

SUMMARY
The molecular basis by which receptor tyrosine kinases (RTKs) recruit and phosphorylate Src Homology 2 (SH2) domain-containing substrates has remained elusive. We used X-ray crystallography, NMR spectroscopy, and cell-based assays to demonstrate that recruitment and phosphorylation of Phospholipase Cγ (PLCγ), a prototypical SH2 containing substrate, by FGF receptors (FGFR) entails formation of an allosteric 2:1 FGFR-PLCγ complex. We show that the engagement of pTyr-binding pocket of the cSH2 domain of PLCγ by the phosphorylated tail of an FGFR kinase induces a conformational change at the region past the cSH2 core domain encompassing Tyr-771 and Tyr-783 to facilitate the binding/phosphorylation of these tyrosines by another FGFR kinase in trans. Our data overturn the current paradigm that recruitment and phosphorylation of substrates are carried out by the same RTK monomer in cis, and disclose an obligatory role for receptor dimerization in substrate phosphorylation in addition to its canonical role in kinase activation.
INTRODUCTION
Receptor tyrosine kinase (RTK) signaling plays essential roles in human biology and pathology (Hunter, 2000; Lemmon and Schlessinger, 2010). Ligand-induced dimerization of the extracellular domains of RTKs juxtaposes the cytoplasmic kinase domains to enable kinase trans-tyrosine phosphorylation (Chen et al., 2008; Goetz and Mohammadi, 2013; Hubbard, 2004; Hunter, 2002; Jura et al., 2011; Lemmon and Schlessinger, 2010). Phosphorylation of tyrosines in the regulatory kinase A-loop elevates the intrinsic kinase activity in an allosteric fashion (Chen et al., 2007; Pellicena and Kuriyan, 2006; Rajakulendran and Sicheri, 2010). In contrast, phosphorylation on tyrosines in the juxtamembrane region, kinase insert or C-terminal tail creates specific recruitment sites for Src Homology 2 (SH2)– or phosphotyrosine binding (PTB)–containing intracellular substrates including enzymes and adaptor proteins (Lemmon and Schlessinger, 2010; Pawson, 2004; Schlessinger and Lemmon, 2003) to facilitate their phosphorylation, which then triggers activation of specific intracellular signaling pathways leading to distinct biological responses (Rotin et al., 1992; Schlessinger and Lemmon, 2003). For example, phosphorylation of a conserved tyrosine in the C-terminal tail of FGFRs creates a docking site for Phospholipase Cγ1 (PLCγ1), a tandem SH2-containing substrate (Eswarakumar et al., 2005; Mohammadi et al., 1991; Peters et al., 1992). This recruitment plays a dual role in the activation of the PLCγ pathway: 1) it facilitates phosphorylation of PLCγ which relieves PLCγ autoinhibition, resulting in upregulation of the lipase activity of the enzyme (Bunney et al., 2012b; Hajicek et al., 2013; Poulin et al., 2005), 2) it translocates PLCγ to the vicinity of its substrate phosphatidylinositol 4,5-bisphosphate (PIP2) in the plasma membrane, where the activated enzyme can hydrolyze PIP2 leading to the generation of the second messengers DAG and IP3 (Ellis et al., 1998; Schlessinger, 1997).
There is a major gap in our understanding of the molecular mechanism by which SH2– or PTB–mediated recruitment/phosphorylation of substrates takes place. The recent elucidation of the crystal structure of 1:1 complex of the activated FGFR1 kinase with the tandem nSH2-cSH2 domain fragment of PLCγ (PDB ID 3GQI (Bae et al., 2009) has been the closest attempt to address this fundamental process in RTK signaling. In this structure, the tyrosine phosphorylated C-tail of FGFR1 kinase binds the nSH2 domain of PLCγ but none of the three PLCγ phosphorylation sites included in the tandem SH2 construct are engaged by the active site of the FGFR1 kinase leaving it uncertain as to how SH2-mediated recruitment facilitates substrate phosphorylation.
Here, we used an assortment of X-ray crystallography, Nuclear Magnetic Resonance (NMR) spectroscopy, mass spectrometry and other biophysical and cell-based experiments to show how two FGFR kinases act cooperatively to recruit and phosphorylate PLCγ, thus overturning the current paradigm that recruitment and phosphorylation of the substrates are carried out by the same receptor i.e. in cis. In addition, our data unravel the molecular basis by which the phosphorylated substrate is disembarked from the FGFR allowing for next cycles of recruitment and phosphorylation to ensue.
RESULTS AND DISCUSSION
C-terminal SH2 Domain (cSH2) of PLCγ Mediates the Recruitment and Phosphorylation of PLCγ by FGFR, EGFR, PDGFR and VEGFR in Living Cells
PLCγ is a key signaling protein for a number of RTKs including FGFRs, EGFRs, PDGFRs, VEGFRs, and NGFRs (Kim et al., 1991; Lemmon and Schlessinger, 2010; Liu et al., 2012; Pascal et al., 1994). PLCγ possesses tandem SH2 domains, an N-terminal SH2 (nSH2) and a C-terminal SH2 (cSH2), which share about 35% sequence identity. To test the roles of the two SH2 domains in the recruitment and phosphorylation of PLCγ by RTKs in living cells, we ablated the phosphotyrosine (pTyr) binding function of the nSH2 and cSH2 domains individually or in combination by mutating the arginine from the SH2 domain-invariant FLVR motif (Bae et al., 2009; Hidaka et al., 1991) within the pTyr binding pocket (Arg-586 of nSH2 and Arg-694 of cSH2) to alanine (Figure 1A) and transiently transfected the wild-type or mutated PLCγ constructs into PLCγ1−/− mouse embryonic fibroblasts (MEF) (Ji et al., 1997). Conveniently PLCγ1−/− MEF endogenously expresses FGFR1, EGFR and PDGFR obviating the need for transfecting these three RTKs into these cells. However, since PLCγ1−/− MEF do not express VEGFR2, a stable cell line overexpressing VEGFR2 was established. Transfected cells were stimulated with FGF1, EGF, PDGF, or VEGF, and phosphorylation on Tyr-783, one of the three major phosphorylation sites of PLCγ, was analyzed. Inactivation of the cSH2 domain alone (N+C−) or of both SH2 domains (N−C−) abrogated the ability of all four RTKs to phosphorylate PLCγ whereas inactivation of the nSH2 domain alone (N−C+) had no effect (Figure 1B). Consistent with the PLCγ phosphorylation data, the PLCγ construct with ablated cSH2 domain failed to co-precipitate with the activated FGFR1 whereas the PLCγ construct with disabled nSH2 domain co-precipitated with FGFR1 as efficiently as the wild-type PLCγ did (Figure 1C). Moreover, cells transfected with PLCγ devoid of a functional cSH2 domain failed to respond to FGF1 with an increase in IP3 production whereas FGF1 treatment of cells transfected with PLCγ lacking a functional nSH2 domain gave rise to similar level of IP3 release as cells transfected with wild-type PLCγ (Figure 1D). These data demonstrate that the cSH2 domain of PLCγ mediates recruitment and phosphorylation of PLCγ by FGFR, VEGFR, EGFR and PDGFR in living cells.
Figure 1. The cSH2 domain of PLCγ is necessary and sufficient for PLCγ phosphorylation by FGFR, EGFR, PDGFR and VEGFR in living cells.
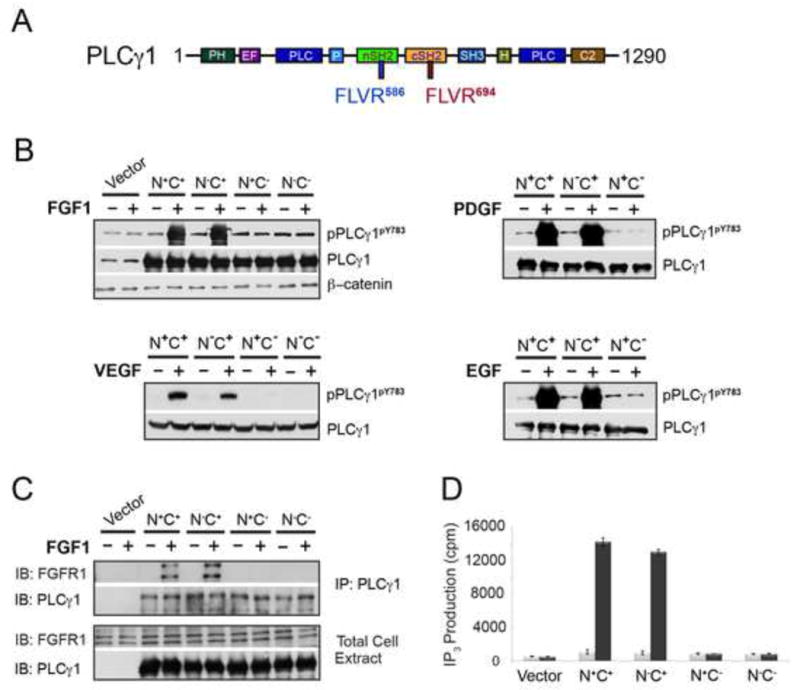
(A) Domain organization of full-length PLCγ. The FLVR motifs in the nSH2 and cSH2 domains are labeled in blue and red, respectively. (B) Immunoblotting analysis of PLCγ phosphorylation by FGFR, EGFR, VEGFR2 and PDGFR in living cells. (C) Co-immunoprecipitation assay using wild-type and mutated PLCγ constructs in living cells. PLCγ1−/− MEF were transfected with the indicated PLCγ constructs and stimulated with FGF1. (D) FGF-induced PI hydrolysis in PLCγ1−/− MEF cells transfected with the indicated wild-type and mutated PLCγ constructs. Data are represented as mean +/− SEM (n=3). See also Figures S1 and S2.
