

Abstract
Overactivation of PI3K/Akt/mTOR is linked with carcinogenesis and serves a potential molecular therapeutic target in treatment of various cancers. Herein, we report the synthesis of trisubstituted-imidazoles and identified 2-chloro-3-(4, 5-diphenyl-1H-imidazol-2-yl) pyridine (CIP) as lead cytotoxic agent. Naïve Base classifier model of in silico target prediction revealed that CIP targets RAC-beta serine/threonine-protein kinase which comprises the Akt. Furthermore, CIP downregulated the phosphorylation of Akt, PDK and mTOR proteins and decreased expression of cyclin D1, Bcl-2, survivin, VEGF, procaspase-3 and increased cleavage of PARP. In addition, CIP significantly downregulated the CXCL12 induced motility of breast cancer cells and molecular docking calculations revealed that all compounds bind to Akt2 kinase with high docking scores compared to the library of previously reported Akt2 inhibitors. In summary, we report the synthesis and biological evaluation of imidazoles that induce apoptosis in breast cancer cells by negatively regulating PI3K/Akt/mTOR signaling pathway.
Introduction
PI3K/Akt/mTOR pathway is a major signaling cascade which operates downstream to the receptor tyrosine kinases such as epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR) and insulin-like growth factor-1 receptor (IGF-1R) [1–3]. The relay of signals from the aforementioned growth factor receptors leads to the activation of Phosphoinositide 3-kinase (PI3K) which catalyses the generation of phosphatidylinositol-3,4,5-triphosphate (PIP3) from phosphatidylinositol-4,5-biphosphate [4]. In turn, PIP3 interacts with pleckstrin homology (PH) domains of phosphoinositide-dependent kinase (PDK) and Akt (Also called as Protein Kinase B) and activates them [5]. In addition, PDK contributes to activation of Akt. Among these two master kinases, Akt acts on wide array of substrates which are involved in regulation of apoptosis, cell cycle, transcription and translation [6, 7]. Therefore, the PI3K/Akt/mTOR pathway plays a central role in regulation of the cell proliferation, survival, migration, angiogenesis and metabolism and extensively contributes to oncogenesis [8–11]. Overactivation of PI3K/Akt/mTOR pathway has been reported in many types of cancers offering a unique therapeutic target to design novel heterocycles against malignancies [12–15].
Imidazole-based compounds have been extensively studied and have been reported to possess good anticancer activity in various types of cancer cells [16, 17]. Previously, we reported the synthesis and antiproliferative activity of imidazole derivatives in various tumor models [18–20]. Studies have suggested that imidazole conjugates possess good PI3K inhibitory activity [21]. SB203580 is a pyridinyl imidazole reported to effectively inhibit PI3K indicate that imidazole-based compounds are pharmacologically important scaffolds to target different type of cancers [21]. Therefore, in continuation our effort to synthesize and explore the various pharmacological properties of heterocycles [22–29], in the present study, we synthesized a series of trisubstituted imidazole derivatives and evaluated their inhibitory efficacy against PI3K/Akt/mTOR signaling pathway in breast cancer cell lines. An in silico target prediction revealed that new compounds target RAC-beta serine/threonine-protein kinase which comprise the Akt and abrogates PI3K/Akt/mTOR pathway. The lead compound, 2-chloro-3-(4, 5-diphenyl-1H-imidazol-2-yl) pyridine (CIP) suppressed the proliferation of breast cancer cells, decreased the phosphorylation of PDK, Akt, mTOR, downregulated the cellular invasion and activated caspases and cleaved PARP to induce apoptosis.
Materials and Methods
All solvents used were of analytical grade and reagents used were purchased from Sigma-Aldrich. 1H and 13C NMR spectra were recorded on a Agilent (400 MHz) spectrometer in CDCl3 or DMSO-d6 as solvent, using TMS as an internal standard and chemical shifts are expressed as ppm. Mass and purity were recorded on a LC-MSD-Trap-XCT. High resolution mass spectra were determined on a Bruker Daltonics instrument. The progress of the reaction was monitored by TLC pre-coated silica gel G plates. The breast cancer MCF-7 and MDA-MB-231 cell lines were obtained from ATCC (Manassas, VA, USA).
General procedure for the synthesis of the trisubstituted imidazoles
The reaction mixture of benzil (1.0 mmol), aldehydes (1.0 mmol), NH4OAc (4.0 mmol) and a metal halide catalyst (0.10 mmol), followed by EtOH (4 ml) was stirred and gently refluxed for 5 h (Fig 1A). The completion of reaction was monitored by TLC and 4 ml of water was added at the end of reaction. The solid was filtered, washed with a mixture of cold EtOH/H2O (1:1, v:v). The crude product was recrystallized from acetone/water 9:1.
Fig 1.

A, Schematic representation for the synthesis of imidazole based small molecules. B, Breast cancer cells (2.5 X 104/mL, MCF-7 & MDA-MB-231) were plated in triplicate, treated with indicated concentrations (0, 10, 25 and 50 μM) of CIP, and then subjected to MTT assay after 24, 48 and 72 h to analyse proliferation of cells. CIP suppresses the viability of various breast cancer cell lines in a dose- and time-dependent manner.
