endothelial HIF1/2 KO mice exposed to vehicle vs. intraperitoneal bleomycin for 2 wk. Head to the left and tail to the right of each image. Green color is indicative of vascular leak and is greatest over the chest and normalized to bladder. E: by quantitation of individual images, and normalized leak intensity (corresponding to increased pulmonary permeability) was greater in control vs. endothelial HIF1/2 KO mice, exposed to bleomycin. n = 6 per group, *P < 0.05.
DISCUSSION
In this study, we have shown that vascular endothelial hypoxic signaling, through HIF, is necessary for the development of PH in both bleomycin-induced pulmonary fibrosis and chronic hypoxia exposure. Importantly, PH development in the bleomycin model was not associated with a change in the degree of parenchymal fibrosis. Although the most striking finding from our studies was that endothelial cell HIF expression regulated the development of PH in vivo, we also explored possible mechanisms by which this occurs. Our cell culture studies point to endothelial cell HIF signaling as being important for CTGF expression, vascular permeability, and endothelium-smooth muscle cell interactions that could promote vascular remodeling. At this point, these in vitro studies are suggestive of mechanisms at play with endothelial cell HIF signaling, but future in vivo studies will be necessary to build on and confirm these initial observations. Together, these findings represent a novel area of research in the treatment of PH, targeting both the vascular endothelium and hypoxic signaling (21).
Consideration of our work in conjunction with other reports in the literature highlights the complexity of interactions between multiple cell types necessary for the development of HIF-mediated PH. The majority of the literature focuses on the role of the PASMC as the main cell type in disease pathogenesis, be it through downstream mediators of HIF signaling (59, 62) or through direct alteration of PASMC apoptosis resistance, enhancing cell proliferation and migration (3). Although the endothelium has been implicated in the chronic hypoxia model of HIF stabilization and signaling (1, 41), our mouse model allows for a specific examination of the role that endothelial HIF signaling plays in the development of PH secondary to fibrosis, with our in vitro studies suggesting that endothelium and pulmonary artery smooth muscle cell-to-cell communication may contribute to the development of PH.
The mechanism of HIF-mediated PH has been shown previously to involve direct and indirect effects on cell ion exchange in the PASMC. For example, HIF is known to influence endothelial cell proliferation and apoptotic resistance, through disruption of mitochondrial and metabolic homeostasis (16). Additionally, examination of fawn-hooded rats showed that an alteration in potassium channel generation and function by HIF (specifically, Kv1.5) contributed to PASMC remodeling (6). This alteration in transmembrane potential is hypothesized to result in an increase in PASMC intracellular calcium flux and tonic vasoconstriction and remodeling of the vasculature (63). Given this known information in PASMCs, we sought to explore whether this same mechanism of alteration in ion transport could possibly play a role in the PMVEC response to hypoxia. Our in vitro studies, examining both ion flux and TEER related to preserved endothelial cell barrier function (36), highlight our findings that there is decreased vascular permeability in our transgenic mouse model upon exposure to bleomycin. This is important, as endothelial barrier dysfunction is known to contribute to vascular remodeling associated with PAH (19). Thus, our data from studies in PMVECs support a role of HIF in the regulation of potassium channel expression and function, calcium flux, and cell monolayer permeability in the vascular endothelium, in a similar manner to PASMCs (54).
Downstream regulation of potassium and calcium channels is only one of the HIF-related mechanisms of PH development that is demonstrated within endothelial cells. A candidate protein in the pathogenesis of fibrosis associated PH is CTGF, a part of the CCN (Cyr61, Ctgf, Nov) family of matricellular regulatory factors that are involved in extracellular signaling. In particular, CTGF (or CCN2) promotes both endothelial adhesion and survival, as well as promoting profibrotic cofactors, such as transforming growth factor-β (TGF-β) (42). Previous work has shown that HIF regulates CTGF via the HRE response (25, 47) and that CTGF has been shown to contribute to lung fibrosis through increased expression of collagen subtypes in a bleomycin-fibrosis model (44). Although our in vitro observations are intriguing, the mechanistic role of endothelial CTGF signaling in the development of Group III PH merits further investigation, including future in vivo studies.
