

Abstract
S-nitrosoglutathione (GSNO) reductase regulates novel endogenous S-nitrosothiol signaling pathways, and mice deficient in GSNO reductase are protected from airways hyperreactivity. S-nitrosothiols are present in the airway, and patients with cystic fibrosis (CF) tend to have low S-nitrosothiol levels that may be attributed to upregulation of GSNO reductase activity. The present study demonstrates that 1) GSNO reductase activity is increased in the cystic fibrosis bronchial epithelial (CFBE41o−) cells expressing mutant F508del-cystic fibrosis transmembrane regulator (CFTR) compared with the wild-type CFBE41o− cells, 2) GSNO reductase expression level is increased in the primary human bronchial epithelial cells expressing mutant F508del-CFTR compared with the wild-type cells, 3) GSNO reductase colocalizes with cochaperone Hsp70/Hsp90 organizing protein (Hop; Stip1) in human airway epithelial cells, 4) GSNO reductase knockdown with siRNA increases the expression and maturation of CFTR and decreases Stip1 expression in human airway epithelial cells, 5) increased levels of GSNO reductase cause a decrease in maturation of CFTR, and 6) a GSNO reductase inhibitor effectively reverses the effects of GSNO reductase on CFTR maturation. These studies provide a novel approach to define the subcellular location of the interactions between Stip1 and GSNO reductase and the role of S-nitrosothiols in these interactions.
Keywords: cystic fibrosis transmembrane conductance regulator, S-nitrosothiols, F508del-CFTR rescue, S-nitrosoglutathione reductase, Hsp70/Hsp90 organizing protein
cystic fibrosis (cf) is a multiorgan system disease associated with mutations in the gene that codes for CF transmembrane conductance regulator (CFTR) protein (5, 14, 26, 33, 43, 48, 53, 60). The majority of wild-type CFTR, and virtually all mutant F508del-CFTR, are degraded before reaching the cell surface (5, 14, 26, 33, 43, 48, 53, 60). Recent studies have shown that inhibiting CFTR ubiquitination and proteosomal degradation with chemical and pharmacological chaperones can promote membrane expression of mutant F508del-CFTR in the cell membrane (1, 17, 25, 41, 55.57, 58). As a result, there is substantial interest in developing compounds that promote F508del-CFTR incorporation into the plasma membranes of CF patients (48, 53).
Molecular chaperones facilitate CFTR folding and suppress its aggregation. They also play a role in recognizing misfolded CFTR and signaling its degradation, thus determining the early fate of the misfolded protein (1, 17, 25, 41, 55.57, 58). Molecular chaperones and cochaperones are involved in CFTR folding and cell-surface expression (6, 27, 47, 48, 64). Hsp70/Hsc70, Hdj-2, Hsp90, cysteine string protein, calnexin, and Hsp70/Hsp90 organizing protein (Hop), or stress-induced phosphoprotein 1 (Stip1), are among the proteins that modulate CFTR folding (44, 62, 63, 64, 65). The interaction of CFTR with Hsc70, coupled to its cochaperone CHIP, leads to degradation of CFTR through the ubiquitin-proteosome pathway (61).
The endogenous S-nitrosothiol S-nitrosoglutathione (GSNO) increases the maturation and function of CFTR in human airway epithelial cells (2, 11, 29, 37, 52, 54, 62–65). GSNO and other S-nitrosothiols have additional beneficial effects on airway function, including relaxation of airway smooth muscle, improvement of ciliary motility, inhibition of amiloride-sensitive sodium transport, and prevention of bacterial and viral infections (3, 16, 19–23, 28, 30, 31, 32, 36, 38, 40, 45–46, 59). Therefore, since S-nitrosothiol levels are low in the CF in the CF airway (24), chronic replacement therapy with a low dose of inhaled GSNO has been proposed as a potential therapy for CF patients (54).
Previous work demonstrates that GSNO is both a corrector and a potentiator: it both activates wild-type (WT) CFTR (11, 37) and redirects F508del-CFTR to the cell membrane (2, 29, 37, 62). The mechanisms involved in its corrector function are complex (36, 61, 62, 63), but a principal effect requires S-nitrosylation of Stip1 at cysteine 403. This signal targets Stip1 for degradation. Loss of Stip1 prevents F508del-CFTR degradation and permits its maturation and cell surface expression (37).
In addition, several lines of evidence suggest that enhanced GSNO metabolism may be relevant to CF (2, 11, 24, 29, 37, 52, 62–65). GSNO reductase, also known as glutathione-dependent formaldehyde dehydrogenase, catalyzes the degradation of GSNO and regulates tissue level of S-nitrosothiols and S-nitrosylated proteins (35, 39, 47). We hypothesized that GSNO reductase activity increases in airway epithelial cells of CF patients and that decreased airway levels of S-nitrosothiols in CF (24) are due, at least in part, to upregulation of GSNO reductase. The present study demonstrates that 1) GSNO reductase activity is elevated in mutant CFBE41o− cells expressing F508del-CFTR compared with the wild-type CFBE41o− cells; 2) GSNO reductase expression level can be increased and is certainly not decreased in the primary human bronchial epithelial cells expressing mutant F508del-CFTR compared with the wild-type cells; 3) GSNO reductase and Stip1 colocalize/interact in CFBE41o− cells; 4) GSNO reductase knockdown with GSNO reductase siRNA duplexes increases the expression of F508del-CFTR, but it decreases Stip1 expression, in CFBE41o− cells; and 5) increasing the cell levels of GSNO reductase expression decreases CFTR maturation, an effect reversed by GSNO reductase inhibitor. These studies suggest a potential roll for GSNO reductase inhibitors in the development of CF therapies.
MATERIALS AND METHODS
Cell culture and reagents.