C-terminal SH2 Domain (cSH2) of PLCγ is Sufficient and Necessary for the Recruitment of PLCγ by the FGFR Kinase in Vitro
Next, we sought to confirm the requirement for the cSH2 domain in recruitment of PLCγ by FGFR kinase in vitro. To this end, recombinant wild-type tandem SH2 fragment of PLCγ (nSH2-cSH2) and the corresponding three mutated fragments (nSH2R586A-cSH2, nSH2-cSH2R694A, and nSH2R586A-cSH2R694A) were made (Figure 2A). As for the recombinant FGFR kinase, the kinase domain of FGFR2 including C-terminal Tyr-769, whose phosphorylation is required for PLCγ recruitment and phosphorylation, was made. To generate a homogeneously phosphorylated kinase sample, all of the autophosphorylation sites of FGFR2 kinase except for Tyr-769 were mutated to non-phosphorylatable residues. To drive the kinase into the active state conformation in the absence of A-loop tyrosine phosphorylation, two pathogenic gain-of-function mutations, Glu565Ala (E565A) and Lys659Glu (K659E) (Chen et al., 2013; Chen et al., 2007; Naski et al., 1996), were introduced into the kinase (Figure 2A). Binding of wild-type or mutated tandem nSH2-cSH2 fragments of PLCγ (nSH2-cSH2, nSH2R586A-cSH2, nSH2-cSH2R694A, and nSH2R586A-cSH2R694A) to the activated FGFR2 kinase, which is mono-phosphorylated on Tyr-769 (FGFR2KpY769), was studied using isothermal titration calorimetry (ITC), surface plasmon resonance (SPR) spectroscopy (Figures 2B–2E), size exclusion chromatography (SEC) and native gel electrophoresis (Figures S1A–1H). In ITC experiments, the nSH2R586A-cSH2 construct bound FGFR2KpY769 with a comparable affinity as the wild-type nSH2-cSH2 construct did whereas both nSH2-cSH2R694A, and nSH2R586A-cSH2R694A failed to bind FGFR2KpY769 (Figures 2B–2E). Consistent with the ITC data, SPR experiments showed that inactivation of nSH2 had a subtle effect on the binding of tandem SH2 to the immobilized FGFR2KpY769 whereas the cSH2-inactivated nSH2-cSH2R694A construct showed a major loss in binding to the immobilized FGFR2KpY769 (Figures 2B–2E). To further corroborate these data, binding interactions of FGFR2KpY769 with the individual nSH2 and cSH2 domains were also studied. As shown in Figure 2F, cSH2 domain bound FGFR2KpY769 with a comparable affinity as the wild-type tandem nSH2-cSH2 construct whereas only weak binding of nSH2 domain could be observed at very high concentrations (Figure 2G, insert panel). Lastly, we also examined binding interactions of the individual SH2 domains with FGFR2KpY769 using solution NMR spectroscopy. Uniformly 15N-labeled nSH2 and cSH2 domains were prepared and their HSQC spectra in the absence and presence of 0.5, 1, and 2 molar equivalents of unlabeled FGFR2KpY769 were acquired (Figures S1I–1L). Titration of cSH2 domain with a one-half molar equivalent of FGFR2KpY769 led to major chemical shift perturbations in the cSH2 domain and resulted in the appearance of a new set of peaks representing the complexed form (i.e., slow chemical exchange) (Figures S1I and 1K). Upon further addition of FGFR2KpY769 to give a 1:1 molar ratio, only the peaks corresponding to the bound form of cSH2 were observed, which shows that cSH2 domain and FGFR2KpY769 form a tight 1:1 complex in solution. To the contrary, FGFR2KpY769 did not induce any new peaks in the spectrum for the nSH2 domain when titrated at 1:1 molar ratio (Figures S1J and 1L). Only reductions in peak intensity and insignificant chemical shift perturbations were observed for some of the resonances which likely stemmed from intermediate and fast exchange, respectively, indicative of poor binding affinity between nSH2 domain and FGFR2KpY769 relative to the cSH2 domain.
Figure 2. The cSH2 domain of PLCγ mediates the recruitment of PLCγ by FGFR kinase in vitro.
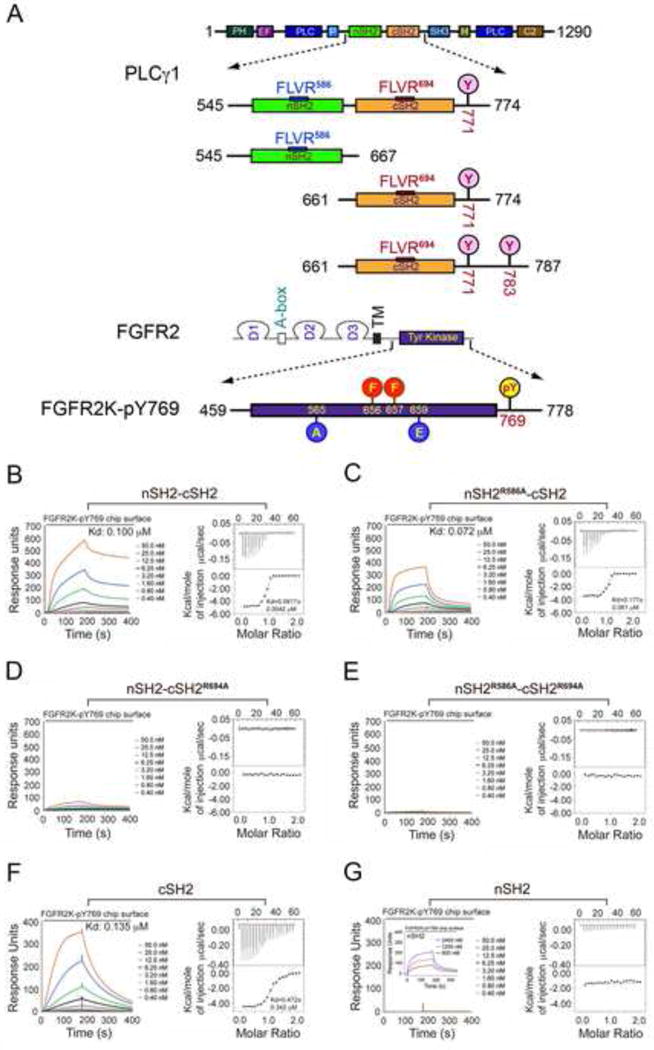
(A) Domain organization of PLCγ and FGFR2 as well as the design of the engineered PLCγ and FGFR2 fragments used in this study. Phosphorylation tyrosine sites included in the tandem SH2 and isolated cSH2 fragments are marked with pink circles. The phosphorylation sites in the A-loop of FGFR2 kinase (Tyr-656 and Tyr-657), which were mutated to phenylalanine, are indicated with red circles. The location of the two gain-of-function mutations introduced to force the kinase into the active state is indicated with blue circles. Tyr-769, the single remaining phosphorylation site in the FGFR2K is highlighted with a yellow circle. (B–G) Analysis by SPR and ITC of the recruitment of wild-type tandem nSH2-cSH2 fragment of PLCγ (B), nSH2R586A-cSH2 (C), nSH2-cSH2R694A (D), nSH2R586A-cSH2R694A (E), cSH2 domain (F) and nSH2 domain (G) of PLCγ by activated FGFR2KpY769. Insert panel of Panel G is SPR binding analysis of nSH2 domain at high concentrations with activated FGFR2KpY769. See also Figures S1 and S2.
To exclude the possibility that the presence of the gain-of-function mutations may have biased the binding of FGFR2KpY769 towards cSH2 domain, we also analyzed binding interactions of phosphorylated wild-type FGFR2K with wild-type and mutated tandem nSH2-cSH2 fragments using native gel electrophoresis (Figure S2). This analysis showed that the phosphorylated wild-type FGFR2K also binds selectively to the cSH2 domain. Lastly we also tested whether cSH2-mediated recruitment of PLCγ is applicable to all FGFRs by analyzing the binding interactions of phosphorylated wild-type FGFR1, FGFR3, and FGFR4 kinases with wild-type or mutated tandem nSH2-cSH2 fragments using native gel electrophoresis (Figure S2). The results clearly show that all four human FGFRs recruit PLCγ via its cSH2 domain.