2-(3, 4-Dimethoxyphenyl)-4, 5-diphenyl-1H-imidazole (1)
1H NMR (CDCl3, 400 MHz) δ: 12.454 (s, NH), 6.878–7.631 (m, 13H), 3.961 (s, 6H). 13C NMR: 149.7, 148.9, 147.9, 136.7, 134.9, 131.4, 131.3, 129.9, 129.3, 129.1, 128.2, 126.6, 126.4, 123.7, 122.1, 113.2, 111.7, 55.7, 55.6. (MM—ES + APCI) m/z: 357.12 [M+H] +. MP 218–219°C
2-Bromo-5-(4, 5-diphenyl-1H-imidazol-2-yl) pyridine (2)
1H NMR (CDCl3, 400 MHz) δ: 13.205(s, NH), 8.777–8.761 (d, J = 4 Hz, 1H), 8.141–8.111 (dd, J1 = 4 Hz, J2 = 4 Hz, 1H), 7.483–7.318 (m, 5H), 7.314–7.241 (m, 6H). 13C NMR: 148.9, 145.3, 141.9, 134.9, 130.4, 130. 5, 129.9, 129. 5, 129.1, 128.2, 126. 9, 126. 3, 123.7, 122.2. (MM—ES + APCI) m/z: 377.04 [M+H] +, HRMS (ESI–TOF) Calculated for C20H14N3Br [M + Na], 398.0263: found 398.0266. MP 248–249°C
2-(4-(Methylsulfonyl) phenyl)-4, 5-diphenyl-1H-imidazole (3)
1H NMR (CDCl3, 400 MHz) δ: 13.092 (s, NH), 8.391–8.371 (d, J = 8 Hz, 2H), 8.095–8.075 (d, J = 8 Hz, 2H), 7.596–7.377 (m, 10H), 3.391 (s, 3H). 13C NMR: 147.6, 146.9, 131.8, 131.7, 129.8, 129.7, 129.5, 128.9, 126.9, 126.8 123.7, 122.1, 113.2, 111.7, 55.4. (MM-ES+APCI) m/z: 375.11 [M+H] +, HRMS (ESI–TOF) Calculated for C22H18N2O2S [M+Na], 397.0981: found 397.0983. MP 256–257°C
2-(4-Butoxyphenyl)-4, 5-diphenyl-1H-imidazole (4)
1H NMR (CDCl3, 400 MHz) δ: 13.20 (s, NH), 8.095–8.075 (d, J = 8 Hz, 2H), 7.923–7.765 (m, 2H) 7.483–7.324 (m, 10H), 3.941–3.867 (m, 2H), 2.171–1.322 (m, 6H). 13C NMR: 146.9, 137.6, 133.7, 129.5, 129.5, 129.4, 128.9, 126.5, 126.0, 124.7 122.7, 114.7, 65.6, 31.8, 24.1, 17.18. (MM—ES + APCI) m/z: 369.17 [M+H] +. MP 229–230°C
3-(4, 5-Diphenyl-1H-imidazol-2-yl)-1H-indole (5)
1H NMR (CDCl3, 400 MHz) δ: 12.4 (s, 1H), 11.404 (s, 1H), 8.461–8.441 (d, J = 8 Hz, 1H), 8.016–8.006 (d, J = 4 Hz, 1H), 7.587–7.128 (m, 13H). 13C NMR: 144.1, 136.7, 128.9, 128.5, 127.6, 125.1, 124.5, 122.4, 121.9, 120.4, 112.1, 106.9. (MM-ES+APCI) m/z: 336.47 [M+H]+. MP 290–291°C
3-(4, 5-Diphenyl-1H-imidazol-2-yl)-2-methyl-1H-indole (6)
1H NMR (CDCl3, 400 MHz) δ: 11.553 (s, 1H, NH), 10.07 (s, 1H, NH), 8.147–7.484 (m, 4H), 7.419–7.102 (m, 10H), 2.214 (s, 3H). 13C NMR: 143.9, 136.7, 136.4, 134.98, 132.6, 128.8, 128.5, 127.6, 126.8, 126.4, 125.69, 125.31, 124.7, 122.6, 121.8, 120.7, 110.6, 102.8, 14.71. (MM-ES+APCI) m/z: 350.16 [M+H] +. MP 298–299°C.
4, 5-Diphenyl-2-(thiophen-2-yl)-1H-imidazole (7)
1H NMR (CDCl3, 400 MHz) δ: 12.79 (s, 1H, NH), 7.15–7.13 (dd, J1 = 4 Hz, J2 = 4 Hz, 1H), 7.69–7.68 (d, J = 4 Hz, 1H), 7.51–7.25 (m, 11H). 13C NMR: 141.5, 136.6, 134.7, 133.9, 130.8, 128.6, 128.1, 127.8, 127.0, 126.5, 124.1. (MM-ES+APCI) m/z: 303.37 [M+H] +. MP 255–256°C
4, 5-Diphenyl-2-(thiophen-3-yl)-1H-imidazole (8)
1H NMR (CDCl3, 400 MHz) δ: 12.63 (s, 1H, NH), 8.04–8.02 (dd, J1 = 4 Hz, J2 = 4 Hz, 1H), 7.71–7.68 (dd, J1 = 4Hz, J2 = 4 Hz, 1H), 7.65–7.63 (dd, J1 = 4 Hz, J2 = 4 Hz, 1H), 7.31–7.53 (m, 10H), 13C NMR: 121.8, 124.2, 126.5, 127.0, 127.7, 128.1, 128.4, 130.8, 132.5, 134.7, 136.6, 142.7 (MM—ES + APCI) m/z: 303.37 [M+H] +. MP 257–259°C
2-Chloro-3-(4, 5-diphenyl-1H-imidazol-2-yl) pyridine (9, CIP)
1H NMR (CDCl3, 400 MHz) δ: 12.591 (1H, NH), 8.841–8.811 (dd, J1 = 4 Hz, J2 = 4 Hz, 1H), 8.390–8.370 (dd, J1 = 4 Hz, J2 = 4 Hz, 1H), 7.567–7.560 (m, 4H), 7.406–7.307 (m, 7H). 13C NMR: 148.7, 140.1, 139.9, 133.9, 130.4, 131. 0, 129.9, 122.2, 129. 5, 129.1, 128.2, 126. 9, 126. 3, 123.7, (MM-ES+APCI) m/z: 332.09 [M+H]+, HRMS (ESI–TOF) Calculated for C20H14ClN3 [M + Na], 354.0768: found 354.0766. MP 228–229°C
2-(4-Chloro-1, 3-dimethyl-2, 5-dihydro-1H-pyrazol-5-yl)-4, 5-diphenyl-1H-imidazole (10)
1H NMR (CDCl3, 400 MHz) δ: 12.591(1H, NH), 7.571–7551 (m, 4H), 7.352–7.283 (m, 6H), 4.348 (s, 1H), 3.829 (s, 3H), 2.609 (s, 3H), 2.254 (s, 1H). 13C NMR: 149.7, 140.3, 139.9, 134.9, 130.4, 131. 0, 129.9, 129. 5, 129.1, 128.2, 126. 9, 76. 3, 43.7, 12.2, (MM-ES+APCI) m/z: 351.17 [M+H]+, HRMS (ESI–TOF) Calculated for C20H19ClN4 [M+Na], 373.1190: found 373.1193. MP 218–219°C.