The pseudostratified cells were primary human bronchial epithelial cells were obtained from bronchi of human lung tissue under a protocol approved by the University of North Carolina Medical School Institutional Review Board. Primary CF human bronchial epithelial cells were seeded at passage 2 on collagen-coated Millicell CM inserts (Millipore) and maintained at an air-liquid interface at 37°C in 5% CO2 for 3–4 wk, which allowed the cells to become fully differentiated (18). The remainder of the cells were CFBE41o− cell lines expressing wild-type and mutant F508del-CFTR and were provided by Dr. Eric Sorscher (University of Alabama). CFBE41o− cells were grown in DMEM, as previously described (37, 62). Media for CFBE41o− cells contained 10% (vol/vol) fetal calf serum and 1% (vol/vol) penicillin/streptomycin (Life Technologies, Gaithersburg, MD). Cells were maintained at 37°C in a humidified atmosphere of 5% carbon dioxide and passaged at confluence approximately every 4 days. All reagents were from Bio-Rad (Hercules, CA) and Sigma Chemical (St. Louis, MO) unless otherwise stated. GSNO was prepared as previously described (37, 63, 64). Note that we have previously published that these cells form tight junctions with high transepithelial resistance if grown in transwells (63).
Immunoblotting.
This was performed as described previously (37, 62–65). Briefly, whole cell extracts from CFBE41o− and primary human bronchial airway epithelial cells expressing wild-type and mutant F508del-CFTR were prepared in 1% NP-40 lysis buffer (50 mM Tris·HCl pH 8.0, 1% NP-40, 150 mM NaCl, 2 μM leupeptin, 1 μM aprotinin, and 1 μM pepstatin, 1 mM DTT, 1 μM PMFS, and 2 μM Na3VO4). Insoluble material from NP-40 was recovered and sheared by passage through a 25-gauge needle. Protein was quantitated by the Lowry assay by using protein assay kit (Sigma Chemical). One hundred micrograms of protein were fractionated on a 6% SDS polyacrylamide gel in 1× electrode buffer (25 mM Tris, 192 mM glycine, and 0.1% SDS at pH 8.3). The fractionated proteins were transferred to nitrocellulose membranes (Bio-Rad) using an electrophoretic transfer cell with Tobin transfer buffer (25 mM Tris, 192 mM glycine, and 20% methanol at pH 8.3). Blots were blocked in Tris-buffered saline-Tween 20 (TBS-T: 10 mM Tris·HCl pH 8.0, 150 mM NaCl, and 0.05% Tween 20) containing 5% nonfat dried milk. Blots were probed with a 1:1,000 dilution of anti-CFTR mAb 596 antibody (a gift from Dr. J. R. Riordan) and a monoclonal anti-Stip1 (Stressgen, Victoria, BC, Canada). Rabbit polyAb anti-ADH5 (GSNO reductase) was obtained from Proteintech Group (Chicago, IL) and incubated in TBS-T containing 5% nonfat dried milk for 45 min at room temperature. The following isotype control antibodies were used: mouse IgG1 and rabbit polyclonal IgG (dilution 1:100). The mouse and rabbit isotypes were used as a negative control. Additionally, all slides were stained with DAPI to image nucleus. Representative images are shown from triplicate experiments. Blots were washed several times in TBS-T and incubated for 30 min with a 1:2,000 dilution of HRP-conjugated anti-mouse antibody (Bio-Rad) in TBS-T containing 5% nonfat dried milk for 30 min. CFTR proteins were visualized using Super Signal chemiluminescent substrate (Pierce, Rockford, IL) and the Versal Imaging System (Bio-Rad). Quantification of protein expression was performed using densitometry software on the Versa Doc (Quality One; Bio-Rad).
Immunoprecipitation.
Immunoprecipitation was performed as described previously (62–65). Whole cell extracts were prepared in 1% NP-40 lysis buffer (50 mM Tris·HCl pH 8.0, 1% NP-40, 150 mM NaCl, 2 μM leupeptin, 1 μM aprotinin, and 1 μM pepstatin, 1 mM DTT, 1 μM PMFS, and 2 μM Na3VO4). Insoluble material from NP-40 was recovered and sheared by passage through a 25-gauge needle. Protein was quantitated by the Lowry assay by using a protein assay kit (Sigma Chemical). Ten microliters of primary Stip1 antibody (anti-Stip1; Stressgen) were added to each fraction and incubated overnight at 4°C with gentle shaking. Supernatant antibody mixtures were treated with 70 μl of Protein A (Boehringer Mannheim, Indianapolis, IN) and incubated for another 4 h. Then, samples underwent centrifugation (1 min) and proteins that were not bound to the beads were removed by washing beads twice with RIPA buffer. Proteins were then eluted from beads by incubation with 100 μl sample buffer at room temperature with continuous mixing for 1 h. Fifty micrograms of protein were fractionated on a 6% SDS polyacrylamide gel. The fractionated proteins were transferred to nitrocellulose membranes using an electrophoretic transfer cell with Tobin transfer buffer. Blots were blocked in Tris buffered saline-Tween 20. Blots were probed with a 1:1,000 dilution of anti-rabbit polyAb anti-ADH5 (GSNO reductase) obtained from Proteintech Group and incubated in TBS-T containing 5% nonfat dried milk for 45 min at room temperature. Blots were washed several times in TBS-T, incubated for 30 min with a 1:2,000 dilution of HRP-conjugated anti-mouse antibody (Bio-Rad) in TBS-T containing 5% nonfat dried milk for 30 min. GSNO reductase proteins were visualized using Super Signal chemiluminescent substrate (Pierce) and the Versal Imaging System (Bio-Rad). Quantification of protein expression was performed using densitometry software on the Versa Doc (Quality One; Bio-Rad).
Enzymatic activity of GSNO reductase in CFBE41o− cells.
The enzymatic activity of GSNO reductase was measured as described previously (35, 47). Briefly, cell lysates were incubated with NADH in reaction buffer (20 mM Tris·HCl pH 8.0) and 0.5 mM EDTA containing 0 or 100 μM GSNO at room temperature. NADH fluorescence (absorption at 340 nm and emission at 455 mm) was measured over time in a FLUOstar Omega (BMG Labtech, Offenberg, Germany). The concentrations of NADH in cell samples were determined using a standard curve (35, 47).