Extracellular matrix remodeling pathways as a potential source of intervention in PH is interesting in light of a recent study that demonstrated, in a rabbit model of pulmonary artery banding, adverse ventricular remodeling induced by isolated RV afterload increase, which was mediated through a TGF-β/CTGF mechanism (18). Interestingly, other TGF-β mediators, such as Twist 1, have been shown to be overexpressed involved in the pathobiology of PAH development and progression (46). With respect to this effect of cardiac remodeling, our data are consistent with a prior study that showed endothelial initiated fibroblast activation as being necessary for the development of cardiomyopathy (68). This may partially explain the difference in findings of RV remodeling in our study compared with a recently published article that examined HIF deletion within smooth muscle cells using an inducible Cre-system (3). Additionally, HIF overexpression in the vascular endothelium has been shown to be protective against the development of cardiac remodeling in mice (64), suggesting the pathophysiology of HIF in the heart is likely much more complex than currently hypothesized.
Of interest, prior work has demonstrated that mice with endothelial cell-specific deletion of HIF develop an age-related increase in vessel permeability (“leakiness”), and associated spontaneous PH (54), a finding seemingly at odds with the conclusion of this study. It is important to note that in the study by Skuli et al. (54), the average age of mice were 9 mo of age and older, while mice reported in this article were on average 4–5 mo of age or younger. In addition, that group examined HIF2-α deletion specific to vascular endothelial cells, whereas our study looked at both HIF1 and HIF2 genetic knockout. Future research should explore the difference between HIF homologs in the vascular endothelium, given the widely disparate response of some cells to stabilization of HIF1-α vs. HIF2-α, such as in macrophage-mediated angiogenesis (14).
Thus, our study demonstrates that the vascular endothelium regulates the development of PH through HIF signaling pathways, and our results suggest multiple potential mechanistic targets that warrant future study to better understand this relationship. The critical role of HIF and HRE targets in the development of both pathologic vascular and cardiac remodeling in PH should prompt further investigation into the use of pharmacologic inhibitors that are already in various stages of development (17) for use in this patient population without any alternatives for treatment.
GRANTS
The study was funded by Grants from the National Institutes of Health (NIH) National Heart, Lung, and Blood Institute HL-095797 (to J. D. West), HL-105479 (to W. E. Lawson), HL-85317 (to T. S. Blackwell), HL-92870 (to T. S. Blackwell), HL-87738 (to A. J. Bryant); P30 AG028740 (to A. J. Bryant); NIH NCRR UL1 RR-024975 and NCRR UL1 TR000064 (to A. J. Bryant); American Thoracic Society/Pulmonary Hypertension Association Fellowship Research Grant (to A. J. Bryant); Gatorade Fund (to A. J. Bryant); and the Department of Veterans Affairs BX001988 (to W. E. Lawson), and BX002378 (to T. S. Blackwell).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.J.B., R.P.C., E.J.C., S.M.M., J.P.F., V.H.H., J.D.W., T.S.B., and W.E.L. conception and design of research; A.J.B., R.P.C., M.E.M., B.R.J., S.D.S., C.S.M., T.R.B., S.G., N.L.P., A.B., H.T., A.K.K., M.A.T., L.A.G., E.J.C., C.G., S.M.M., J.P.F., J.D.W., T.S.B., and W.E.L. performed experiments; A.J.B., R.P.C., S.D.S., N.L.P., A.B., V.V.P., M.A.T., H.-J.D., E.J.C., C.G., S.M.M., J.P.F., V.H.H., J.D.W., T.S.B., and W.E.L. analyzed data; A.J.B., R.P.C., A.R.H., H.-J.D., L.A.G., E.J.C., E.W.S., S.M.M., J.P.F., V.H.H., J.D.W., T.S.B., and W.E.L. interpreted results of experiments; A.J.B., J.D.W., T.S.B., and W.E.L. prepared figures; A.J.B. and W.E.L. drafted manuscript; A.J.B., R.P.C., S.D.S., H.T., A.R.H., V.V.P., M.A.T., C.G., E.W.S., S.M.M., J.P.F., V.H.H., J.D.W., T.S.B., and W.E.L. edited and revised manuscript; A.J.B., E.W.S., J.P.F., J.D.W., T.S.B., and W.E.L. approved final version of manuscript.