2-(Naphthalen-2-yl)-4, 5-diphenyl-1H-imidazole (11)
1H NMR (CDCl3, 400 MHz) δ: 12.71 (s, NH), 8.69 (s, 1H), 8.26–8.24 (d, J = 8 Hz, 2H), 7.967–7.501 (m, 7H), 7.498–7.284 (m, 7H). 13C NMR: 145.4, 137.9, 137.0, 135.2, 133.5, 130.9, 130.2, 129.4, 128.7, 128.4, 128.2, 128.0, 127.7, 127.5, 127.3, 127.0, 126.5, 126.4, 126.3, 125.9, 125.0 (MM—ES + APCI) m/z: 347.20 [M+H]+. MP 273–275°C.
Pharmacology
MTT Assay
The antiproliferative effect of newly synthesized compounds against breast cancer cells was determined by the MTT dye uptake method as described previously [30]. The breast cancer MCF-7 and MDA-MB-231 cell lines were obtained from ATCC (Manassas, VA, USA). Briefly, breast cancer cells (2.5 X 104/ml) were incubated in triplicate in a 96-well plate, in the presence of varying compound concentrations at a volume of 0.2 ml, for different time intervals at 37°C. Thereafter, a 20 μl MTT solution (5 mg/ml in PBS) was added to each well. After a 2 h incubation at 37°C, a 0.1 ml lysis buffer (20% SDS, 50% dimethylformamide) was added; incubation was performed for 1 h at 37°C, and the optical density (OD) at 570 nm was measured by Tecan plate reader. 0.01% DMSO was used as the negative control and 0.01% MTT was used as a control agent.
Flow cytometric analysis
Flow cytometric analysis was performed to determine whether CIP can induce apoptosis of tumor cells as described earlier [31]. Briefly, MDA-MB-231 breast cancer cells (5 x 105) were plated in petri dish and 24 h later the cells were exposed to compound CIP (50 μM) for 0, 24, 36 and 48 h. Thereafter cells were washed, fixed with 70% ethanol, and incubated for 30 min at 37°C with 0.1% RNase A in PBS. Cells were washed again, resuspended, and stained with PBS containing 25 μg/ml propidium iodide (PI) for 15 min at room temperature. The cell cycle distribution across the various phases was analyzed with a CyAn ADP flow cytometer (Dako Cytomation).
Caspase 3/7 activity assay
MDA-MB-231 cells (5 x 105) were plated in 6-well plates and allowed to adhere for 24 h. The cells were then exposed to CIP (50 μM) for 0, 24, 36 and 48 h. Thereafter, the Caspase-3/7 activities were determined by a Caspase-Glo® 3/7 assay kit (Promega, Madison, USA) according to the manufacturer’s instructions.
Target prediction-based enrichment of Imidazole series
In order to understand the mode-of-action of trisubstituted imidazoles towards the inhibition of human breast cancer cells, we applied an in silico target prediction tool, which was developed using the Bernoulli Naïve Bayes (BNB) algorithm, as implemented in Scikit-learn [32]. This method has been validated previously by Mervin et al [33]. Active compounds were extracted from ChEMBL [34], using pChEMBL values larger than 5 (10 μM) and a confidence score cut-off of 5 to define activity. Inactive data points were extracted from PubChem [35] for compounds declared inactive from screens stored within BioAssay [36]. The BNB model was trained on these compounds using the 2048 bit Morgan fingerprints, with a radius of 2, generated by RDKit [37]. Stringent class-specific thresholds were applied to binarize predictions for the Imidazole series.
In order to improve the statistical significance of this analysis, predictions for the imidazole sets are compared to predictions generated by chance for 1,000 sets of randomly selected compounds from the PubChem repository. An Average Ratio enrichment metric is calculated from this analysis (Eq 1), which reflects the average of the series of predictions in the background (Ri), divided by the frequency of predictions in the imidazole series (Fimidazole). A lower score represents targets that are more enriched, and hence found to be significant for the imidazole series.
| (1) |
Western blotting
Western blotting analysis was performed as described earlier [38]. Briefly, For detection of various proteins, CIP-treated whole-cell extracts were lysed in a lysis buffer (20 mM Tris (pH 7.4), 250 mM NaCl, 2 mM EDTA (pH 8.0), 0.1% TritonX-100, 0.01 mg/ml aprotinin, 0.005 mg/ml leupeptin, 0.4 mM PMSF,and 4 mM NaVO4). Lysates were then spun at 14,000 rpm for 10 mins to remove insoluble material, and then resolved on a 7.5% SDS gel. After electrophoresis, the proteins were electrotransferred to a nitrocellulose membrane, blocked with 5% non-fat milk, and probed with various antibodies (1:1000) overnight at 4°C. The blot was washed, exposed to HRP-conjugated secondary antibodies for 1 h, and finally examined by chemiluminescence (ECL; GE Healthcare, Little Chalfont, Buckinghamshire, UK).
Invasion Assay
A BD Biocoat™ Matrigel™ invasion chamber with 8-μm pores in the light-tight polyethylene terephthalate membrane and was coated with a reconstituted basement membrane gel (BD Biosciences). 2 X 105 cells were suspended in serum-free DMEM and seeded into the Matrigel transwell chambers. The cells were incubated with CIP for 8 h. After incubation, the outer surfaces of the transwell chambers were wiped with cotton swabs, and the invading cells were fixed and stained with crystal violet solution. The invading cells were then counted in five randomly selected areas under microscopic observation.
Molecular docking analysis
The trisubstituted imidazoles were docked to the crystal structure of human RAC-Beta Serine/Threonine-Protein Kinase (Akt2) complexed with an inhibitor (PDB: 2JDR) [39] using MOE [40]. Protonation states of amino acids were assigned using protonate3D in MOE [41]. Ligands were ionized at physiological pH using MOE. The binding site was defined by the position of the co-crystalized ligand. Docking calculations were carried out using MOE’s induced fit protocol that treats amino acid side-chains near the binding site as flexible.