Immunohistochemistry of GSNO reductase in CFBE41o− cells.
CFBE41o− cells expressing wild-type and mutant F508del-CFTR were grown to confluence on six-well plates. The culture medium was pipetted off and replaced with HBSS for 30 s with gentle agitation. The HBSS was then decanted, and 10% phosphate-buffered formalin was slowly added to each culture. The cultures were fixed at 4°C for several days, after which time immunostaining for GSNO reductase was performed using rabbit polyclonal anti-GSNO reductase antibody (1:50; Proteintech Group) as the primary antibody, and a biotinylated goat anti-rabbit antibody was used as the secondary antibody. The mean surface of GSNO reductase staining from CFBE41o− cells expressing wild-type and F508del-CFTR was calculated from three different experiments by image analysis using ImageJ software (National Institutes of Health).
Immunofluorescence and confocal laser scanning microscopy.
CFBE41o− cells expressing wild-type and mutant F508del-CFTR were grown to confluence on glass coverslips, washed (2×) with 5 ml of complete DPBS, fixed with 3.7% paraformaldehyde (5 min), washed (2×) with DPBS, permeabilized with 0.1% Triton-X100 in DPBS (vol/vol) (10 min), washed (2×) with DPBS, incubated with a blocking solution with a composition of 5% normal goat serum (NGS) and 0.01% Triton-X100 in 1× DPBS (PBS-T-NGS) for 45 min at room temperature, and then incubated overnight at 4°C with primary anti-Stip1 (Hop) antibody (1:100, mouse monoclonal; Stressgen) or with primary anti-GSNO reductase antibody (1:150 dilution, rabbit polyclonal; Abcam). The cells were then washed (2× for 15 min) with blocking solution and incubated at room temperature for 45 min with a secondary antibody to GSNO reductase (1:500 dilution, Alexa Fluor 568; Molecular Probes, Invitrogen) or Stip1(Hop) (1:500 dilution, Alexa Flour 488; Molecular Probes, Invitrogen), made in blocking solution. The cells were washed (2× for 15 min) in blocking solution with 0.1% NGS and stained with Dapi for nuclear staining, followed by washing in DPBS. As negative control an isotype control for mouse and rabbit (BD Pharmingen) were, respectively, used at the same concentration as Hop and GSNO reductase primary antibodies followed by labeling with the respective secondary antibodies. The cells were mounted and visualized using ×63 NA1.4 oil immersion lens on Leica TCS SP5 for laser scanning confocal microscopy. The white light lasers of 488 nm (20%) and 568 nm (65%) were used for excitation of Hop-Alexa488 (emission: 500–550 nm) and GSNO reductase-Alexa568 (emission: 580–640 nm), respectively. Identical imaging parameters were used for isotype controls.
Small interfering RNA knockdown of GSNO reductase.
Small interfering (si)RNA sequences were synthesized and purified by matrix-assisted laser desorption ionization/time-of-flight spectrometric analysis (Ambion, Austin, TX). GSNO reductase siRNA duplexes were >90% pure, as measured by HPLC, with sense 5′-3′-AAAUCAACAAAGCCU UUGAtt and antisense 5′-3′-UCAAAGGCUUUGUUGAUUUca. Scrambled GSNO reductase siRNA was used as a control. CFBE41o− cells expressing F508del-CFTR were transfected with 50, 75, and 100 nM of GSNO reductase construct using INTERFERin (Polyplus-Transfection SA). After 48 h, cells were rinsed (3×) with ice-cold PBS and lysed. Proteins were analyzed after 48 h of transfection by Western blot analysis using rabbit polyclonal anti-GSNO reductase antibody.
Statistics.
We conducted two-way ANOVA for each experiment. In each model, we included the main effects of treatment and band and their interaction. The statistical analyses were performed with SAS 9.1 (SAS Institute, Cary, NC). Multiple comparisons were adjusted by the Dunnett's method. P < 0.05 was considered statistically significant.
RESULTS
GSNO reductase activity differences in CFBE41o− cells transfected with either wild-type or mutant F508del-CFTR.
Immunostaining studies demonstrated that GSNO reductase is significantly expressed in CFBE41o− cells transfected with F508del-CFTR compared with CFBE41o− cells transfected with wild-type CFTR (Fig. 1, A and B; n = 3; P < 0.05). Furthermore, GSNO reductase activity was elevated approximately twofold in the mutant CFBE41o− cells expressing F508del-CFTR compared with wild-type CFBE41o− cells (Fig. 1C; n = 3; P < 0.002).
Fig. 1.
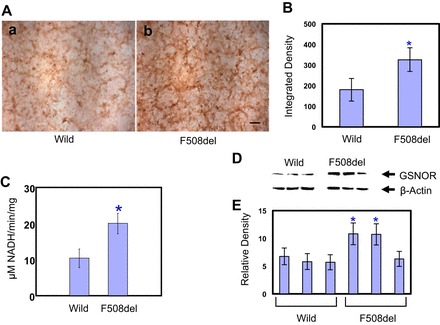
A: S-nitrosoglutathione reductase (GSNOR) immunostaining in CFBE41o− cells expressing wild-type and mutant F508del-CFTR. GSNO reductase is expressed minimally in the wild-type CFTR (a), whereas it is significantly expressed in the mutant F508del-CFTR CFBE41o− cells (b). Representative images are shown from triplicate experiments (scale bar = 250 μm). B: integrated density of GSNO reductase significantly elevated in F508del-CFTR compared with wild-type CFTR in CFBE41o− cells. The results are expressed as means ± SD (n = 3; *P < 0.05). C: GSNO reductase activity differences in wild-type and F508del-CFTR in CFBE41o− cells expressing wild-type and mutant F508del-CFTR. GSNO-dependent NADH consumption: cell lysate was incubated with NADH in a reaction buffer (20 mM Tris HCl pH 8.0) and 0.5 mM EDTA containing 0 or 100 μM GSNO at room temperature. NADH fluorescence (absorption at 340 nm and emission at 455 mm) was measured over time in a FLUOstar Omega. Concentration of NADH was determined from a standard curve as described previously. GSNO reductase activity was significantly elevated (2-fold; n = 3; *P < 0.02). D: GSNO reductase expression level in 3 different clones of primary human bronchial airway epithelial cells expressing wild-type and mutant F508del-CFTR. The membrane was stripped and reprobed with β-actin to verify that equal amounts of protein were added. E: blots were repeated 3 times and scanned, and densitometry was performed for quantification. Data are means ± SD (n = 3; *P < 0.05 compared with mean wild-type density).