Crystal Structure of FGFR2KpY769 in Complex with the PLCγ cSH2 Domain Reveals a 2:1 FGFR-PLCγ Stoichiometry
Having demonstrated that the cSH2 domain is necessary and sufficient for the recruitment and phosphorylation of PLCγ, we then solved the crystal structure of FGFR2KpY769 in complex with a PLCγ cSH2 fragment consisting of residues Asn-661 to Leu-774 in the presence of a non-hydrolyzable ATP analog (AMP-PCP) at 2.6 Å resolution (Figures 3 and 4). This cSH2 construct includes the cSH2 domain and Tyr-771, the first phosphorylation site of PLCγ past the cSH2 domain.
Figure 3. Recruitment and phosphorylation of cSH2 cannot be accomplished by the same kinase in cis.
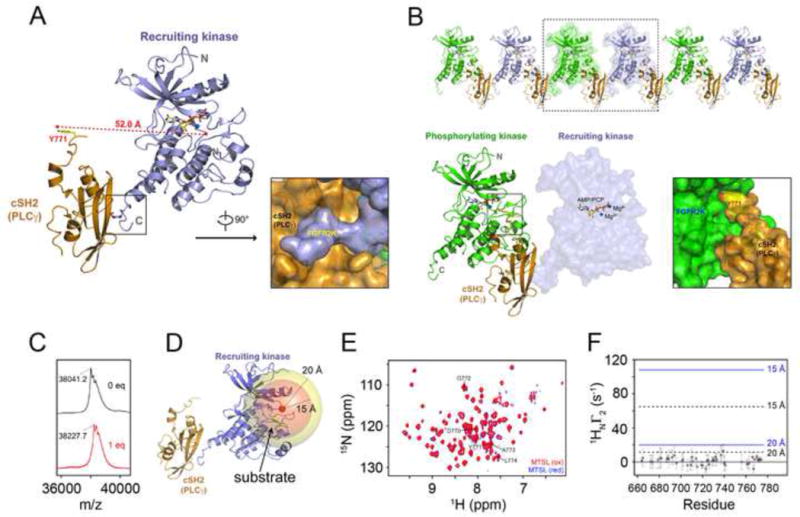
(A) Ribbon diagram of the crystal structure of the complex between mutationally-activated monophosphorylated FGFR2KpY769 in complex with PLCγ cSH2 domain. Surface representation showing insertion of phosphorylated C-terminal tail of recruiting kinase into the phosphotyrosine binding pocket of the PLCγ cSH2 domain. (B) 2:1 FGFR2 kinase-PLCγ cSH2 complex is observed in the crystal lattice. Surface representation showing insertion of Tyr-771 phosphorylation site from the PLCγ cSH2 domain into the active site of the phosphorylating kinase. The phosphorylating kinase and the recruiting kinase are colored green and lightblue, respectively. The PLCγ cSH2 domain is colored orange. The ATP analogue (AMP-PCP) and magnesium ions are rendered as sticks and blue spheres, respectively. (C) MALDI-TOF results of MTSL labeling of Cys-491 in the glycine-rich loop of FGFR2KpY769 (FGFR2K-pY769MTSL). The observed mass/charge difference upon labeling with MTSL was 186.5, which agrees closely with the expected value of 186.3. (D) Crystal structure of recruiting FGFR2K-pY769 bound to cSH2 showing the Cβ of Cys-491 (red sphere) and two spheres of 15 Å and 20 Å surrounding this site. A bound substrate tyrosine in the catalytic pocket, shown in green color stick, would be expected to lie within the 15 Å sphere. (E) 1H/15N TROSY spectra of 1:1 samples of cSH2:FGFR2K-pY769MTSL before (intact MTSL; red) and after treatment with ascorbic acid (reduced MTSL; blue). The cSH2 samples were 70% deuterated. (F) 1H relaxation enhancement (Γ2) due to the presence of the MTSL spin label probed using a two-point TROSY method where the difference in the relaxation delays were 10 msec (Iwahara et al., 2007). Y771 is shown as a red circle. Distances of 15 Å and 20 Å between the spin label and amide proton are shown on the plot using correlation times of 15 nsec (black, dotted line) and 25 nsec (blue, solid line). A Γ2 value close to zero indicates a distance > ~25 Å from the spin label. Based on the crystal structures of FGFR kinases-substrate complexes, the distance between the tyrosine to be phosphorylated and adjacent residues ranges from ~10 to 13 Å. See also Figures S3, S4, S5 and S8.
Figure 4. Crystal structure of 2:1 FGFR2 kinase-PLCγ cSH2 complex.
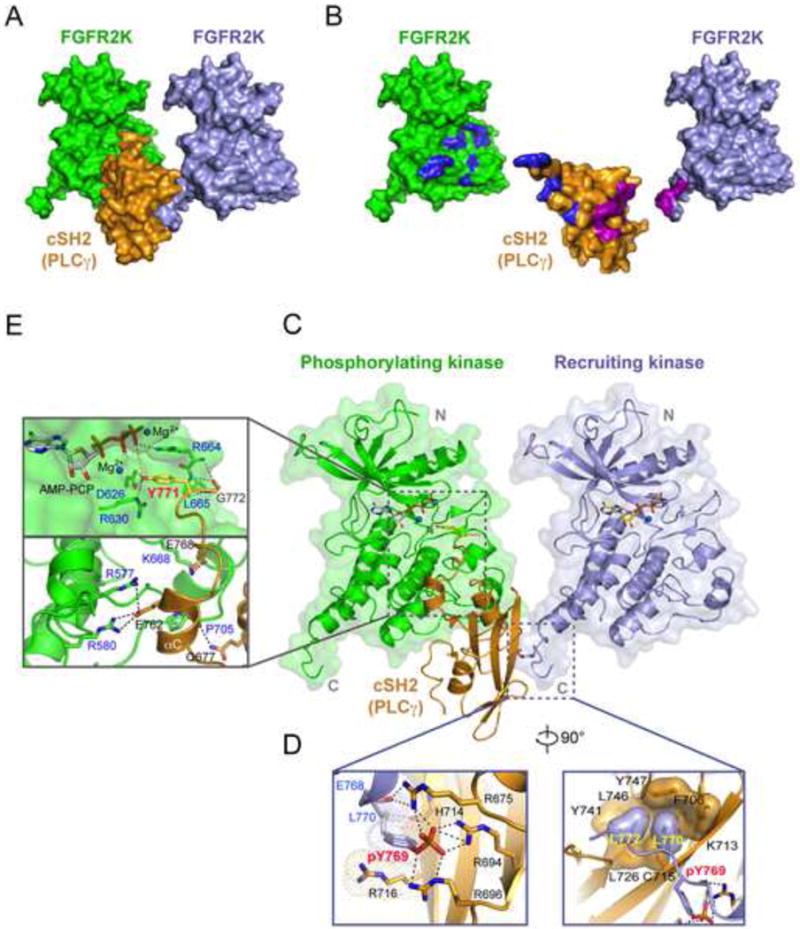
(A) Whole view of the 2:1 FGFR2 kinase-PLCγ cSH2 complex. (B) The binding interfaces between the cSH2 domain and the recruiting kinase are colored magenta and those between the cSH2 domain and the phosphorylating kinase are colored blue. (C) The phosphotyrosine binding pocket of the PLCγ cSH2 domain (in orange) is engaged by the phosphorylated tyrosine (pTyr-769) at the C-terminal tail of the recruiting kinase (in lightblue) while Tyr-771 of PLCγ is docked into the active site of the phosphorylating kinase (in green). (D) Close-up views of the interactions between the recruiting kinase and the cSH2 domain. (E) Close-up views of the interactions between the phosphorylating kinase and the PLCγ cSH2 domain. In panel D and E, side chains of key interacting residues are shown as sticks. Hydrogen bonds are shown as dashed lines. Oxygen atoms are colored red and nitrogen atoms are colored blue. The ATP analogue (AMP-PCP) and magnesium ions are rendered as sticks and blue spheres, respectively. See also Figures S4, S6, S8 and S9.