Results and Discussion
Chemical synthesis of trisubstituted imidazoles
We previously reported the microwave assisted synthesis and crystal structure studies of 2-butyl-4-chloro-imidazole-5-carbaldehyde [19, 20, 42]. In addition, solvent free and microwave-assisted synthesis of 4,5-disubstituted imidazoles have been reported using 1,2-diketones and urotropine in the presence of ammonium acetate [43]. In this report, we prepared 1,2,4-trisubstituted-1H-imidazoles by heating a mixture of benzil, an aldehyde and ammonium acetate using ethanol as solvent. The obtained imidazoles (3a-k) were characterized by melting point, LC-MS, 1H NMR, and 13C NMR analysis. The appearance of a 1H NMR peak at δ value of 12.5–12.7 for the HN-proton of imidazole confirms the ring formation. Detailed physical parameters of all the compounds synthesized is provided as supplemental information (S1 Table).
Trisubstituted imidazoles elicit an antiproliferative effect against human breast cancer cells
Imidazole derivatives are known for their antiproliferative effect towards various cancer models. We initially evaluated the effect of novel trisubstituted imidazoles against MDA-MB-231 cells using MTT assay as described earlier [44, 45]. Among the newly synthesized structures, compound 1, 2 and CIP displayed antiproliferative effect with the IC50 values of 21.1, 17.8 and 24.1 μM respectively. Further investigation of the compound 1, 2 and CIP against mammary epithelial cells (MCF-10A) revealed that, CIP selectively induce cytotoxicity in breast cancer cells and did not exhibit significant cytotoxicity against normal cells. To confirm the antiproliferative potential of the trisubstituted imidazoles, we further investigated the effect of compounds on hepatocellular carcinoma (HepG2) and normal (LO2) cells and found CIP as a lead anticancer compound with minimal cytotoxicity towards non-cancerous cells. The cytotoxicity profile of all the compounds is provided as supplementary information (S1 Table). Thereafter, we investigated the effect of CIP on MCF-7 and MDA-MB-231 breast cancer cells at different doses (0, 10, 25 & 50 μM) and time-points (24, 48 & 72 h). Upon treatment with CIP, we observed a significant decline in the viability of both the cell lines in a dose and time dependent manner (Fig 1B).
Investigation of structure-activity relationship (SAR) of the tested molecules suggests that introducing small sized and electron withdrawing atom containing heterocyclic groups at the position 2 in the imidazole ring would favour the specificity for cancer cells. Molecules having these SAR features better match the polarity of the Akt2 binding surface, since polar region in the Akt2 binding pocket is small.
CIP significantly accumulates MDA-MB-231 cells in sub-G1 phase and increases the activity of executioner caspases
We next investigated the potential of CIP to induce apoptosis in breast cancer MDA-MB-231 cells. We first observed that CIP induced significant apoptosis in a time dependent manner as evidenced by increased accumulation of cells in Sub-G1 phase of the cell cycle (Fig 2A). We further noted that exposure of the CIP also caused a significant increase in the activity of effector caspases -3 and -7 (Fig 2B), thereby indicating the apoptotic potential of CIP against breast cancer cells.
Fig 2.
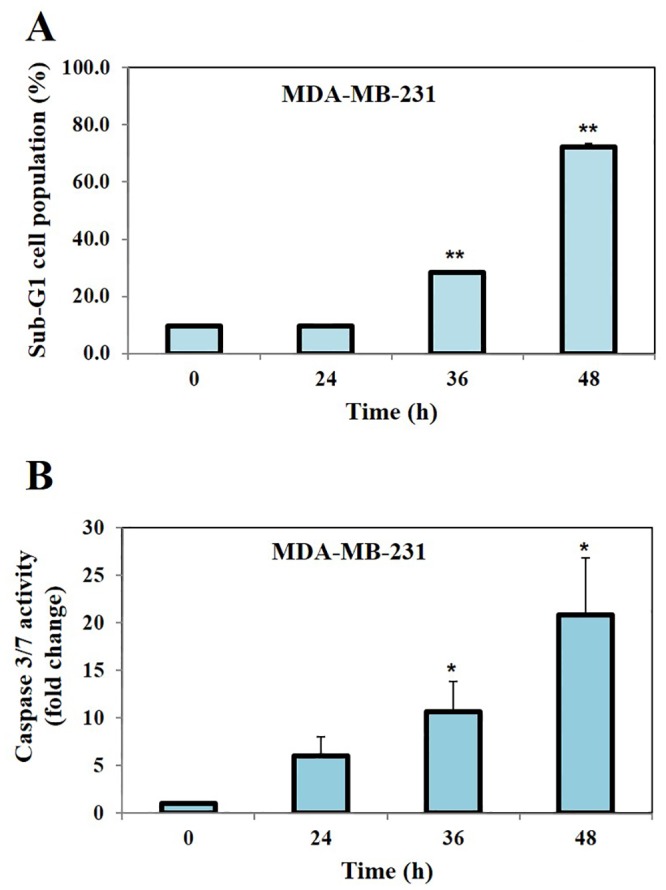
A, The analysis of cell cycle distribution following treatment with CIP was performed using flow cytometry. MDA-MB-231 cells were exposed to compound CIP (50 μM) for indicated time intervals (0, 24, 36 and 48 h), after which the cells were harvested and stained with PI. The cell distribution across the various phases of the cell cycle was analyzed with a flow cytometer. We observed that CIP induced significant apoptosis in a time dependent manner as evidenced by increased accumulation of cells in Sub-G1 phase of the cell cycle. B, MDA-MB-231 cells were exposed to compound CIP (50 μM) at indicated time intervals (0, 24, 36 and 48 h), after which they were harvested and caspase3/7 activity was measured using Caspase-Glo® 3/7 assay kit. We found that treatment of MDA-MB-231 cells with CIP caused the significant increase in the caspases-3/7 activity. * for p<0.05, ** for p<0.005.