GSNO reductase expression level differences in primary human bronchial epithelial cells expressing wild-type and mutant F508del-CFTR.
GSNO reductase protein expression levels were elevated approximately twofold in two of the three mutant F508del-CFTR donors. The third clone did not show any changes compared with primary wild-type human bronchial epithelial cells (Fig. 1, D and E).
Cellular colocalization of GSNO reductase and Stip1 in CFBE41o− cells.
We have demonstrated cellular colocalization of GSNO reductase and Stip1 in human bronchial airway epithelial cells by immunoprecipitation (Fig. 2C; n = 2), and we showed that exogenous GSNO can reduce excess GSNO reductase to wild-type levels (Fig. 2C). In addition, our confocal microscopy results suggest that GSNO reductase is present in CFBE41o− cells and colocalizes with Stip1 (Fig. 2A). Isotope controls were negative (Fig. 2B). Taken together, these data reveal that GSNO reductase and cochaperone Stip1 interact directly in CFBE41o− cells.
Fig. 2.
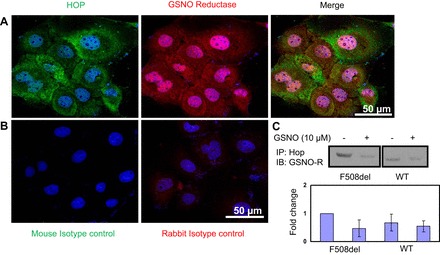
A and B: cellular colocalization of GSNO reductase and Stip1 in CFBE41o− cells expressing mutant F508del-CFTR. Cells were fixed with 3.7% paraformaldehyde for 5 min, permeabilized by 0.1% Triton X-100, and incubated with primary anti-GSNO reductase and anti-Stip1 antibody at 4°C overnight. Cells were incubated with secondary antibodies for 45 min at room temperature and then mounted coverslips and visualized with the Leica laser scanning confocal microscopy (TCS-SP5X; ×63 NA1.4 Oil immersion lens). The mouse and rabbit isotypes were used as a negative control. Additionally, all slides were stained with DAPI to image the nucleus. Representative images are shown from triplicate experiments. Confocal laser scanning microscopy shows colocalization of Stip1 (A, left, and B, left) and GSNO reductase (A, middle, and B, right) in the cytosolic and nucleoplasmic regions in the merged image (A, right) and nuclei stained with DAPI (B). Merged images with DAPI for mouse (B, left) isotype controls for Hop and rabbit (B, right) isotype controls for GSNO reductase were concentration matched and used under identical imaging conditions on Leica TCS SP5, ×63 NA1.4. C: colocalization of Hop and GSNO reductase in CFBE41o− ΔF508 cells. WT, wild type; IB, immunoblot; IP, immunoprecipitation.
GSNO reductase knockdown with siRNA increases expression of F508del- CFTR.
To confirm the role of GSNO reductase in F508del-CFTR maturation, we knocked out endogenous GSNO reductase by transfecting mutant CFBE41o− cells with 50, 75, and 100 nM of GSNO reductase siRNA. After 48 h of transfection with 50 nM of siRNA, duplexes for GSNO reductase minimally decreased endogenous GSNO reductase levels. Also, there was no effect on CFTR levels. However, at concentration of 100 nM of siRNA duplexes for GSNO reductase, endogenous levels of GSNO reductase expression were markedly decreased, which produced a significant increase in levels of immature and fully mature forms of F508del-CFTR (Fig. 3; second row, lanes 4 and 5). Interestingly, the increase of both immature and mature forms of CFTR levels was associated with decreased Stip1 expression (Fig. 3; third row, lane 5; n = 3; *P < 0.02, **P < 0.001).
Fig. 3.
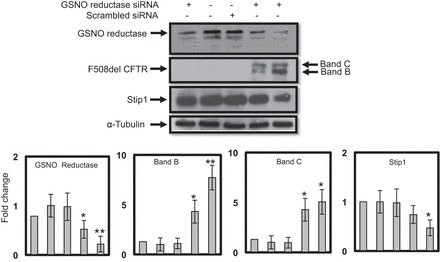
GSNO reductase knockdown with siRNA increases expression of the CFTR. CFBE41o− cells expressing mutant F508del-CFTR were transfected with 50 (lane 1), 75 (lane 4), and 100 nM (lane 5) of siRNA duplexes specific for GSNO reductase using Lipofectamine 2000 (Invitrogen). After 48 h of transfection, cells were rinsed 3 times with PBS and lysed directly on plates using lysis buffer containing protease inhibitors. Scrambled GSNO reductase siRNA was used as a control. Immunoblotting was performed on whole cell lysates (100 μg protein per lane). The membrane was stripped and reprobed with α-tubulin to verify that equal amounts of protein were added. The optical densities of the bands were quantified by densitometry. Data are means ± SD; n = 3; *P < 0.02; **P < 0.001.
GSNO reductase inhibits maturation of CFTR and GSNO reductase inhibitor reverses this effect.
Increasing endogenous GSNO reductase levels by transfection with FLAG-tagged GSNO reductase (2 μM; 48 h) caused an effective decrease (2.2-fold) in immature band (band B) and 1.8-fold decrease in mature band (band C) of CFTR (Fig. 4; lane 3) compared with control. However, the GSNO reductase inhibitor C3 (1 μM; 4 h) effectively reversed the inhibitory effects of FLAG-tagged GSNO reductase, thereby causing an increase of immature band by 2.1-fold (P < 0.02) and an increase of mature band by 2.0-fold (P < 0.02) of CFTR in wild-type CFBE41o− cells (Fig. 4; lane 2; n = 4).