ACKNOWLEDGMENTS
The excellent professional assistance of John H. Newman M.D. is greatly appreciated. This work was supported by the Gatorade Trust through funds distributed by the University of Florida, Department of Medicine. The research is also supported by the Claude D. Pepper Older Americans Independence Centers at the University of Florida (1 P30 AG028740) and Clinical and Translational Science Institute [National Center for Research Resources (NCRR) UL1TR000064]. In addition, this study utilized biological specimens and data provided by the Lung Tissue Research Consortium (LTRC) supported by the National Heart, Lung, and Blood Institute (NHLBI).
REFERENCES
- 1.Ahmad A, Ahmad S, Malcolm KC, Miller SM, Hendry-Hofer T, Schaack JB, White CW. Differential regulation of pulmonary vascular cell growth by hypoxia-inducible transcription factor-1α and hypoxia-inducible transcription factor-2α. Am J Respir Cell Mol Biol 49: 78–85, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alva JA, Zovein AC, Monvoisin A, Murphy T, Salazar A, Harvey NL, Carmeliet P, Iruela-Arispe ML. VE-Cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn 235: 759–767, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Ball MK, Waypa GB, Mungai PT, Nielsen JM, Czech L, Dudley VJ, Beussink L, Dettman RW, Berkelhamer SK, Steinhorn RH, Shah SJ, Schumacker PT. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1α. Am J Respir Crit Care Med 189: 314–324, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baran CP, Opalek JM, McMaken S, Newland CA, O'Brien JM Jr, Hunter MG, Bringardner BD, Monick MM, Brigstock DR, Stromberg PC, Hunninghake GW, Marsh CB. Important roles for macrophage colony-stimulating factor, CC chemokine ligand 2, and mononuclear phagocytes in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med 176: 78–89, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bohr DF, Webb RC. Vascular smooth muscle function and its changes in hypertension. Am J Med 77: 3–16, 1984. [DOI] [PubMed] [Google Scholar]
- 6.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1α-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 113: 2630–2641, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Boutou AK, Pitsiou GG, Trigonis I, Papakosta D, Kontou PK, Chavouzis N, Nakou C, Argyropoulou P, Wasserman K, Stanopoulos I. Exercise capacity in idiopathic pulmonary fibrosis: the effect of pulmonary hypertension. Respirology 16: 451–458, 2011. [DOI] [PubMed] [Google Scholar]
- 8.Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, Carmeliet P. Heterozygous deficiency of hypoxia-inducible factor-2α protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest 111: 1519–1527, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burton VJ, Ciuclan LI, Holmes AM, Rodman DM, Walker C, Budd DC. Bone morphogenetic protein receptor II regulates pulmonary artery endothelial cell barrier function. Blood 117: 333–341, 2011. [DOI] [PubMed] [Google Scholar]
- 10.Capparelli C, Whitaker-Menezes D, Guido C, Balliet R, Pestell TG, Howell A, Sneddon S, Pestell RG, Martinez-Outschoorn U, Lisanti MP, Sotgia F. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle 11: 2272–2284, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen WL, Huang XQ, Zhao LY, Li J, Chen JW, Xiao Y, Huang YY, Liu J, Wang GL, Guan YY. Involvement of Kv1.5 protein in oxidative vascular endothelial cell injury. PLoS One 7: e49758, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Degryse AL, Tanjore H, Xu XC, Polosukhin VV, Jones BR, Boomershine CS, Ortiz C, Sherrill TP, McMahon FB, Gleaves LA, Blackwell TS, Lawson WE. TGFβ signaling in lung epithelium regulates bleomycin-induced alveolar injury