In silico mode-of-action analysis for CIP that inhibits the growth of human breast cancer cells
In order to understand the molecular mechanism of CIP responsible for the antiproliferative effect in human breast cancer cells, we carried out an in silico target prediction analysis. Enrichment for the targets from the in silico target prediction analysis are shown in supplementary table (S2 Table). The top 20 targets from the 1,080 models are shown. “RAC-Beta Serine/Threonine-Protein Kinase” (Ranked 2nd) and “RAC-Alpha Serine/Threonine-Protein Kinase” (Ranked 8th), comprise the Akt protein, relevant for this study due to its involvement in the PI3K/Akt/mTOR pathway, and are highly enriched in this analysis.
CIP downregulates phosphorylation of PDK, Akt and mTOR in breast cancer cells
Based on the in silico prediction, we next analysed whether CIP modulates phosphorylation of Akt, PDK and mTOR in MDA-MB-231 cells using western blotting as described previously [46, 47]. As observed by western blot analysis, we found that, CIP downregulated the phosphorylation of Akt, PDK and mTOR in a dose-dependent manner (Fig 3) and at the same time Akt, PDK and mTOR protein expression remained unaltered.
Fig 3. MDA-MB-231 cells were treated with CIP at indicated concentrations (0, 10, 15, 25 and 50 μM) for 8 h and levels of phospho-PDK, phospho-Akt, phospho-mTOR and PDK, Akt, mTOR was analysed using western blotting.
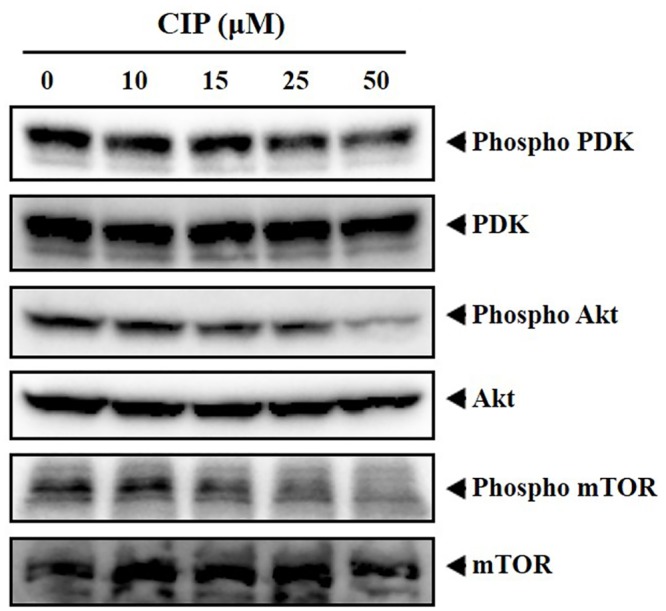
We found that, CIP downregulated the phosphorylation of Akt, PDK and mTOR in a dose-dependent manner without any change in the expression levels of Akt, PDK and mTOR proteins.
CIP induces apoptosis of human breast cancer cells
Cell shrinkage, formation of apoptotic bodies, activation of caspases, and cleavage of chromosomal DNA mediated by caspase dependent nucleases are the major events in the cells undergoing apoptosis [48]. In the event of cell committed to apoptosis, cascade of reactions occurs ultimately leading to the activation of caspase-3 which further cleaves full length PARP into fragments [49, 50]. Therefore, we first investigated the effect of CIP on procaspase-3 and found the activation of procaspase-3 in a time time-dependent manner (Fig 4A). Similarly, we observed the subsequent decrease in full length PARP with increase in cleaved fragment. We further determined the effect of CIP on the expression of various proliferative and survival proteins such as cyclin D1, VEGF, survivin and Bcl-2 in MDA-MB-231 cells. Fig 4B demonstrates a substantial decline in the expression of cyclin D1, VEGF, survivin and Bcl-2 proteins in the time-dependent manner.
Fig 4. A and B, MDA-MB-231 cells were treated with CIP (25 μM) for indicated time intervals (0, 12, 24, 36 and 48 h) and expression of apoptotic markers (PARP, Procaspase-3), antiapoptotic proteins (Bcl-2, Survivin, VEGF), cell cycle regulator (Cyclin D1) was profiled using western blot analysis.
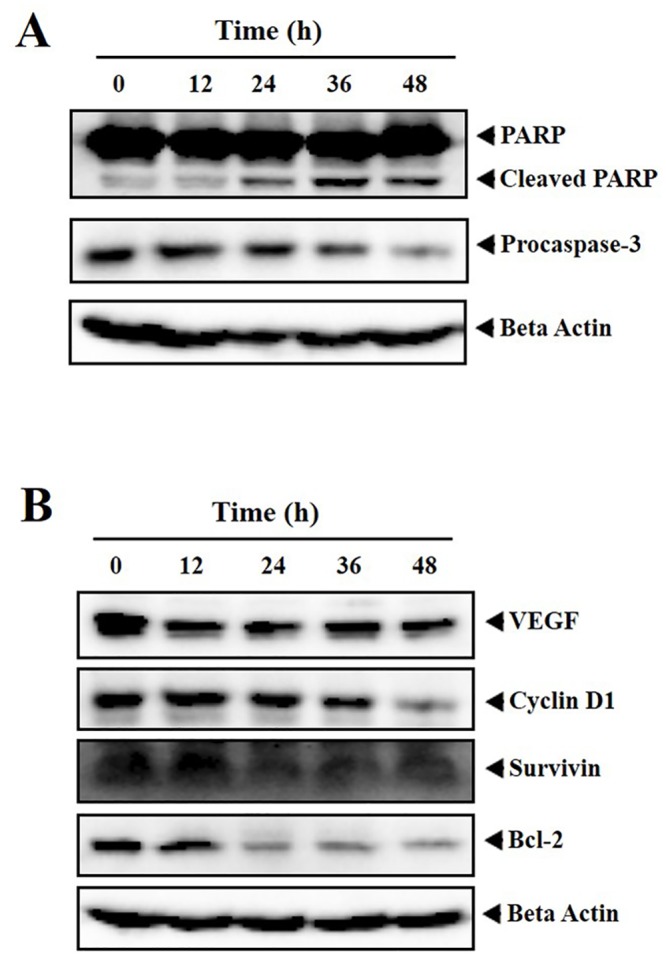
We observed the significant decline in the expression of PARP, procaspase-3, cyclin D1, VEGF, survivin and Bcl-2 proteins in the time-dependent manner without alteration in the levels of beta actin.