The asymmetric unit of the crystal contains a 1:1 FGFR2KpY769-PLCγ cSH2 complex mediated by contacts between the phosphorylated C-tail of FGFR2KpY769 and the pTyr binding pocket of the PLCγ cSH2 domain (Figure 3A) (Table S1). Despite lacking the A-loop tyrosines, FGFR2KpY769 adopts the active state conformation and contains an AMP-PCP molecule in its ATP binding cleft demonstrating that the E565A and K659E gain-of-function mutations have forced the kinase into active state without A-loop tyrosine phosphorylation (Figure S3). Surprisingly, however, the phosphorylatable Y771 of the PLCγ cSH2 domain is not bound in cis into the active site of the kinase in the 1:1 complex (Figure 3A). Instead Y771 binds in trans into the active site of the neighboring kinase in the crystal lattice where it is poised to be phosphorylated (Figure 3B). Hence, the FGFR2KpY769-PLCγ cSH2 complex structure implies that PLCγ recruitment and phosphorylation involves a 2:1 receptor-substrate complex wherein one receptor acts as “recruiter” and the other receptor serves as “phosphorylater”. In fact, structural analysis shows that due to spatial constraints, recruitment and phosphorylation of the PLCγ cSH2 domain cannot be carried out by the same kinase, that is, in cis. Tyr-771 is only five residues away from the end of the rigid core of PLCγ cSH2 domain, which is not enough to provide sufficient flexibility for Tyr-771 to reach the active site of the kinase (Figure S4A). Likewise, Tyr-769 of FGFR2 is only two residues away from the end of the αI helix (Figure S4A), which also does not provide enough C-terminal flexibility to position the cSH2 domain closer to the active site of the FGFR kinase such that phosphorylation of Tyr-771 can occur in cis. Nevertheless, to rule out the possibility that crystal packing contacts may have hindered binding in cis of Tyr-771 into the active site of the recruiting kinase, we performed paramagnetic relaxation enhancement (PRE) experiments to obtain proximity information between Tyr-771 of the cSH2 domain and the substrate tyrosine binding pocket of FGFR2KpY769. For this purpose, we attached an MTSL, a commonly used nitroxide paramagnetic spin label, to Cys-491 in the Gly-rich loop of the FGFR2KpY769 (Figures 3C and 3D). Conveniently, this naturally occurring cysteine is about 10 Å away from the catalytic base of the kinase which is well within the PRE transfer range of MTSL (Figure 3D). Following a series of triple resonance experiments to assign the backbone amide 1H/15N resonances of the cSH2 domain (Figure 3E), a 1:1 complex of 70% deuterated, 15N-labeled cSH2 with the spin-labeled FGFR2K-pY769MTSL was prepared and 1H T2 relaxation enhancements in the cSH2 domain was probed using a two-point TROSY method (Iwahara et al., 2007) where the difference in the relaxation delays was 10 msec. Following collection of the data on the paramagnetically labeled FGFR2K-pY769MTSL, ascorbic acid was added to reduce the nitroxide allowing us to measure relaxation rates in the absence of the paramagnetic state. As shown in Figure 3F, the corresponding 1H PRE Γ2 rates for C-terminal residues 760–773 of cSH2 which include the phosphorylatable tyrosine 771 are close to zero. Moreover, there are no discernable spectral differences between 15N-HSQC paramagnetic and diamagnetic spectra or spectra of the native complex prepared in the absence of MTSL incorporation. Taken together, these NMR PRE data show that Tyr-771 of cSH2 and its surrounding sequences are more than ~20 Å away from the spin label. To ensure that our PRE assay is capable of detecting spectral changes/rate enhancements within ~20 Å range, we performed a control PRE experiment wherein the minimal kinase domain of FGFR2 (residues 459 to 768; FGFR2Kshort) was isotopically enriched with 15N and labeled with MTSL on Cys-491 in the same manner as FGFR2KpY769. A comparison of paramagnetic and diamagnetic spectra of this construct shows several missing peaks in the former consistent with the distance dependent perturbation of the spin label on the Gly-rich loop (Figure S5). Taken together, these PRE experiments support the crystal structure showing that recruitment and phosphorylation cannot be accomplished by the same kinase in cis.
Interactions at the Interfaces between cSH2 and the Recruiting and Phosphorylating FGFR2 Kinases
The interface between the cSH2 molecule and the recruiting kinase molecule buries a total of 967 Å2 of solvent exposed surface area (Figures 4A and 4B). At this interface, the phosphorylated Tyr-769 (pTyr-769) and the following Leu-770 and Leu-772 of the recruiting kinase plug into the pTyr binding pocket of the cSH2 domain using the canonical “two-prong in socket” mechanism with pTyr-769 engaging the hydrophilic hole, and Leu-770 and Leu-772 engaging the hydrophobic hole of the socket (Figures 4C and 4D). pTyr-769 engages in a total of seven hydrogen bonds: two each with both Arg-675 and Arg-696, and three with Arg-694 of the PLCγ cSH2 domain. In addition, backbone atoms of Glu-768 and Leu-770 of the recruiting kinase molecule make hydrogen bonds with the side chain of Arg-675 and backbone carbonyl oxygen of His-714 of the PLCγ cSH2 domain. Another notable contact at this hydrophilic hole of the socket is the π-cation interaction between the phenyl ring of pTyr-769 and Arg-716 of the PLCγ cSH2 domain (Figure 4D). In the second hydrophobic hole of the socket, Leu-770 and Leu-772 of the recruiting kinase are immersed in hydrophobic contacts with Phe-706, Cys-715, Leu-726, Tyr-741, Leu-746, and Tyr-747 of the PLCγ cSH2 domain (Figure 4D).
The interface between the cSH2 molecule and the phosphorylating kinase molecule buries a total of 1246 Å2 of solvent exposed surface area (Figures 4A and 4B), and harbors several canonical kinase-substrate contacts at the enzyme active site (Figures 4C and 4E). The hydroxyl moiety of Tyr-771 makes two short hydrogen bonds with the catalytic base (Asp-626) of the kinase and is also engaged in a π-cation interaction with Arg-664 from the A-loop of the phosphorylater kinase. In addition to these canonical contacts proximal to the enzyme active site, the phosphorylating kinase and the PLCγ cSH2 domain engage each other in six hydrogen bonding contacts away from the enzyme active site. These distal contacts include the bidentate hydrogen bonds between Glu-762 of PLCγ and Arg-580 of the kinase, two hydrogen bonds between Glu-768 of PLCγ and Lys-668 of the kinase, and one hydrogen bond between Gln-677 of PLCγ and the kinase backbone oxygen of Pro-705 (Figure 4E). Harmonious with the requirement for the cSH2 domain in mediating PLCγ phosphorylation by FGFR kinase, Gln-677, Glu-762, and Glu-768 are fully conserved among the PLCγ orthologs across species (Figure S6A).
The cSH2 Recruitment by FGFR2KpY769 Induces a Conformational Change at the C-tail of cSH2
Based on the structure, we inferred that the engagement of pTyr binding pocket of the cSH2 domain by the recruiting kinase might induce conformational changes in the cSH2 that facilitate its phosphorylation on Tyr-771 by the phosphorylating kinase. Hence we employed NMR spectroscopy to probe the conformational changes of the cSH2 domain upon recruitment. To this end, chemical shift perturbation of perdeuterated, 15N-labeled cSH2 in the presence of FGFR2KpY769 were calculated and plotted as function of residue (Figure 5A). Mapping the perturbed residues onto the crystal structure revealed three clusters of residues in the cSH2 domain that incur significant chemical shifts/signal attenuation in the presence of FGFR2KpY769 (Figures 5B–5D). One cluster is comprised of residues within/or close to the pTyr binding pocket that binds the phosphorylated C-terminal tail of the recruiting kinase in the crystal structure. Two other regions with significant chemical shift perturbations included the core and C-terminal tail of cSH2 domain that face the phosphorylating kinase opposite to the pTyr binding pocket. Interestingly, we observed a new set of resonances for the C-terminal residues of cSH2 domain including Tyr-771 (Figures 5B and 5C) indicating that the C-terminus of the cSH2 domain undergoes a conformational change when the pTyr pocket of cSH2 is engaged by the phosphorylated C-tail of FGFR2KpY769.
Figure 5. Upon recruitment via its pTyr binding pocket to the phosphorylated tail of FGFR2KpY769, the cSH2 domain experience global conformational changes that facilitate the phosphorylation of C-terminal Tyr-771 and Tyr-783 of cSH2 by another FGFR2KpY769 in trans.