and fibroblast recruitment. Am J Physiol Lung Cell Mol Physiol 300: L887–L897, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dikalov SI, Li W, Doughan AK, Blanco RR, Zafari AM. Mitochondrial reactive oxygen species and calcium uptake regulate activation of phagocytic NADPH oxidase. Am J Physiol Regul Integr Comp Physiol 302: R1134–R1142, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eubank TD, Roda JM, Liu H, O'Neil T, Marsh CB. Opposing roles for HIF-1α and HIF-2α in the regulation of angiogenesis by mononuclear phagocytes. Blood 117: 323–332, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farkas L, Farkas D, Ask K, Moller A, Gauldie J, Margetts P, Inman M, Kolb M. VEGF ameliorates pulmonary hypertension through inhibition of endothelial apoptosis in experimental lung fibrosis in rats. J Clin Invest 119: 1298–1311, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fijalkowska I, Xu W, Comhair SA, Janocha AJ, Mavrakis LA, Krishnamachary B, Zhen L, Mao T, Richter A, Erzurum SC, Tuder RM. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol 176: 1130–1138, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fraisl P, Aragones J, Carmeliet P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat Rev Drug Discov 8: 139–152, 2009. [DOI] [PubMed] [Google Scholar]
- 18.Friedberg MK, Cho MY, Li J, Assad RS, Sun M, Rohailla S, Honjo O, Apitz C, Redington AN. Adverse biventricular remodeling in isolated right ventricular hypertension is mediated by increased TGFβ1 signaling and is abrogated by angiotensin receptor blockade. Am J Respir Cell Mol Biol 49: 1019–1028 2013. [DOI] [PubMed] [Google Scholar]
- 19.Good RB, Gilbane AJ, Trinder SL, Denton CP, Coghlan G, Abraham DJ, Holmes AM. Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary arterial hypertension. Am J Pathol 185: 1850–1858, 2015. [DOI] [PubMed] [Google Scholar]
- 20.Greenspon J, Li R, Xiao L, Rao JN, Sun R, Strauch ED, Shea-Donohue T, Wang JY, Turner DJ. Sphingosine-1-phosphate regulates the expression of adherens junction protein E-cadherin and enhances intestinal epithelial cell barrier function. Dig Dis Sci 56: 1342–1353, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guignabert C, Tu L, Girerd B, Ricard N, Huertas A, Montani D, Humbert M. New molecular targets of pulmonary vascular remodeling in pulmonary arterial hypertension: importance of endothelial communication. Chest 147: 529–537, 2015. [DOI] [PubMed] [Google Scholar]
- 22.Hamada K, Nagai S, Tanaka S, Handa T, Shigematsu M, Nagao T, Mishima M, Kitaichi M, Izumi T. Significance of pulmonary arterial pressure and diffusion capacity of the lung as prognosticator in patients with idiopathic pulmonary fibrosis. Chest 131: 650–656, 2007. [DOI] [PubMed] [Google Scholar]
- 23.Hassan M, Riley J, Chernomordik V, Smith P, Pursley R, Lee SB, Capala J, Gandjbakhche AH. Fluorescence lifetime imaging system for in vivo studies. Mol Imag 6: 229–236, 2007. [PMC free article] [PubMed] [Google Scholar]
- 24.Hemnes AR, Brittain EL, Trammell AW, Fessel JP, Austin ED, Penner N, Maynard KB, Gleaves L, Talati M, Absi T, Disalvo T, West J. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med 189: 325–334, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Higgins DF, Biju MP, Akai Y, Wutz A, Johnson RS, Haase VH. Hypoxic induction of Ctgf is directly mediated by Hif-1. Am J Physiol Renal Physiol 287: F1223–F1232, 2004. [DOI] [PubMed] [Google Scholar]