CIP suppresses CXCL12-induced MDA-MB-231 Cell invasion
Akt-targeted gene products are known to be involved in motility of cancer cells [5]. Therefore, we evaluated potential of CIP to modulate on cellular invasion according to the method described previously [51]. In addition, several studies have demonstrated the critical role and interaction of CXCL12 with CXCR4 in cancer cells which contributes largely to tumor metastasis [52]. Fig 5 indicates the movement of the cells in the presence and absence of CIP and CXCL12. CIP was found to significantly suppress the invasion of MDA-MB-231 cells by nearly 50%, both in the presence and absence of CXCL12, compared with the respective controls, suggesting that CIP interferes with invasive ability of breast cancer cells.
Fig 5. MDA-MB-231 cells were suspended in serum-free DMEM and seeded into the Matrigel transwell chambers and were incubated with CIP (25 μM) for 8 h.

The invading cells were fixed and stained with crystal violet solution and invaded cells were counted in five randomly selected areas. CIP significantly suppressed the cell invasion both in the presence and absence of CXCL12 suggesting that CIP interferes with invasive potential of breast cancer cells.
Molecular docking analysis
Based on the in silico and in vitro results, we investigated the binding of the imidazole series compounds to the RAC-Beta Serine/Threonine-Protein Kinase (Akt2 kinase) using MOE platform. Our docking calculations revealed that trisubstituted imidazoles bind with high docking scores:the average of the MOE scores for the compounds is found to be -8.38. To validate our in silico docking results, we have docked previously reported compounds that have low micro molar affinity for Akt2 Kinase (92 molecules having IC50 value of 1–50 μM extracted from ChEMBL database) using the same docking protocol. The average of the docking scores for this data is -8.62 which is very close to the average score for the imidazole series.
Docking of the trisubstituted imidazoles reveal common binding modes as shown in Fig 6. Phenyl moieties at positions 4 and 5 perfectly fit in the hydrophobic profile of the surface of the deep binding pocket (Fig 6A and 6B). Fragments at position 2, which contain electronegative atoms, match to the polar surface of the binding pocket (Fig 6A and 6B).
Fig 6. In silico molecular docking studies of trisubstituted imidazoles against kinase domain of Akt2:

Common binding poses of trisubstituted imidazoles towards the Akt2 kinase domain. The molecular surface of the protein is represented based on the surface polarity; green, pink and red colours show hydrophobic, polar and solvent exposed regions, respectively. For the sake of better visualisation of the binding pocket surface of Akt2, molecular surface was rendered in two panels (A and B) in which ligands were rotated 180 degrees.
Statistical analysis
Student t-test was used to analyze the data. * for p<0.05 was considered statistical significant. * for p<0.05; ** for p<0.005.
Discussion
The present report demonstrates the anticancer activity of trisubstituted imidazole that targets PI3K/Akt/mTOR pathway. CIP, the lead molecule among the synthesized imidazole derivatives supressed the proliferation of breast cancer (MDA-MB-231, MCF-7) and hepatocellular carcinoma (HepG2) cells and did not induce significant cytotoxicity against normal (MCF-10A, LO2) cells. CIP caused the accumulation of breast cancer cells in Sub-G1 phase, increased the executioner caspase-3/7 activity, downregulated the phosphorylation of PDK, Akt and mTOR in breast cancer cells. Further, it modulated the levels of various proteins involved in cell cycle regulation (Cyclin D1), angiogenesis (VEGF), apoptosis (Caspase-3, PARP), and survival (Survivin, Bcl-2). Further, CIP inhibited the cell invasion indicating its inhibitory role in cell motility.
One of the major events in apoptosis is the activation of caspases thereby cleavage of PARP and activation of caspase-activated deoxyribonuclease (CAD). In the event of cell undergoing apoptosis, caspase-3 activates CAD, and CAD in turn degrades genomic DNA into oligomers [53, 54]. The cells with lesser DNA content are often termed as hypodiploid cells which are detected as Sub-G1 cell population. The lead compound CIP caused the accumulation of cells in Sub-G1 phase demonstrating that CIP induce caspase mediated apoptosis. These results were confirmed and correlated by the cleavage of PARP, a prominent DNA repair enzyme and activation of executioner caspase-3 and caspase-7.
We performed cheminformatics-based target enrichment studies to predict the probable molecular therapeutic target of the lead compound and found a number of targets to be enriched, including Akt protein, and other proteins belonging to the MAPK pathway. Although, the results of the study presented multiple targets, we analysed the effect of CIP on PI3K/Akt/mTOR signalling pathway, because, previous studies have highlighted imidazole derivatives as inhibitors of PI3K/Akt/mTOR proteins [55–57]. In silico predicted target was experimentally validated by in vitro investigation in breast cancer cell lines. Several studies have showed that Akt pathway plays pivotal role in cell survival, growth and proliferation [58]. In order to demonstrate the effect of CIP on Akt signalling pathway, we investigated the effect of CIP on the phosphorylation of Akt pathway proteins including PDK1 (Ser-241), Akt (Ser-473) and mTOR. We observed the downregulation in the phosphorylation of proteins involved in Akt signalling cascade. Therefore, it is evident that, CIP mediates its anticancer activity at least partly via blockade of PI3K/Akt/mTOR signalling pathway. This finding opens an avenue for the development of novel trisubstituted imidazole based small molecules as therapeutic agents that target PI3K/Akt/mTOR signalling pathway in human diseases.
Conclusion
In conclusion, we synthesized trisubstituted imidazoles, identified the bioactive cytotoxic lead structure, predicted the likely target and demonstrated in vitro efficacy of lead compound to abrogate the activation of the PI3K/Akt/mTOR pathway in human breast cancer cells. Therefore, designing of CIP-based small molecule inhibitors to abrogate PI3K/Akt/mTOR pathway may serve as an effective therapeutic strategy to fight against various types of cancer.