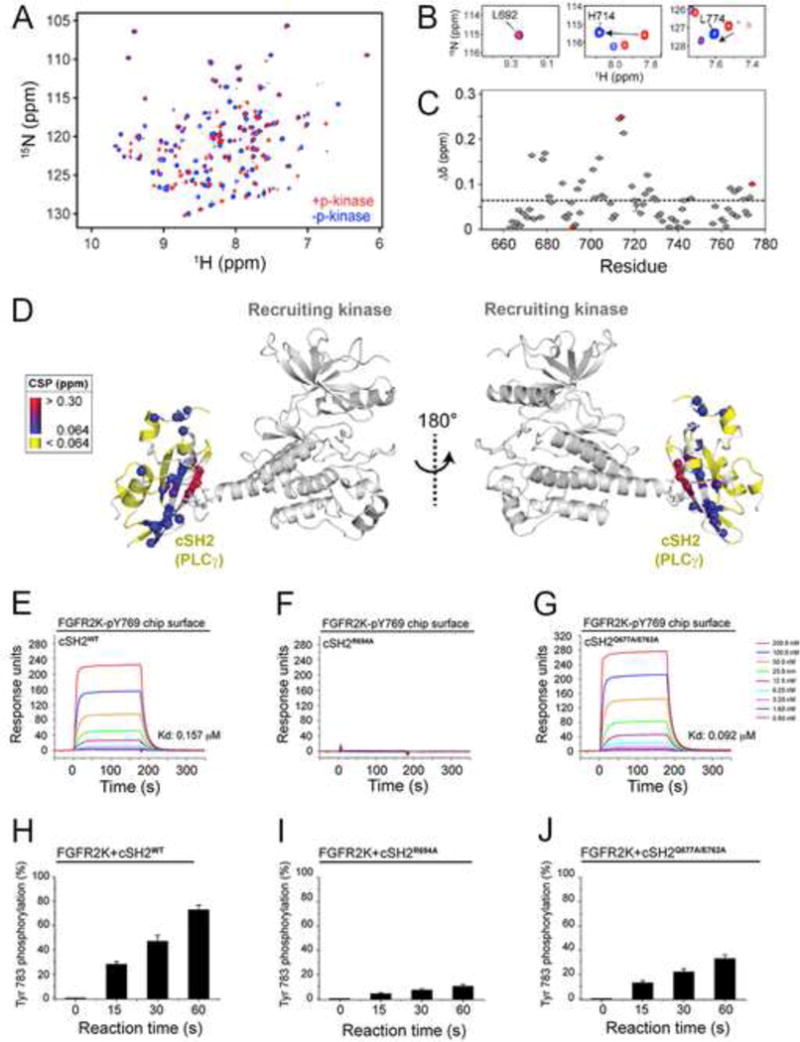
(A) Overlay of 1H/15N TROSY spectra of free cSH2 and a 1:1 complex of cSH2:FGFR2KpY769. (B) Examples of specific residues experiencing chemical shift perturbations from panel A. (C) Combined chemical shift perturbation plot showing the difference between free and bound cSH2 as a function of residue. The red circles correspond to the three residues shown in panel B. (D) Chemical shift perturbations from panel C mapped onto the structure. (E–G) Comparison of binding interactions of wild-type cSH2 domain and mutants thereof harboring R694A single and Q677A/E762A double mutations by SPR. (H–J) Comparison of phosphorylation of wild-type cSH2 domain and mutants thereof harboring R694A single and Q677A/E762A double mutations using in vitro kinas assay. In panels H–J, wild-type and mutated cSH2 domains were offered as substrate to the FGFR2KpY769 in the presence of ATP:MgCl2 and phosphorylation of cSH2 domain on Tyr-783 was quantitated using mass spectrometry. Data are represented as mean ± SEM (n=3). See also Figure S7.
To functionally test this possible conformational rearrangement in the cSH2 domain, we compared phosphorylation of the wild-type cSH2 and the cSH2R694A mutant on Tyr-783 by FGFR2KpY769 in vitro (Figures 5E and 5F, 5H and 5I). The cSH2R694A mutant harbors an alanine in place of the critical arginine from the FLVR motif which disables cSH2 domain from binding to FGFR2KpY769 as determined by SPR (Figure 5F). Compared to the wild-type cSH2, the cSH2R694A mutant was phosphorylated to a significantly lower amount (Figure 5I) supporting our NMR data that recruitment of cSH2 domain to one kinase molecule facilitates the trans-phosphorylation of the tyrosine located in the C-terminal tail of cSH2 domain by another kinase.
To provide further evidence for the 2:1 FGFR2KpY769-cSH2 complex in solution, we studied the impact of Glu-762-Ala and Gln-677-Ala mutations on the recruitment and trans-phosphorylation of the cSH2 domain on Tyr-783 by the FGFR2KpY769 in vitro. As these two residues interact with the “phosphorylater” kinase (Figure 4), we reasoned that their mutations should impair phosphorylation of the cSH2 domain on its tail tyrosine without impacting recruitment of cSH2 domain to FGFR2KpY769. Consistent with the crystal structure, binding analysis by SPR spectroscopy shows that the cSH2Q677A/E762A mutant binds with comparable affinity to the FGFR2KpY769 as the wild-type cSH2 does (Figure 5G). Despite retaining the full capacity to be recruited, the cSH2Q677A/E762A mutant was phosphorylated to a much lesser degree than the wild-type cSH2 domain (Figure 5J). Lastly, we also devised an in vitro kinase complementation to demonstrate that phosphorylation of cSH2 domain on tyrosines occurs in trans. In this experiment, which is shown schematically in Figure S7, an enzymatically-dead but recruitment-able version of FGFR2K harboring the K514M mutation was monophosphorylated on Tyr-769 (pY769-FGFR2Kdead) by an FGFR2K lacking Tyr-769 (FGFR2Kshort) in trans which was subsequently complexed with either wild-type cSH2 domain or the cSH2Q677A/E762A. Purified 1:1 pY769-FGFR2Kdead:cSH2 and pY769-FGFR2Kdead:cSH2Q677A/E762A were then mixed with the recruitment-deficient FGFR2Kshort and time-dependent phosphorylation on Tyr-771 of cSH2 was quantitated by mass spectrometry and expressed as % total tryptic peptide containing Tyr-771. As shown in Figure S7, the cSH2Q677A/E762A mutant was also phosphorylated to a much lesser degree than the wild-type cSH2 domain. Taken together with the NMR results, these data provide functional evidence for formation of an allosteric 2:1 FGFR2KpY769-cSH2 complex that is necessary for phosphorylation of cSH2 domain on tyrosine in solution.
Cell-based Experiments Validate the Structurally-deduced 2:1 RTK-PLCγ Phosphorylation Model
To test the biological relevance of our structurally-deduced 2:1 RTK-substrate phosphorylation model (Figure 6A), we also examined the impact of mutating Glu-762 and Gln-677 of PLCγ to alanine on the phosphorylation of PLCγ by activated RTKs in living cells. Consistent with the results of in vitro transphosphorylation, phosphorylation of PLCγQ677A/E762A in cells was also significantly reduced even though PLCγQ677A/E762A was effectively co-precipitated with the activated RTKs such as FGFR2, PDGFR and VEGFR2 (Figures 6B–6D). Our ability to uncouple recruitment and phosphorylation of PLCγ from one another both in vitro and in living cells provides strong evidence in support of the 2:1 RTK-substrate model whereby PLCγ is engaged by two RTKs with one receptor serving as “recruiter” and the other acting as the “enzyme”.
Figure 6. Mutations of PLCγ residues that interface with the “phosphorylating” kinase impair PLCγ phosphorylation without impacting PLCγ recruitment to the FGFR, PDGFR and VEGFR.
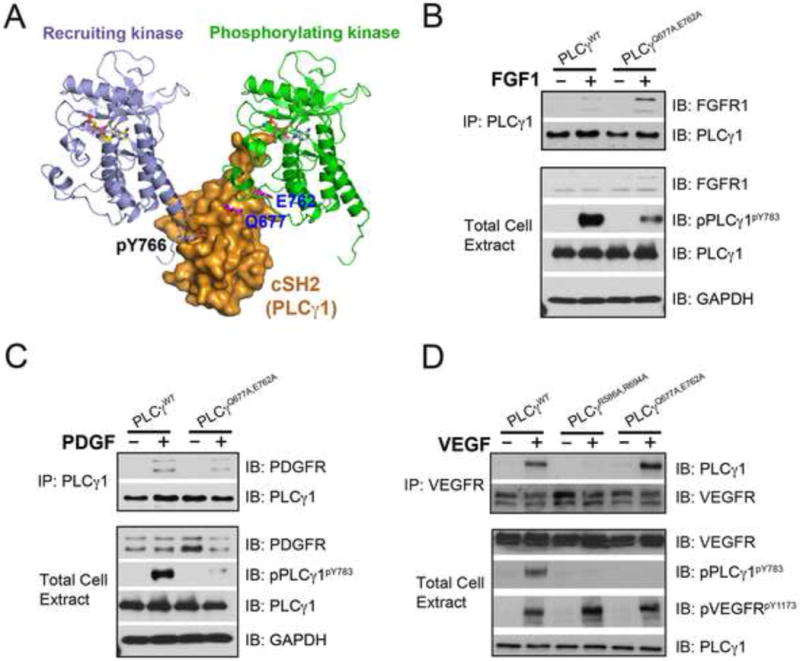
(A) Gln-677 and Glu-762 of PLCγ and the phosphotyrosine binding pocket of cSH2 domain lie on the opposing faces of cSH2 domain. (B–D) PLCγ null fibroblasts were transfected with expression vectors for wild-type PLCγ, and the indicated PLCγ mutants. Following cell stimulation with 50 ng/ml FGF1, PDGF or VEGF, cell lysates were blotted with the indicated antibodies. See also Figure S7.