- 26.Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest 117: 3810–3820, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirose Y, Johnson ZI, Schoepflin ZR, Markova DZ, Chiba K, Toyama Y, Shapiro IM, Risbud MV. FIH-1-Mint3 axis does not control HIF-1α transcriptional activity in nucleus pulposus cells. J Biol Chem 289: 20,594–20,605, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Idiopathic Pulmonary Fibrosis Clinical Research Network, Zisman DA, Schwarz M, Anstrom KJ, Collard HR, Flaherty KR, Hunninghake GW. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med 363: 620–628, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Irwin D, Helm K, Campbell N, Imamura M, Fagan K, Harral J, Carr M, Young KA, Klemm D, Gebb S, Dempsey EC, West J, Majka S. Neonatal lung side population cells demonstrate endothelial potential and are altered in response to hyperoxia-induced lung simplification. Am J Physiol Lung Cell Mol Physiol 293: L941–L951, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Karmouty-Quintana H, Zhong H, Acero L, Weng T, Melicoff E, West JD, Hemnes A, Grenz A, Eltzschig HK, Blackwell TS, Xia Y, Johnston RA, Zeng D, Belardinelli L, Blackburn MR. The A2B adenosine receptor modulates pulmonary hypertension associated with interstitial lung disease. FASEB J 26: 2546–2557, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel NF. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest 106: 1311–1319, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kimura M, Taniguchi H, Kondoh Y, Kimura T, Kataoka K, Nishiyama O, Aso H, Sakamoto K, Hasegawa Y. Pulmonary hypertension as a prognostic indicator at the initial evaluation in idiopathic pulmonary fibrosis. Respiration 85: 456–463, 2013. [DOI] [PubMed] [Google Scholar]
- 33.Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, Newcomb DC, Jones BR, Roldan J, Lane KB, Morrisey EE, Beers MF, Yull FE, Blackwell TS. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci USA 108: 10,562–10,567, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lawson WE, Polosukhin VV, Stathopoulos GT, Zoia O, Han W, Lane KB, Li B, Donnelly EF, Holburn GE, Lewis KG, Collins RD, Hull WM, Glasser SW, Whitsett JA, Blackwell TS. Increased and prolonged pulmonary fibrosis in surfactant protein C-deficient mice following intratracheal bleomycin. Am J Pathol 167: 1267–1277, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Majka S, Hagen M, Blackwell T, Harral J, Johnson JA, Gendron R, Paradis H, Crona D, Loyd JE, Nozik-Grayck E, Stenmark KR, West J. Physiologic and molecular consequences of endothelial Bmpr2 mutation. Respir Res 12: 84, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Makarenko VV, Usatyuk PV, Yuan G, Lee MM, Nanduri J, Natarajan V, Kumar GK, Prabhakar NR. Intermittent hypoxia-induced endothelial barrier dysfunction requires ROS-dependent MAP kinase activation. Am J Physiol Cell Physiol 306: C745–C752, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mandegar M, Remillard CV, Yuan JX. Ion channels in pulmonary arterial hypertension. Prog Cardiovasc Dis 45: 81–114, 2002. [DOI] [PubMed] [Google Scholar]
- 38.Mandegar M, Yuan JX. Role of K+ channels in pulmonary hypertension. Vasc Pharmacol 38: 25–33, 2002. [DOI] [PubMed] [Google Scholar]
- 39.Nathan SD, Shlobin OA, Ahmad S, Koch J, Barnett SD, Ad N, Burton N, Leslie K. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respiration 76: 288–294, 2008. [DOI] [PubMed] [Google Scholar]
- 40.Olschewski H, Ghofrani HA, Walmrath D, Schermuly R, Temmesfeld-Wollbruck B, Grimminger F, Seeger W. Inhaled prostacyclin and iloprost in severe pulmonary hypertension secondary to lung fibrosis. Am J Respir Crit Care Med 160: 600–607, 1999. [DOI] [PubMed] [Google Scholar]
- 41.Palmer LA, Semenza GL, Stoler MH, Johns RA. Hypoxia induces type II NOS gene expression in pulmonary artery endothelial cells via HIF-1. Am J Physiol Lung Cell Mol Physiol 274: L212–L219, 1998. [DOI] [PubMed] [Google Scholar]