Fig. 4.
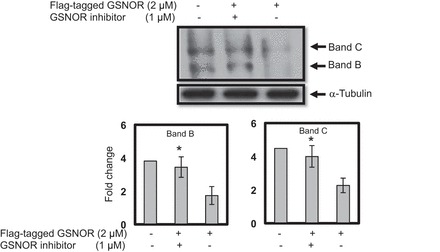
Endogenous GSNO reductase significantly inhibits CFTR expression and maturation, and the GSNO reductase inhibitor increases CFTR expression and maturation. The endogenous FLAG-tagged GSNO reductase (2 μM) inhibits the maturation of CFTR. However, in the presence of the GSNO reductase inhibitor C3 (Chem Div) at concentration of 2 μM effectively reverses the GSNO reductase inhibitory effects on CFTR expression and maturation in wild-type CFBE41o−cells. The membrane was stripped and reprobed with α-tubulin to verify that equal amounts of protein were added. The optical densities of the bands were quantified by densitometry. Data are means ± SD; n = 4; *P < 0.02.
DISCUSSION
CF is caused by mutations in the CFTR gene that impair synthesis and membrane trafficking CFTR and/or reduce Cl− channel activity of the protein (5, 14, 26, 33, 43, 48, 53, 60). The most common mutation associated with CF, F508del-CFTR, is a single amino acid, phenylalanine deletion (5, 14, 48). The majority of wild-type CFTR proteins, and virtually all F508del-CFTR proteins, are degraded before reaching the cell surface (26, 33, 43, 53). However, F508del-CFTR can function as a cAMP-activated Cl−-chloride channel if it indeed reaches the plasma membrane. Hence, there is an interest in identifying pharmacological agents that could promote F508del-CFTR membrane insertion in vivo. A number of molecules have been identified that modestly correct expression of F508del-CFTR, making it potentially functional in plasma membranes (6, 15, 27, 49, 50, 66); however, their precise mechanisms of action are unknown.
One class of F508del-CFTR correctors for which a mechanism of action is established is the class of compounds known as S-nitrosothiols (2, 29, 37, 62–65). Note in this regard that GSNO and other S-nitrosothiol species in this class are in transnitrosation equilibria in vivo. S-nitrosothiols increase CFTR maturation by S-nitrosylation of specific cysteine residues of molecular chaperones involved in regulating CFTR biogenesis and membrane trafficking. These include cysteine string protein (Csp), heat shock cognate 70 (Hsc70), and Stip1 (7, 10, 13, 37, 42, 56, 61, 63). Cochaperone Stip1 is a critical component for efficient maturation of steroid receptor complexes (8, 9, 12). Stip1 provides an important link between Hsp70 and Hsp90 and coordinates Hsp actions in folding protein substrates. Stip1 also plays an active role in functional maturation of Stip1-complexes (9). The molecular structure of Stip1 has two reduced cysteines (C26 and C403) located in tyrosine-cysteine “YC” S-nitrosylation motifs (8). We have reported that Stip1 plays a central role in the mechanism by which S-nitrosothiols increase CFTR maturation (37): it coimmunoprecipitates with CFTR (37), and its expression, free and bound to CFTR, is decreased by GSNO through an effect of GSNO (and other S-nitrosothiols) to S-nitrosylate C403, targeting Stip1 for degradation and permitting CFTR maturation (37). Indeed, Stip1 siRNA also enhances CFTR maturation.
In addition, S-nitrosothiols have CFTR-independent beneficial effects that may promote airway function, including relaxing airway smooth muscle, improving ciliary motility, inhibiting amiloride-sensitive sodium transport, and preventing bacterial and viral infections (3, 16, 19–23, 28, 30, 31, 34, 36, 37, 40, 59). Because levels of S-nitrosothiols are low in the CF airway (24), chronic therapy with inhaled GSNO is therefore appealing as a potential therapy for patients carrying F508del-CFTR mutation (54). Dose-response experiments in monolayer cell cultures indicate that 5–10 μM of S-nitrosothiols may be optimal for increase F508del- CFTR function (52, 62, 63, 65).
However, high concentrations of GSNO may have undesirable effects in the airway (64). Furthermore, GSNO did not restore Cl− current in full-thickness human pseudostratified columnar epithelial cell cultures or in airway epithelial cells expressing only F508del-CFTR (37). S-nitrosothiols with higher membrane permeability, such as S-nitrosoglutathione diethyl ester (GNODE) and S-nitroso-N-acetyl cysteine (SNO-NAC), are more efficient in increasing the expression, maturation, and function of F508del-CFTR in airway epithelial cells (60). These, coupled with inhibitors of S-nitrosothiol catabolism, are proposed as complementary therapies for patients expressing F508del-CFTR.
GSNO reductase, also known as alcohol dehydrogenase 5 (ADH5) or glutathione-dependent formaldehyde dehydrogenase, is widely expressed in tissues and may protect cells from nitrosative stress (35, 37, 47). GSNO reductase decreases S-nitrosothiol levels and protein S-nitrosylation signaling via catabolism of GSNO, which is in transnitrosation equilibrium with most cellular S-nitrosothiols (37). Mice deficient in GSNO reductase have increased concentrations of S-nitrosothiol in their lungs, whereas GSNO reductase activity is elevated in a murine model of allergic asthma (35). The importance of GSNO reductase in regulating the levels of S-nitrosylated Stip1 and the influence this has on the maturation and trafficking of CFTR are unknown. In this study we sought to determine the effects of loss of GSNO reductase, on maturation of F508del-CFTR in CFBE41o− cells. Interestingly, we found that GSNO reductase expression may be higher in some F508del-CFTR expressing primary human airway cells compared with matched wild-type cells, although it will be worthwhile in the future to study a larger number of clones. These results may suggest that increased GSNO reductase activity in F508del-CFTR cells may enhance Stip1 denitrosylation in those cells and inhibit the various beneficial effects of endogenous S-nitrosothiols in the airway epithelium.