To further validate the 2:1 RTK-PLCγ recruitment/phosphorylation model, we devised a complementation assay in living cells. In this assay, two full-length RTK mutants are made as follows: one containing mutation of the lysine from the ATP binding cleft, and another carrying mutation of the C-terminal tyrosine, whose phosphorylation is necessary for PLCγ recruitment, to phenylalanine. Both mutants are defective in PLCγ phosphorylation either due to lack of phosphotransfer activity (“kinase-dead”) or inability to recruit PLCγ. We reasoned that if our 2:1 model holds true, then these receptor mutants should be able to complement each other in phosphorylating PLCγ when co-expressed on the surface of cells and treated with ligand. Specifically, ligand treatment should induce heterodimerization between half of these mutants (the other half being inactive mutant homodimers) allowing the enzymatically active but recruitment-deficient RTK to phosphorylate the “kinase-dead” on the C-terminal tyrosine, thereby creating a docking site for the cSH2 domain of PLCγ on the “kinase-dead” RTK. The phosphorylated “kinase-dead” RTK would then recruit PLCγ offering the substrate for phosphorylation in trans to the recruitment-deficient but kinase active RTK in the context of heterodimer (Figure 7A). We chose to test this using FGFR1 and VEGFR2 as examples. A lentiviral expression system was used to express the FGFR1 mutants in BaF3 cells, which lack endogenous FGFR expression. Indeed, as shown in Figure 7B, treatment of cells co-infected with both constructs resulted in phosphorylation of PLCγ, whereas no PLCγ phosphorylation was observed in cells infected with either of the two FGFR1 mutants individually. As predicted from our 2:1 model, the phosphorylation level of PLCγ in the co-transfected cells was about 50% of that observed in cells transfected with wild-type FGFR1 (Figures 7B and 7C). As for VEGFR2, porcine aortic endothelial (PAE) cell lines stably expressing kinase-dead (VEGFR2K866R) or recruitment-deficient (VEGFR2Y1173F) VEGFR2 mutants were made. The VEGFR2Y1173F expressing cells were then used to infect with retrovirus expression vector for recruitment-deficient VEGFR2K866R. Upon treatment with VEGF there was no detectable phosphorylation of PLCγ in the VEGFR2K866R or VEGFR2Y1173F cells. In contrast, in VEGFR2Y1173F cells infected with retroviral vector for VEGFR2K866R, phosphorylation of PLCγ was induced upon ligand treatment (Figures 7D and 7E). These cell-based data unambiguously demonstrate that substrate phosphorylation occurs in the context of an FGFR or VEGFR dimer wherein one receptor monomer serves as the recruiter and the other acts as the phosphorylating enzyme.
Figure 7. Receptor complementation experiment confirms the validity of the 2:1 RTK-substrate model in living cells.
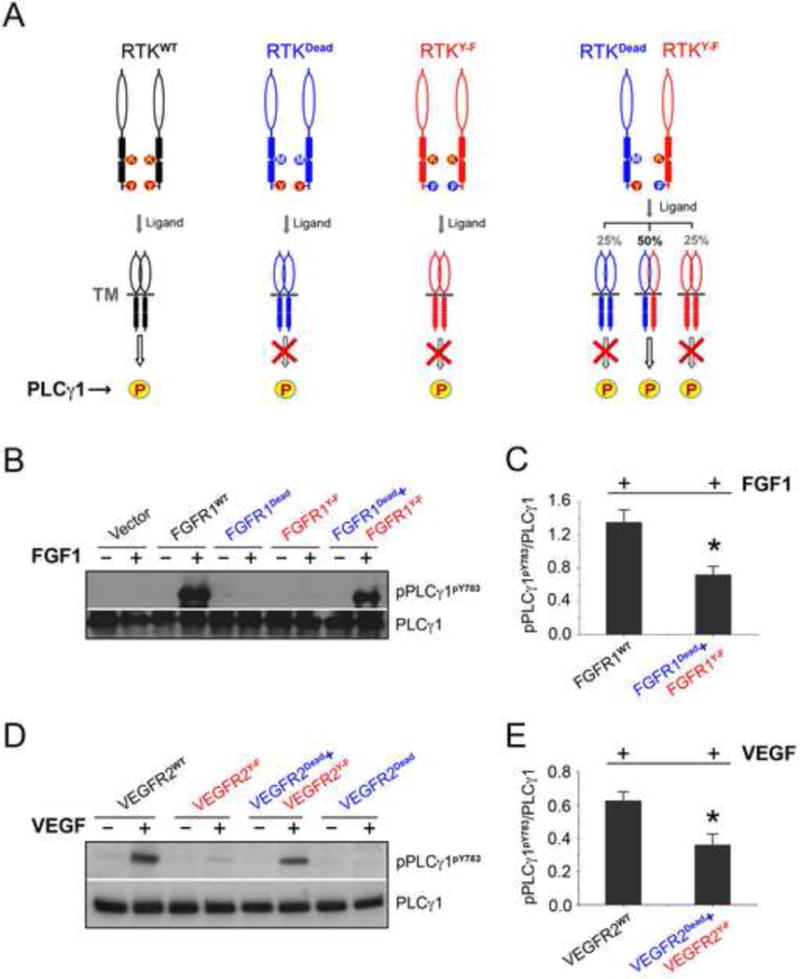
(A) Schematic of the receptor complementation experiment used to validate the 2:1 RTK-substrate model in living cells. RTKdead is devoid of kinase activity and hence cannot phosphorylate PLCγ. RTKY−F is catalytically active but is also defective in phosphorylating PLCγ as it cannot recruit PLCγ. (B&C) FGFR1Dead(FGFR1K514M) and FGFR1Y−F(FGFR1Y766F) receptors complement each other in phosphorylating PLCγ. BaF3 cells were infected with lentiviral expression vectors for FGFR1WT, FGFR1K514M, and FGFR1Y766F individually or co-infected with FGFR1K514M and FGFR1Y766F. Cells were stimulated with FGF1, and cell lysates were analyzed by Western blotting with the indicated antibodies (B). Semi-quantitation of the phosphorylation level of PLCγ was from Western blotting result (C). Data are represented as mean ± SEM (n=3), *p<0.01. (D&E) VEGFR2Dead(VEGFR2K866R) and VEGFR2Y−F(VEGFR2Y1173F) receptors complement each other in phosphorylating PLCγ. Cells were stimulated with VEGF, and cell lysates derived from PAE cells stably expressing wild-type VEGFR2, VEGFR-2Y1173F alone or co-expressing VEGFR2Y1173F with kinase dead receptor, VEGFR2K866R were blotted for total PLCγ1 and phospho-PLCγ1 (D). Semi-quantitation of the phosphorylation level of PLCγ was from Western blotting result (E). Data are represented as mean ± SEM (n=3), *p<0.01. See also Figures S7 and S9.
Our identification of cSH2 domain as the principal mediator of PLCγ recruitment to FGFR2 is diametrically opposed to the previously reported crystal structure of the activated FGFR1 kinase complexed with the tandem nSH2-cSH2 domain fragment of PLCγ (PDB ID 3GQI (Bae et al., 2009)) showing that this tandem construct is recruited via nSH2 domain. Notably, 3GQI does not provide a satisfactory explanation as how the nSH2-mediated recruitment would facilitate PLCγ phosphorylation as none of the phosphorylation sites that follow the cSH2 domain are engaged by the FGFR1 kinase. In fact, the PLCγ phosphorylation site (Tyr-771) is over 60 Å away from the active site of FGFR1 kinase (Bae et al., 2009) (Figure S8). These disparities prompted us to carefully inspect the 3GQI (Bae et al., 2009) structure. Upon close inspection, it appears that the presence of decavanadate complex ions in the crystallization conditions together with crystal packing contacts (Figure S8) have contributed to non-physiological binding of the nSH2 domain of PLCγ to the activated FGFR1 kinase.
Insights into the preferential binding of FGFR and other RTKs to the cSH2 domain of PLCγ can be gleaned through sequence alignment of PLCγ recruitment sites from several RTKs that utilize PLCγ as their common intracellular substrate. Relative to cSH2, the hydrophobic hole of the socket in nSH2 appears narrower due to insertion of three residues in the loop between αB helix and βG strand (Figures S6A and S6B). Additionally, substitution of Arg-696 of cSH2 domain with Ser-588 in nSH2 domain in the hydrophilic hole of socket should also reduce the overall affinity of nSH2 domain for phosphotyrosine containing peptides. As shown in Figure 4D, Arg-696 of cSH2 domain, which makes two tight hydrogen bonds with phosphate moiety of pTyr-769, is not conserved in the nSH2 domain of PLCγ (Ser-588 in nSH2) (Figure S6A). Interestingly, in all RTKs, the PLCγ recruitment sites maps to the C-terminal tail of the receptor not too far from the αI helix, the last secondary structure element of the kinase domain. In fact, in FGFRs, VEGFRs, TRKs, C-KIT, CSF1R, and RET the recruitment sites are nearly equidistance from the predicted end of kinase domain (Figure S6C). Based on these observations it is likely that additional selectivity for cSH2 domain may be achieved due to steric factors in the quaternary structure that are more permissible for cSH2 than nSH2 domain.