- 42.Perbal B. CCN proteins: multifunctional signalling regulators. Lancet 363: 62–64, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Pi L, Xia H, Liu J, Shenoy AK, Hauswirth WW, Scott EW. Role of connective tissue growth factor in the retinal vasculature during development and ischemia. Invest Ophthalmol Vis Sci 52: 8701–8710, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ponticos M, Holmes AM, Shi-wen X, Leoni P, Khan K, Rajkumar VS, Hoyles RK, Bou-Gharios G, Black CM, Denton CP, Abraham DJ, Leask A, Lindahl GE. Pivotal role of connective tissue growth factor in lung fibrosis: MAPK-dependent transcriptional activation of type I collagen. Arthritis Rheum 60: 2142–2155, 2009. [DOI] [PubMed] [Google Scholar]
- 45.Raghu G, Behr J, Brown KK, Egan JJ, Kawut SM, Flaherty KR, Martinez FJ, Nathan SD, Wells AU, Collard HR, Costabel U, Richeldi L, de Andrade J, Khalil N, Morrison LD, Lederer DJ, Shao L, Li X, Pedersen PS, Montgomery AB, Chien JW, O'Riordan TG, Artemis-IPF Investigators . Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med 158: 641–649, 2013. [DOI] [PubMed] [Google Scholar]
- 46.Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Pechoux C, Bogaard HJ, Dorfmuller P, Remy S, Lecerf F, Plante S, Chat S, Fadel E, Houssaini A, Anegon I, Adnot S, Simonneau G, Humbert M, Cohen-Kaminsky S, Perros F. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 131: 1006–1018, 2015. [DOI] [PubMed] [Google Scholar]
- 47.Rimon E, Chen B, Shanks AL, Nelson DM, Sadovsky Y. Hypoxia in human trophoblasts stimulates the expression and secretion of connective tissue growth factor. Endocrinology 149: 2952–2958, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sedgwick JB, Menon I, Gern JE, Busse WW. Effects of inflammatory cytokines on the permeability of human lung microvascular endothelial cell monolayers and differential eosinophil transmigration. J Allergy Clin Immunol 110: 752–756, 2002. [DOI] [PubMed] [Google Scholar]
- 49.Semenza GL. Involvement of hypoxia-inducible factor 1 in pulmonary pathophysiology. Chest 128: 592S–594S, 2005. [DOI] [PubMed] [Google Scholar]
- 50.Shimoda LA, Manalo DJ, Sham JS, Semenza GL, Sylvester JT. Partial HIF-1α deficiency impairs pulmonary arterial myocyte electrophysiological responses to hypoxia. Am J Physiol Lung Cell Mol Physiol 281: L202–L208, 2001. [DOI] [PubMed] [Google Scholar]
- 51.Shimoda LA, Semenza GL. HIF and the lung: role of hypoxia-inducible factors in pulmonary development and disease. Am J Respir Crit Care Med 183: 152–156, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shorr AF, Wainright JL, Cors CS, Lettieri CJ, Nathan SD. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur Respir J 30: 715–721, 2007. [DOI] [PubMed] [Google Scholar]
- 53.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, Elliott CG, Gaine SP, Gladwin MT, Jing ZC, Krowka MJ, Langleben D, Nakanishi N, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 54: S43–S54, 2009. [DOI] [PubMed] [Google Scholar]
- 54.Skuli N, Liu L, Runge A, Wang T, Yuan L, Patel S, Iruela-Arispe L, Simon MC, Keith B. Endothelial deletion of hypoxia-inducible factor-2alpha (HIF-2α) alters vascular function and tumor angiogenesis. Blood 114: 469–477, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21: 70–71, 1999. [DOI] [PubMed] [Google Scholar]