Furthermore, we found that GSNO reductase knockdown results in enhanced CFTR maturation and decreased Stip1 expression. Indeed, we found that GSNO reductase and Stip1 colocalize and directly interact in CFBE41o− cells (Fig. 2C). Because S- nitrosylation of Stip1 causes decreased interaction between Stip1 and CFTR, permitting CFTR maturation (37), denitrosylation of Stip1 by GSNO reductase inhibits F508del-CFTR correction.
We determined that increasing expression of endogenous GSNO reductase led to lower CFTR expression. We found that cells transfected with FLAG-tagged GSNO reductase had significantly decreased maturation of CFTR and that GSNO reductase inhibition effectively reversed the effect of GSNO reductase on CFTR expression. It is important to note that inhibitors of GSNO reductase are in development for treatment of asthma and CF (3, 47, 51). Our current results support the notion that augmented GSNO reductase activity increases CFTR maturation in full-thickness airway epithelial cultures cells. These findings will help to optimize the dosing of S-nitrosothiols and GSNO reductase inhibitors that potentially could be used as F508del-CFTR correctors. They may also provide a marker (increased GSNO reductase activity) for those CF patients most likely to benefit from GSNO reductase inhibition.
GRANTS
This work was supported by the Cystic Fibrosis Foundation and National Heart, Lung, and Blood Institute Grants 1P0-HL-101871-01A1 and 3R0-1HL-59337.
DISCLOSURES
B. Gaston is a consultant for Nivalis Therapeutics. No other conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
K.Z., M.S.F., L.P., A.P., S.H.R., and S.J.L. conception and design of research; K.Z., V.S., A.Z., M.B., D.B., P.G., M.Z., Z.G., M.S.F., S.R., V.J., K.D., L.S., D.A.C., A. Straub, F.S., L.P., S.H.R., T.J.K., S.J.L., and B.G. performed experiments; K.Z., V.S., M.S.F., A. Sattar, L.S., and L.P. analyzed data; K.Z., V.S., M.S.F., and B.G. interpreted results of experiments; K.Z., V.S., P.G., and S.R. prepared figures; K.Z., F.S., L.P., S.J.L., and B.G. drafted manuscript; K.Z., V.S., P.G., F.S., L.P., and B.G. edited and revised manuscript; K.Z. and B.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Eric Sorscher for providing CFBE41o− cell lines expressing wild-type and mutant F508del-CFTR. Also, we thank Dr. John Riordan for providing the monoclonal anti-CFTR antibody.
REFERENCES
- 1.Amaral MD. CFTR and chaperones. J Mol Neurosci 23: 41–47, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Andersson C, Gaston B, Roomans G. S-Nitrosoglutathione induces functional ΔF508 CFTR in cultured airway epithelial cells. Biochem Biophys Res Commun 297: 552–557, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Angers R, Mutka S, Bove PF, Gabriel SE. Pharmacological correction and acute inhibition of GSNOR results in improved in vitro CFTR function. Pediatr Pulmonol 38: 241, 2014. [Google Scholar]
- 4.Asano K, Chee C, Gaston B, Lilly C, Gerard C, Drazen J, Stamler JS. Constitutive and induible nitric oxide synthase gene expression, regulation and activity in human lung epithelial cells. Proc Natl Acad Sci USA 91: 10089–10093, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boucher RC. An overview of the pathogenesis of cystic fibrosis lung disease. Adv Drug Deliv Rev 54: 1359–1371, 2002. [DOI] [PubMed] [Google Scholar]
- 6.Bradbury NA. Focus on sodium 4-phenylbutyrate downregulates Hsc70 implications for intracellular trafficking of ΔF508 CFTR. Am J Physiol Cell Physiol 278: C257–C258, 2000. [DOI] [PubMed] [Google Scholar]
- 7.Braun JE, Wilbanks SM, Scheller RH. The cysteine string secretory vesicle protein activates Hsc70 ATPase. J Biol Chem 271: 25989–25993, 1996. [DOI] [PubMed] [Google Scholar]
- 8.Carrigan P, Riggs D, Chinkers M, Smith D. Functional comparison of human and Drosophilia Stip 1 reveals novel role in steroid receptor maturation. J Biol Chem 280: 8906–8911, 1998. [DOI] [PubMed] [Google Scholar]
- 9.Carrigan P, Sikkink L, Smith D, Ramirez-Alvardo M. Domain: domain interactions with Stip 1 the Hsp70/Hsp90 organizing protein are required for protein stability and structure. Protein Sci 15: 522–532, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chamberlain L, Burgoyne R. Cysteine-string protein: the chaperone at the synapse. J Neurochem 74: 1781–1789, 2000. [DOI] [PubMed] [Google Scholar]
- 11.Chen L, Patel RP, Teng X, Bosworth CA, Lancaster JR Jr, Matalon S. Mechanisms of cystic fibrosis transmembrane conductance regulator activation by S-nitrosoglutathione. J Biol Chem 281: 9190–9199, 2006. [DOI] [PubMed] [Google Scholar]
- 12.Chen S, Sullivan W, Smith D. Differential inter-actions of p23 and the TPR-containing proteins Stip 1, cyp40, FKBP52 and FKBP51 with Hsp90 mutants. Cell Stress Chaperones 3: 118–129, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collawn JF, Bebok Z, Matalon S. Search and rescue: finding ways to correct ΔF508 CFTR. Am J Respir Cell Mol Biol 40: 385–387, 2009. [DOI] [PubMed] [Google Scholar]
- 14.Davis PB. Cystic fibrosis since 1938. Am J Respir Crit Care Med 173: 475–482, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Egan ME, Pearson M, Weiner SA, Rajendran V, Rubin D, Glöckner-Pagel J, Canny S, Du K, Lukacs GL, Caplan MJ. Curcumin: a major constituent of turmeric corrects cystic fibrosis defects. Science 304: 600–602, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Fang K, Johns R, Macdonald T, Kinter M, Gaston B. S-nitrosoglutathione breakdown prevents airway smooth muscle relaxation in the guinea pig. Am J Physiol Lung Cell Mol Physiol 279: L716–L721, 2000. [DOI] [PubMed] [Google Scholar]