Structural Basis for the Post-phosphorylation Dissociation Step of PLCγ from RTKs
Once phosphorylation of PLCγ is completed, the phosphorylated PLCγ must dissociate from the recruiting receptor kinase so that the next cycle of recruitment and phosphorylation can ensue. Phosphorylated Tyr-783 has been shown to fold over and bind in cis to the cSH2 domain of PLCγ (Bunney et al., 2012a; DeBell et al., 2007; Poulin et al., 2005). This intramolecular interaction induces a major structural rearrangement in PLCγ, which relieves PLCγ autoinhibition thereby elevating the lipase activity of the enzyme (Bunney et al., 2012a; Poulin et al., 2000, 2005). According to our structure, such a cis interaction would compete with binding of pTyr-769 of the recruiting FGFR2 kinase to the cSH2 domain thus forcing the phosphorylated PLCγ to come off the recruiting receptor. To test this hypothesis, we prepared cSH2 domain phosphorylated on Tyr783 (pY783-cSH2) and examined its interaction with FGFR2KpY769 using SPR spectroscopy. As shown in Figure S9A, pY783-cSH2 failed to bind FGFR2KpY769 supporting the model that the intramolecular interaction between phosphorylated Tyr-783 and the cSH2 domain of PLCγ, competes with pTyr-769 of the FGFR2 kinase for binding to the cSH2 domain. Hence our data provide a plausible molecular basis for how PLCγ is disengaged from the activated FGFR once phosphorylation of PLCγ is completed (Figure S9B). Notably, our structural data exposes the presence of an elegant coupling mechanism between phosphorylation-induced disengagement of PLCγ from FGFR and phospholipase activation of PLCγ.
In summary, the structural and biochemical data presented in this manuscript clearly establish that the recruitment and phosphorylation of SH2-containing substrates, a fundamental process in RTK signaling, is achieved in the context of a receptor dimer wherein one monomer recruits the substrate and offers it to the second monomer that acts as the “enzyme”. Thus, our findings overturn the current paradigm that SH2-mediated recruitment and phosphorylation are carried out by the same receptor i.e. in cis. Moreover, our 2:1 trans model suggests that SH2 binding specificity is achieved at the quaternary level within an RTK dimer thus challenging the current view regarding determinants of specificity of substrate recognition by an RTK. Our data identify an unprecedented role for receptor dimerization in substrate phosphorylation in addition to its canonical role in kinase activation. In addition to providing the molecular basis for one of the fundamental steps in RTK signaling, our data will have major impact on future drug discovery efforts in the RTK field. Essentially all of the current RTK inhibitors target the ATP binding pockets and often are cross-reactive between RTKs due to the significant sequence conservation of ATP binding pockets among RTKs. In addition, these inhibitors indiscriminately block all the pathways downstream of RTKs including those that may not be involved in a particular disease. Based on our model, it would be now possible to discover drugs that interfere with binding of PLCγ to the “phosphorylating” FGFR. Such drugs would selectively inhibit the PLCγ pathway in human diseases such as myeloproliferative syndrome (Emsley and Cowtan), where PLCγ signaling has been shown to be a critical downstream effector of the ZNF198-FGFR1 fusion gene (Roumiantsev et al., 2004).
EXPERIMENTAL PROCEDURES
Cell Culture and Immunoblotting Experiments
Transient transfection of PLCγ1−/− mouse embryo fibroblasts (MEF) (generous gift from Dr. Graham Carpenter) with pRK5 expression vectors for PLCγ1 (N+C+), PLCγ1R586A (N−C+), PLCγ1R694A (N+C−), and PLCγ1R586A,R694A (N−C−) was done using lipofectamine 2000 according to manufacturer’s protocol. The FUCRW lentiviral vector (Memarzadeh et al., 2007) was used to express wild-type and mutated FGFR2 and VEGFR2 molecules in BaF3 and porcine aortic endothelial (PAE) cell lines, respectively, for receptor complementation experiments. Full details are described in Supplemental Experimental Procedures.
Phosphatidylinositol Hydrolysis Assay in PLCγ1−/− MEF Cells
IP3 formation was measured as previously described(Everett et al., 2009) and detailed in Supplemental Experimental Procedures.
Protein Expression and Purification
All the wild-type and mutated FGFR kinases, wild-type and mutated tandem nSH2-cSH2 domains and individual nSH2 and cSH2 domains including the isotopically enriched cSH2 (U-15N, U-15N/13C, U-2H/15N) were expressed in E. coli (BL21) and purified as described in Supplemental Experimental Procedures.
In Vitro Binding Assays
The isothermal titration calorimetry (ITC), surface plasmon resonance (SPR) spectroscopy, size exclusion chromatography (SEC), native gel electrophoresis and nuclear magnetic resonance (NMR) spectroscopy were used to determine the binding selectivity of the PLCγ SH2 domains towards FGFR2KpY769. For full experimental details please refer to the Supplemental Experimental Procedures.
Paramagnetic Relaxation Enhancement (PRE) Experiments
Prior to incorporation of the spin label, FGFR2KpY769 was treated with 20 mM DTT for 15 min in a buffer containing 20 mM Tris-HCl (pH 7.5) and 150 mM NaCl. The protein was then exchanged into a buffer containing 20 mM Tris-HCl (pH 6.5), 150 mM NaCl and diluted to a concentration of 40 μM. A 150 mM solution of S-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate (MTSL) in acetonitrile was added in half-molar equivalent portions every 10 min on ice. The progress of the reaction was monitored using a Bruker ultrafleXtreme™ MALDI-TOF. Fully labeled FGFR2KpY769 was buffer exchanged into 25 mM HEPES (pH 7.5), 150 mM NaCl 0.1% NaN3, 5% D2O, and added to a sample of 70% deuterated cSH2 to give a final molar ratio of 1.4:1, FGFR2K-pY769:cSH2. A two-point 1H/15N TROSY method was used to calculate 1HN T2 relaxation enhancement values (Γ2) as previously described (Iwahara et al., 2007) and detailed in Supplemental Experimental Procedures.
Crystallization and Structure Determination
Crystals of FGFR2KpY769-cSH2 complex were grown by hanging drop vapor diffusion at 4 °C using crystallization buffer composed of 25 mM HEPES (pH 7.5), PEG20000 (12%–18%) and 2% (w/v) Benzamidine hydrochloride. Diffraction data were processed using HKL2000 Suite (Otwinowski and Minor, 1997). Molecular replacement solutions for the FGFR2 kinase and the cSH2 domain were found by the program Phaser in CCP4 Suite (1994) using the crystal structure of FGFR2 kinase (PDB ID: 2PVY (Chen et al., 2007)) and the crystal structure of cSH2 domain (PDB ID: 3GQI (Bae et al., 2009)) as the search model, respectively. Model building was carried out using Coot (Emsley and Cowtan, 2004), and iterative positional and B-factor refinements were done using PHENIX (Adams et al., 2002). The refined structure shows good geometry and Ramachandran statistics. Data collection and structure refinement statistics are listed in Table S1. Full details are described in Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
The authors are thankful to Dr. Regina Goetz, Mr. Yang Liu for critically reading the manuscript and making thoughtful suggestions. This work was supported by the U.S. National Institutes of Health NIDCR Grant DE13686 (to M.M.), NIH/NEI (R21CA191970 and R21CA193958) (to N.R.), NINDS grant P30 NS050276 (to T.A.N.), grants from Natural Science Foundation of China 81473261, 31270789 (to Z.H. and H.C.) and the St Baldrick’s Foundation (to A.M.), and start-up funded from NYU (to N.J.T.). The NMR data collected at NYU was supported by an NIH S10 grant (OD016343) while the data collected at NYSBC was made possible by a grant from NYSTAR and ORIP/NIH facility improvement grant CO6RR015495. The 900 MHz NMR spectrometer was purchased with funds from NIH grant P41GM066354, the Keck Foundation, New York State Assembly, and U.S. Dept. of Defense. The coordinates and structure factors for the complex will be deposited in the RCSB Protein Data Bank under released upon acceptance of the manuscript.