- 56.Tanjore H, Cheng DS, Degryse AL, Zoz DF, Abdolrasulnia R, Lawson WE, Blackwell TS. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem 286: 30,972–30,980, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanjore H, Degryse AL, Crossno PF, Xu XC, McConaha ME, Jones BR, Polosukhin VV, Bryant AJ, Cheng DS, Newcomb DC, McMahon FB, Gleaves LA, Blackwell TS, Lawson WE. β-catenin in the alveolar epithelium protects from lung fibrosis after intratracheal bleomycin. Am J Respir Crit Care Med 187: 630–639, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tanjore H, Xu XC, Polosukhin VV, Degryse AL, Li B, Han W, Sherrill TP, Plieth D, Neilson EG, Blackwell TS, Lawson WE. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med 180: 657–665, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.ten Freyhaus H, Dagnell M, Leuchs M, Vantler M, Berghausen EM, Caglayan E, Weissmann N, Dahal BK, Schermuly RT, Ostman A, Kappert K, Rosenkranz S. Hypoxia enhances platelet-derived growth factor signaling in the pulmonary vasculature by down-regulation of protein tyrosine phosphatases. Am J Respir Crit Care Med 183: 1092–1102, 2011. [DOI] [PubMed] [Google Scholar]
- 60.Tuder RM, Archer SL, Dorfmuller P, Erzurum SC, Guignabert C, Michelakis E, Rabinovitch M, Schermuly R, Stenmark KR, Morrell NW. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol 62: D4–D12, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Varia MA, Bundy BN, Deppe G, Mannel R, Averette HE, Rose PG, Connelly P. Cervical carcinoma metastatic to para-aortic nodes: extended field radiation therapy with concomitant 5-fluorouracil and cisplatin chemotherapy: a Gynecologic Oncology Group study. Int J Rad Oncol Biol Physics 42: 1015–1023, 1998. [DOI] [PubMed] [Google Scholar]
- 62.Veith C, Marsh LM, Wygrecka M, Rutschmann K, Seeger W, Weissmann N, Kwapiszewska G. Paxillin regulates pulmonary arterial smooth muscle cell function in pulmonary hypertension. Am J Pathol 181: 1621–1633, 2012. [DOI] [PubMed] [Google Scholar]
- 63.Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 98: 1528–1537, 2006. [DOI] [PubMed] [Google Scholar]
- 64.Wei H, Bedja D, Koitabashi N, Xing D, Chen J, Fox-Talbot K, Rouf R, Chen S, Steenbergen C, Harmon JW, Dietz HC, Gabrielson KL, Kass DA, Semenza GL. Endothelial expression of hypoxia-inducible factor 1 protects the murine heart and aorta from pressure overload by suppression of TGF-β signaling. Proc Natl Acad Sci USA 109: E841–E850, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65.Weitzenblum E, Sautegeau A, Ehrhart M, Mammosser M, Pelletier A. Long-term oxygen therapy can reverse the progression of pulmonary hypertension in patients with chronic obstructive pulmonary disease. Am Rev Respir Dis 131: 493–498, 1985. [DOI] [PubMed] [Google Scholar]
- 66.West J, Harral J, Lane K, Deng Y, Ickes B, Crona D, Albu S, Stewart D, Fagan K. Mice expressing BMPR2R899X transgene in smooth muscle develop pulmonary vascular lesions. Am J Physiol Lung Cell Mol Physiol 295: L744–L755, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.West JD, Austin ED, Gaskill C, Marriott S, Baskir R, Bilousova G, Jean JC, Hemnes AR, Menon S, Bloodworth NC, Fessel JP, Kropski JA, Irwin DC, Ware LB, Wheeler LA, Hong CC, Meyrick BO, Loyd JE, Bowman AB, Ess KC, Klemm DJ, Young PP, Merryman WD, Kotton D, Majka SM. Identification of a common Wnt-associated genetic signature across multiple cell types in pulmonary srterial hypertension. Am J Physiol Cell Physiol 307: C415–C430, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Widyantoro B, Emoto N, Nakayama K, Anggrahini DW, Adiarto S, Iwasa N, Yagi K, Miyagawa K, Rikitake Y, Suzuki T, Kisanuki YY, Yanagisawa M, Hirata K. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation 121: 2407–2418, 2010. [DOI] [PubMed] [Google Scholar]
- 69.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT, Semenza GL. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α. J Clin Invest 103: 691–696, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]