- 17.Farinha CM, Noguerira P, Mendes F, Penque D, Amaral MD. The human DnaJ homologue (Hdj)-1/heat-shock protein (Hsp) 40 co-chaperone is required for the in vivo stabilization of the cystic fibrosis transmembrane conductance regulator by Hsp70. Biochem J 366: 797–806, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fulcher ML, Gabriel SE, Burns KA, Yankaskas JR, Randell S. Well-differentiated human airway epithelial cell cultures. Methods Mol Med 107: 183–206. [DOI] [PubMed] [Google Scholar]
- 19.Gaston B. Nitric oxide and thiol groups. Biochim Biophys Acta 1411: 323–333, 1999. [DOI] [PubMed] [Google Scholar]
- 20.Gaston B, Doctor A, Singel D, Stamler JS. S-nitrosothiol signaling in respiratory biology. Am J Respir Crit Care Med 173: 1186–1193, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaston B, Ratjen F, Vaughan NR, Malhotra JW, Canady RG, Snyder AH, Hunt JF, Gaertig S, Goldberg JB. Nitrogen redox balance in the cystic fibrosis airway: effects of anti-pseudomonal therapy. Am J Respir Crit Care Med 165: 387–390, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Gaston B, Reilly J, Drazen JM, Fackler J, Ramden P, Arnelle D, Mullins ME, Sugarbaker D, Singel D, Loscalzo Stamler JS J. Endogenous nitrogen oxides and bronchodilator S-nitrosothiols in human airways. Proc Natl Acad Sci USA 90: 10957–10961, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaston B, Sears S, Woods J, Hunt J, Ponaman J, McMahon T, Stamler J. Bronchodilator S-nitrosothiol deficiency in asthmatic respiratory failure. Lancet 351: 1317–1319, 1998. [DOI] [PubMed] [Google Scholar]
- 24.Grasemann H, Gaston B, Fang K, Paul K, Ratjen F. Decreased levels of nitrosothiols in the lower airways of patients with cystic fibrosis and normal pulmonary function. J Pediatr 135: 770–772, 1999. [DOI] [PubMed] [Google Scholar]
- 25.Grove DE, Rosser MF, Ren HY, Naren AP, Cyr DM. Mechanisms for rescue of correctable folding defects in CFTR ΔF508. Mol Biol Cell 20: 4059–4069, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guggino WB, Stanton BA. New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat Rev Mol Cell Biol 7: 426–436, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Heda GD, Marino CR. Surface expression of the cystic fibrosis transmembrane conductance regulator mutant deltaF508 is markedly upregulated by combination treatment with sodium butyrate and low temperature. Biochem Biophys Res Commun 271: 659–664, 2000. [DOI] [PubMed] [Google Scholar]
- 28.Hogg N, Singh R, Konorev E, Joseph J, Kalyanaraman B. S-Nitrosoglutathione as a substrate for γ-glutamyl transpeptidse. Biochem J 323: 477–481, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howard M, Fischer H, Roux J, Santos B, Gullans S, Yancey P, Welch W. Mammalian osmolytes and S-nitrosoglutathione promote F508 CFTR protein maturation and function. J Biol Chem 278: 35159–35167, 2003. [DOI] [PubMed] [Google Scholar]
- 30.Jilling T, Haddad I, Cheng S, Matalon S. Nitric oxide inhibits heterologous CFTR expression in polarized epithelial cells. Am J Physiol Lung Cell Mol Physiol 277: L89–L96, 1999. [DOI] [PubMed] [Google Scholar]
- 31.Johnson MA, Macdonald TL, Mannick JB, Conaway MR, Gaston B. Accelerated S-nitrosothiol breakdown by amyotrophic lateral sclerosis mutant copper, zinc-superoxide dismutase. J Biol Chem 276: 39872–39878, 2001. [DOI] [PubMed] [Google Scholar]
- 32.Kamosinska B, Radomski MW, Duszyk M, Radomski A, Man SF. Nitric oxide activates chloride currents in human lung epithelial cells. Am J Physiol Lung Cell Mol Physiol 272: L1098–L1104, 1997. [DOI] [PubMed] [Google Scholar]
- 33.Kopito RR. Biosynthesis and degradation of CFTR. Physiol Rev 79: S167–S172, 1999. [DOI] [PubMed] [Google Scholar]
- 34.Lancaster J, Gaston B. NO and nitrosothiols: spatial confinement and free diffusion. Am J Physiol Lung Cell Mol Physiol 287: L465–L466, 2004. [DOI] [PubMed] [Google Scholar]
- 35.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JA. metabolic enzyme for S.-nitrosothiol conserved from bacteria to humans. Nature 410: 490–496, 2001. [DOI] [PubMed] [Google Scholar]
- 36.Mannick J, Schonhoff C, Papeta N, Ghafourifar P, Szibor M, Fang K, Gaston B. S-Nitrosylation of mitochondrial caspases. J Cell Biol 154: 1111–1116, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marozkina N, Yemen S, Borowitz M, Liu L, Plapp M, Sun F, Islam R, Erdmann-Gilmore P, Townsend R, Lichti C, Manti S, Clapp P, Randell S, Gaston B, Zaman K. Hsp70/Hsp90 organizing protein as a nitrosylation target in cystic fibrosis. Proc Natl Acad Sci USA 107: 11393–11398, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marozkina NV, Gaston B. S-nitrosylation signaling regulates cellular protein interactions. Biochim Biophys Acta 820: 722–729, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marozkina NV, Wei C, Yemen S, Wallrabe H, Nagji AS, Liu L, Marozkina T, Jones DR, Gaston B. S-nitrosoglutathione reductase in human lung cancer. Am J Respir Cell Mol Biol 46: 63–70, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marshall H, Que L, Stamler JS, Gaston B. S-nitrosothiols in lung inflammation. Therapeutic targets of airway inflammation. In: Lung Biology in Health and Disease, edited by Eissa A. New York: Marcel Dekker, 2003, p. 123–134. [Google Scholar]