Supporting Information
(DOC)
(DOC)
Acknowledgments
This research was supported by University Grants Commission (41-257-2012-SR), Vision Group Science and Technology, Department of Science and Technology (NO. SR/FT/LS-142/2012) to Basappa. KSR thanks Department of Science and Technology Indo-Korea [INT/Indo-Korea/122/2011-12] and Institution of Excellence, University of Mysore for financial support and instrumentation facility. CDM thanks the University of Mysore for Department of Science and Technology-Promotion of University Research and Scientific Excellence (DST-PURSE) Research Associate fellowship. This work was supported by NUHS Bench-to-Bedside-To-Product grant to GS. Deanship of Scientific Research, College of Science Research Centre, King Saud University, Kingdom of Saudi Arabia is also acknowledged. SP thanks the Netherlands Organisation for Scientific Research (NWO, grant number NWO-017.009–065) and the Prins Bernhard Cultuurfonds for funding.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This research was supported by University Grants Commission (41-257-2012-SR), Vision Group Science and Technology, and Department of Science and Technology (NO. SR/FT/LS-142/2012) to Basappa. KSR thanks Department of Science and Technology Indo-Korea [INT/Indo-Korea/122/2011-12] and Institution of Excellence, University of Mysore for financial support and instrumentation facility. CDM thanks the University of Mysore for Department of Science and Technology-Promotion of University Research and Scientific Excellence (DST-PURSE) Research Associate fellowship. This work was supported by NUHS Bench-to-Bedside-to-Product grant to GS. Deanship of Scientific Research, College of Science Research Centre, King Saud University, Kingdom of Saudi Arabia is also acknowledged. SP thanks the Netherlands Organisation for Scientific Research (NWO, grant number NWO-017.009-065) and the Prins Bernhard Cultuurfonds for funding. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Ludwig JA, Lamhamedi-Cherradi S-E, Lee H-Y, Naing A, Benjamin R. Dual Targeting of the Insulin-Like Growth Factor and Collateral Pathways in Cancer: Combating Drug Resistance. Cancers. 2011;3(3):3029–54. 10.3390/cancers3033029. PMC3759185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arcaro A, Guerreiro AS. The Phosphoinositide 3-Kinase Pathway in Human Cancer: Genetic Alterations and Therapeutic Implications. Current Genomics. 2007;8(5):271–306. 10.2174/138920207782446160. PMC2652403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Makinoshima H, Takita M, Saruwatari K, Umemura S, Obata Y, Ishii G, et al. Signaling through the Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) Axis Is Responsible for Aerobic Glycolysis mediated by Glucose Transporter in Epidermal Growth Factor Receptor (EGFR)-mutated Lung Adenocarcinoma. Journal of Biological Chemistry. 2015;290(28):17495–504. 10.1074/jbc.M115.660498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keane KN, Cruzat VF, Carlessi R, de Bittencourt PIH, Newsholme P. Molecular Events Linking Oxidative Stress and Inflammation to Insulin Resistance and β-Cell Dysfunction. Oxidative Medicine and Cellular Longevity. 2015;2015:15 10.1155/2015/181643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug resistance updates: reviews and commentaries in antimicrobial and anticancer chemotherapy. 2008;11(1–2):32–50. Epub 2008/01/02. 10.1016/j.drup.2007.11.003 ; PubMed Central PMCID: PMCPmc2442829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amaravadi R, Thompson CB. The survival kinases Akt and Pim as potential pharmacological targets. Journal of Clinical Investigation. 2005;115(10):2618–24. 10.1172/JCI26273. PMC1236693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes & development. 1999;13(22):2905–27. Epub 1999/12/02. . [DOI] [PubMed] [Google Scholar]
- 8.Martelli AM, Evangelisti C, Chiarini F, Grimaldi C, Manzoli L, McCubrey JA. Targeting the PI3K/AKT/mTOR signaling network in acute myelogenous leukemia. Expert Opinion on Investigational Drugs. 2009;18(9):1333–49. 10.1517/14728220903136775 . [DOI] [PubMed] [Google Scholar]
- 9.Brader S, Eccles SA. Phosphoinositide 3-kinase signalling pathways in tumor progression, invasion and angiogenesis. Tumori. 2004;90(1):2–8. [DOI] [PubMed] [Google Scholar]
- 10.Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29(36):4989–5005. 10.1038/onc.2010.236 [DOI] [PubMed] [Google Scholar]
- 11.Taniguchi CM, Kondo T, Sajan M, Luo J, Bronson R, Asano T, et al. Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3-kinase via Akt and PKCλ/ζ. Cell Metabolism. 2006;3(5):343–53. 10.1016/j.cmet.2006.04.005 [DOI] [PubMed] [Google Scholar]
- 12.Yap TA, Garrett MD, Walton MI, Raynaud F, de Bono JS, Workman P. Targeting the PI3K–AKT–mTOR pathway: progress, pitfalls, and promises. Current Opinion in Pharmacology. 2008;8(4):393–412. [DOI] [PubMed] [Google Scholar]
- 13.McAuliffe PF, Meric-Bernstam F, Mills GB, Gonzalez-Angulo AM. Deciphering the Role of PI3K/Akt/mTOR Pathway in Breast Cancer Biology and Pathogenesis. Clinical Breast Cancer. 2010;10, Supplement 3:S59–S65. 10.3816/CBC.2010.s.013 [DOI] [PubMed] [Google Scholar]
- 14.Ching CB, Hansel DE. Expanding therapeutic targets in bladder cancer: the PI3K/Akt/mTOR pathway. Lab Invest. 2010;90(10):1406–14. 10.1038/labinvest.2010.133 [DOI] [PubMed] [Google Scholar]