References
- 1.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 2.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 3.Bae JH, Lew ED, Yuzawa S, Tome F, Lax I, Schlessinger J. The selectivity of receptor tyrosine kinase signaling is controlled by a secondary SH2 domain binding site. Cell. 2009;138:514–524. doi: 10.1016/j.cell.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bunney TD, Esposito D, Mas-Droux C, Lamber E, Baxendale RW, Martins M, Cole A, Svergun D, Driscoll PC, Katan M. Structural and Functional Integration of the PLCgamma Interaction Domains Critical for Regulatory Mechanisms and Signaling Deregulation. Structure. 2012a;20:2062–2075. doi: 10.1016/j.str.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bunney TD, Esposito D, Mas-Droux C, Lamber E, Baxendale RW, Martins M, Cole A, Svergun D, Driscoll PC, Katan M. Structural and functional integration of the PLCγ interaction domains critical for regulatory mechanisms and signaling deregulation. Structure. 2012b;20:2062–2075. doi: 10.1016/j.str.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen H, Huang Z, Dutta K, Blais S, Neubert TA, Li X, Cowburn D, Traaseth NJ, Mohammadi M. Cracking the molecular origin of intrinsic tyrosine kinase activity through analysis of pathogenic gain-of-function mutations. Cell Rep. 2013;4:376–384. doi: 10.1016/j.celrep.2013.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen H, Ma J, Li W, Eliseenkova AV, Xu C, Neubert TA, Miller WT, Mohammadi M. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol Cell. 2007;27:717–730. doi: 10.1016/j.molcel.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen H, Xu CF, Ma J, Eliseenkova AV, Li W, Pollock PM, Pitteloud N, Miller WT, Neubert TA, Mohammadi M. A crystallographic snapshot of tyrosine trans-phosphorylation in action. Proc Natl Acad Sci U S A. 2008;105:19660–19665. doi: 10.1073/pnas.0807752105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeBell K, Graham L, Reischl I, Serrano C, Bonvini E, Rellahan B. Intramolecular regulation of phospholipase C-gamma1 by its C-terminal Src homology 2 domain. Mol Cell Biol. 2007;27:854–863. doi: 10.1128/MCB.01400-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ellis MV, James SR, Perisic O, Downes CP, Williams RL, Katan M. Catalytic domain of phosphoinositide-specific phospholipase C (PLC). Mutational analysis of residues within the active site and hydrophobic ridge of plcdelta1. J Biol Chem. 1998;273:11650–11659. doi: 10.1074/jbc.273.19.11650. [DOI] [PubMed] [Google Scholar]
- 11.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 12.Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 13.Everett KL, Bunney TD, Yoon Y, Rodrigues-Lima F, Harris R, Driscoll PC, Abe K, Fuchs H, de Angelis MH, Yu P. Characterization of phospholipase Cγ enzymes with gain-of-function mutations. J Biol Chem. 2009;284:23083–23093. doi: 10.1074/jbc.M109.019265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goetz R, Mohammadi M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat Rev Mol Cell Biol. 2013;14:166–180. doi: 10.1038/nrm3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hajicek N, Charpentier TH, Rush JR, Harden TK, Sondek J. Autoinhibition and phosphorylation-induced activation of phospholipase C-γ isozymes. Biochem. 2013;52:4810–4819. doi: 10.1021/bi400433b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hidaka M, Homma Y, Takenawa T. Highly conserved eight amino acid sequence in SH2 is important for recognition of phosphotyrosine site. Biochem Biophys Res Commun. 1991;180:1490–1497. doi: 10.1016/s0006-291x(05)81364-6. [DOI] [PubMed] [Google Scholar]
- 17.Hubbard SR. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5:464–471. doi: 10.1038/nrm1399. [DOI] [PubMed] [Google Scholar]
- 18.Hunter T. Signaling–2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 19.Hunter T. Tyrosine phosphorylation in cell signaling and disease. Keio J Med. 2002;51:61–71. doi: 10.2302/kjm.51.61. [DOI] [PubMed] [Google Scholar]
- 20.Iwahara J, Tang C, Clore GM. Practical aspects of (1)H transverse paramagnetic relaxation enhancement measurements on macromolecules. J Magn Reson. 2007;184:185–195. doi: 10.1016/j.jmr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji QS, Winnier GE, Niswender KD, Horstman D, Wisdom R, Magnuson MA, Carpenter G. Essential role of the tyrosine kinase substrate phospholipase C-gamma1 in mammalian growth and development. Proc Natl Acad Sci U S A. 1997;94:2999–3003. doi: 10.1073/pnas.94.7.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jura N, Zhang X, Endres NF, Seeliger MA, Schindler T, Kuriyan J. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol Cell. 2011;42:9–22. doi: 10.1016/j.molcel.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim HK, Kim JW, Zilberstein A, Margolis B, Kim JG, Schlessinger J, Rhee SG. PDGF stimulation of inositol phospholipid hydrolysis requires PLC-gamma 1 phosphorylation on tyrosine residues 783 and 1254. Cell. 1991;65:435–441. doi: 10.1016/0092-8674(91)90461-7. [DOI] [PubMed] [Google Scholar]
- 24.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu BA, Engelmann BW, Jablonowski K, Higginbotham K, Stergachis AB, Nash PD. SRC Homology 2 Domain Binding Sites in Insulin, IGF-1 and FGF receptor mediated signaling networks reveal an extensive potential interactome. Cell Commun Signal. 2012;10:27. doi: 10.1186/1478-811X-10-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Memarzadeh S, Xin L, Mulholland DJ, Mansukhani A, Wu H, Teitell MA, Witte ON. Enhanced paracrine FGF10 expression promotes formation of multifocal prostate adenocarcinoma and an increase in epithelial androgen receptor. Cancer Cell. 2007;12:572–585. doi: 10.1016/j.ccr.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohammadi M, Honegger AM, Rotin D, Fischer R, Bellot F, Li W, Dionne CA, Jaye M, Rubinstein M, Schlessinger J. A tyrosine-phosphorylated carboxy-terminal peptide of the fibroblast growth factor receptor (Flg) is a binding site for the SH2 domain of phospholipase C-gamma 1. Mol Cell Biol. 1991;11:5068–5078. doi: 10.1128/mcb.11.10.5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naski MC, Wang Q, Xu J, Ornitz DM. Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nat Genet. 1996;13:233–237. doi: 10.1038/ng0696-233. [DOI] [PubMed] [Google Scholar]
- 29.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 30.Pascal SM, Singer AU, Gish G, Yamazaki T, Shoelson SE, Pawson T, Kay LE, Forman-Kay JD. Nuclear magnetic resonance structure of an SH2 domain of phospholipase C-gamma 1 complexed with a high affinity binding peptide. Cell. 1994;77:461–472. doi: 10.1016/0092-8674(94)90160-0. [DOI] [PubMed] [Google Scholar]
- 31.Pawson T. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004;116:191–203. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- 32.Pellicena P, Kuriyan J. Protein-protein interactions in the allosteric regulation of protein kinases. Curr Opin Struct Biol. 2006;16:702–709. doi: 10.1016/j.sbi.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 33.Peters KG, Marie J, Wilson E, Ives HE, Escobedo J, Del Rosario M, Mirda D, Williams LT. Point mutation of an FGF receptor abolishes phosphatidylinositol turnover and Ca2+ flux but not mitogenesis. Nature. 1992;358:678–681. doi: 10.1038/358678a0. [DOI] [PubMed] [Google Scholar]
- 34.Poulin B, Sekiya F, Rhee SG. Differential roles of the Src homology 2 domains of phospholipase C-gamma1 (PLC-gamma1) in platelet-derived growth factor-induced activation of PLC-gamma1 in intact cells. J Biol Chem. 2000;275:6411–6416. doi: 10.1074/jbc.275.9.6411. [DOI] [PubMed] [Google Scholar]
- 35.Poulin B, Sekiya F, Rhee SG. Intramolecular interaction between phosphorylated tyrosine-783 and the C-terminal Src homology 2 domain activates phospholipase C-gamma1. Proc Natl Acad Sci U S A. 2005;102:4276–4281. doi: 10.1073/pnas.0409590102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rajakulendran T, Sicheri F. Allosteric protein kinase regulation by pseudokinases: insights from STRAD. Sci Signal. 2010;3:pe8. doi: 10.1126/scisignal.3111pe8. [DOI] [PubMed] [Google Scholar]
- 37.Rotin D, Honegger AM, Margolis BL, Ullrich A, Schlessinger J. Presence of SH2 domains of phospholipase C gamma 1 enhances substrate phosphorylation by increasing the affinity toward the epidermal growth factor receptor. J Biol Chem. 1992;267:9678–9683. [PubMed] [Google Scholar]
- 38.Roumiantsev S, Krause DS, Neumann CA, Dimitri CA, Asiedu F, Cross NC, Van Etten RA. Distinct stem cell myeloproliferative/T lymphoma syndromes induced by ZNF198-FGFR1 and BCR-FGFR1 fusion genes from 8p11 translocations. Cancer Cell. 2004;5:287–298. doi: 10.1016/s1535-6108(04)00053-4. [DOI] [PubMed] [Google Scholar]
- 39.Schlessinger J. Phospholipase Cgamma activation and phosphoinositide hydrolysis are essential for embryonal development. Proc Natl Acad Sci U S A. 1997;94:2798–2799. doi: 10.1073/pnas.94.7.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schlessinger J, Lemmon MA. SH2 and PTB domains in tyrosine kinase signaling. Science’s STKE: signal transduction knowledge environment. 2003;2003:RE12. doi: 10.1126/stke.2003.191.re12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.