- 41.Meacham GC, Lu King S Z, Sorscher E, Tousson A, Cyr DM. The Hdj-2/Hsc70 chaperone pair facilities early steps in CFTR biogenesis. EMBO J 18: 1492–1505, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meacham GC, Patterson C, Zhang W, Younger J, Cyr D. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol 3: 100–105, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Mogayzel PJ, Flume PA. Update in cystic fibrosis 2009. Am J Respir Crit Care Med 181: 539–544, 2010. [DOI] [PubMed] [Google Scholar]
- 44.Okiyoneda T, Barriere H, Bagdany M, Rabeh WM, Du K, Hohfeld J, Young JC, Lukacs GL. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 329: 805–810, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palmer L, Doctor A, Chhabra P, Sheram ML, Laubach V, Karlinsey MZ, Forbes M, Macdonald T, Gaston B. S-nitrosothiols signal hypoxia-mimetic vascular pathology. J Clin Invest 117: 2592–2601, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perkins W, Pabelick C, Warner DO, Jones KA. CGMP-independent mechanism of airway smooth muscle relaxation induced by S-nitrosoglutathione. Am J Physiol Cell Physiol 275: C468–C474, 1998. [DOI] [PubMed] [Google Scholar]
- 47.Que LG, Liu L, Whitehead GS, Gavett SH, Schwartz DA, Stamler JS. Protection from experimental asthma by an endogenous bronchodilator. Science 308: 1618–1621, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem 77: 701–726, 2008. [DOI] [PubMed] [Google Scholar]
- 49.Rubenstein RC, Egan ME, Zeitlin PL. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J Clin Invest 100: 2457–2465, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rubenstein RC, Zeitlin PL. Sodium 4-phenybutyrate downregulates Hsc70: Implications for intracellular trafficking of ΔF508 CFTR. Am J Physiol Cell Physiol 278: C259–C267, 2000. [DOI] [PubMed] [Google Scholar]
- 51.Sanghani PC, Davis WI, Fears SL, Green SL, Tang Y, Martin E, Bryan NS, Sanghani SP. Kinetic and cellular characterization of novel inhibitors of S-nitrosoglutathione reductase. J Biol Chem 284: 24354–24362, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Servetnyk Z, Krujkova J, Gaston B, Zaman K, Hjelte L, Roomans G, Dragomir A. Activation of delF508 CFTR in CF airway epithelial cell lines and CF nasal epithelial cells by S-nitrosoglutathione. Respir Res 7: 124–130, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Skach WR. CFTR: new members join the fold. Cell 127: 673–675, 2006. [DOI] [PubMed] [Google Scholar]
- 54.Snyder A, McPherson ME, Hunt JF, Stamler JS, Gaston B. Acute effects of aerosolized S-nitrosoglutathione in cystic fibrosis. Am J Respir Crit Care Med 165: 1–5, 2002. [DOI] [PubMed] [Google Scholar]
- 55.Sun F, Mi Z, Condliffe SB, Bertrand CA, Gong X, Lu X, Zhang R, Latoche JD, Pilewski JM, Robbins PD, Frizzell RA. Chaperone displacement from mutant cystic fibrosis transmembrane conductance regulator restores its function in human airway epithelia. FASEB J 22: 3255–3263, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, Zhou J, McCartney J, Arumugam V, Decker C, Yang J, Young C, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu P. Rescue of a CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA 106: 18825–18830, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ward CL, Kopito RR. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem 269: 25710–25718, 1994. [PubMed] [Google Scholar]
- 58.Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin proteasome pathway. Cell 83: 121–127, 1995. [DOI] [PubMed] [Google Scholar]
- 59.Yang Y, Loscalzo J. S-nitrosoprotein formation and localization in endothelial cells. Proc Natl Acad Sci USA 102: 117–122, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Younger JM, Fan CY, Chen L, Rosser MF, Patterson C, Cyr DM. Cystic fibrosis transmembrane conductance regulator as a model substrate to study endoplasmic reticulum protein quality control in mammalian cells. Methods Mol Biol 301: 293–303, 2005. [DOI] [PubMed] [Google Scholar]
- 61.Younger JM, Ren HY, Chen L, Fan CY, Fields A, Patterson C, Cyr DM. A foldable CFTR ΔF508 biogenic intermediate accumulates upon inhibition of the Hsc70-CHIP E3 ubiquitin ligase. J Cell Biol 167: 1075–1085, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zaman K, Bennett D, Fraser-Butler M, Greenberg Z, Getsy P, Sattar A, Smith L, Corey D, Sun F, Hunt J, Lewis SJ, Gaston B. S-nitrosothiols increases cystic fibrosis transmembrane regulator expression and maturation in the cell surface. Biochem Biophys Res Commun 443: 1257–1262, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zaman K, Carraro S, Doherty J, Henderson E, Lendermon E, Liu L, Verghese G, Ross M, Palmer L, Doctor A, Stamler J, Gaston B. A novel class of compounds that increase CFTR expression and maturation in epithelial cells. Mol Pharmacol 70: 1435–1442, 2006. [DOI] [PubMed] [Google Scholar]
- 64.Zaman K, McPherson M, Vaughan J, Hunt J, Mendes F, Gaston B, Palmer L. S-Nitrosoglutathione increases cystic fibrosis transmembrane regulator maturation. Biochem Biophys Res Commun 284: 65–70, 2001. [DOI] [PubMed] [Google Scholar]
- 65.Zaman K, Palmer L, Doctor A, Hunt J, Gaston B. Concentration-dependent effects of endogenous S-nitrosoglutathione on gene regulation by specificity proteins Sp3 and Sp1. Biochem J 380: 67–74, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zeitlin PL, Diener-West M, Rubenstein RC, Boyle MP, Lee C, Brass-Ernst L. Evidence of CFTR function in cystic fibrosis after systemic administration of 4-phenylbutyrate. Mol Ther 6: 119–126, 2002. [DOI] [PubMed] [Google Scholar]