- 15.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–44. 10.1038/nrd2926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rademaker-Lakhai JM, van den Bongard D, Pluim D, Beijnen JH, Schellens JHM. A Phase I and Pharmacological Study with Imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a Novel Ruthenium Anticancer Agent. Clinical Cancer Research. 2004;10(11):3717–27. 10.1158/1078-0432.ccr-03-0746 [DOI] [PubMed] [Google Scholar]
- 17.Baviskar AT, Madaan C, Preet R, Mohapatra P, Jain V, Agarwal A, et al. N-Fused Imidazoles As Novel Anticancer Agents That Inhibit Catalytic Activity of Topoisomerase IIα and Induce Apoptosis in G1/S Phase. Journal of Medicinal Chemistry. 2011;54(14):5013–30. 10.1021/jm200235u [DOI] [PubMed] [Google Scholar]
- 18.Thimmegowda N, Swamy SN, Kumar CA, Kumar YS, Chandrappa S, Yip GW, et al. Synthesis, characterization and evaluation of benzimidazole derivative and its precursors as inhibitors of MDA-MB-231 human breast cancer cell proliferation. Bioorganic & medicinal chemistry letters. 2008;18(1):432–5. [DOI] [PubMed] [Google Scholar]
- 19.Kumar CA, Jayarama S, Basappa, Salimath BP, Rangappa KS. Pro-apoptotic activity of imidazole derivatives mediated by up-regulation of Bax and activation of CAD in Ehrlich Ascites Tumor cells. Investigational new drugs. 2007;25(4):343–50. Epub 2007/03/21. 10.1007/s10637-006-9033-4 . [DOI] [PubMed] [Google Scholar]
- 20.Kumar CA, Swamy SN, Gaonkar SL, Basappa, Salimath BP, Rangappa KS. N-Substituted-2-butyl-5-chloro-3H-imidazole-4-carbaldehyde derivatives as anti-tumor agents against Ehrlich ascites tumor cells in vivo. Medicinal chemistry (Shariqah (United Arab Emirates)). 2007;3(3):269–76. Epub 2007/05/17. . [DOI] [PubMed] [Google Scholar]
- 21.Zhao S, Konopleva M, Cabreira-Hansen M, Xie Z, Hu W, Milella M, et al. Inhibition of phosphatidylinositol 3-kinase dephosphorylates BAD and promotes apoptosis in myeloid leukemias. Leukemia. 2004;18(2):267–75. [DOI] [PubMed] [Google Scholar]
- 22.Kannaiyan R, Manu K, Chen L, Li F, Rajendran P, Subramaniam A, et al. Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-Jun N-terminal kinase and suppression of PI3 K/Akt signaling pathways. Apoptosis. 2011;16(10):1028–41. 10.1007/s10495-011-0629-6 [DOI] [PubMed] [Google Scholar]
- 23.Sugahara K, Thimmaiah KN, Bid HK, Houghton PJ, Rangappa KS. Anti-tumor activity of a novel HS-mimetic-vascular endothelial growth factor binding small molecule. PloS one. 2012;7(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bharathkumar H, Mohan CD, Rangappa S, Kang T, Keerthy H, Fuchs JE, et al. Screening of quinoline, 1, 3-benzoxazine, and 1, 3-oxazine-based small molecules against isolated methionyl-tRNA synthetase and A549 and HCT116 cancer cells including an in silico binding mode analysis. Organic & Biomolecular Chemistry. 2015. [DOI] [PubMed] [Google Scholar]
- 25.Fongmoon D, Shetty AK, Basappa, Yamada S, Sugiura M, Kongtawelert P, et al. Chondroitinase-mediated degradation of rare 3-O-sulfated glucuronic acid in functional oversulfated chondroitin sulfate K and E. The Journal of biological chemistry. 2007;282(51):36895–904. Epub 2007/10/24. 10.1074/jbc.M707082200 . [DOI] [PubMed] [Google Scholar]
- 26.Mantelingu K, Sadashiva M, Rangappa K. A simple and efficient method for the synthesis of 1, 2-benzisoxazoles: a series of its potent acetylcholinesterase inhibitors. Indian journal of chemistry Sect B: Organic chemistry, including medical chemistry. 2004;43(9):1954–7. [Google Scholar]
- 27.Anusha S, Anandakumar B, Mohan CD, Nagabhushana G, Priya B, Rangappa KS. Preparation and use of combustion-derived Bi 2 O 3 for the synthesis of heterocycles with anti-cancer properties by Suzuki-coupling reactions. RSC Advances. 2014;4(94):52181–8. [Google Scholar]
- 28.Sadashiva MP, Basappa S, Nanjundaswamy S, Li F, Manu KA, Sengottuvelan M, et al. Anti-cancer activity of novel dibenzo[b,f]azepine tethered isoxazoline derivatives. BMC chemical biology. 2012;12:5 Epub 2012/10/05. 10.1186/1472-6769-12-5 ; PubMed Central PMCID: PMCPmc3554437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Priya BS, Anil Kumar C, Nanjunda Swamy S, Basappa, Naveen S, Shashidhara Prasad J, et al. 2-(2-(2-Ethoxybenzoylamino)-4-chlorophenoxy)-N-(2-ethoxybenzoyl)benzamine inhibits EAT cell induced angiogenesis by down regulation of VEGF secretion. Bioorg Med Chem Lett. 2007;17(10):2775–80. Epub 2007/03/23. 10.1016/j.bmcl.2007.02.074 . [DOI] [PubMed] [Google Scholar]
- 30.Ashwini N, Garg M, Mohan CD, Fuchs JE, Rangappa S, Anusha S, et al. Synthesis of 1,2-benzisoxazole tethered 1,2,3-triazoles that exhibit anticancer activity in acute myeloid leukemia cell lines by inhibiting histone deacetylases, and inducing p21 and tubulin acetylation. Bioorg Med Chem. 2015;23(18):6157–65. Epub 2015/08/25. 10.1016/j.bmc.2015.07.069 . [DOI] [PubMed] [Google Scholar]
- 31.Anusha S, Mohan CD, Ananda H, Baburajeev CP, Rangappa S, Mathai J, et al. Adamantyl-tethered-biphenylic compounds induce apoptosis in cancer cells by targeting Bcl homologs. Bioorganic & Medicinal Chemistry Letters. 2016;26(3):1056–60. 10.1016/j.bmcl.2015.12.026 [DOI] [PubMed] [Google Scholar]
- 32.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: Machine learning in Python. The Journal of Machine Learning Research. 2011;12:2825–30. [Google Scholar]
- 33.Mervin LH, Afzal AM, Drakakis G, Lewis R, Engkvist O, Bender A. Target prediction utilising negative bioactivity data covering large chemical space. Journal of Cheminformatics. 2015;7:51 10.1186/s13321-015-0098-y. PMC4619454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic acids research. 2012;40(Database issue):D1100–7. Epub 2011/09/29. 10.1093/nar/gkr777 ; PubMed Central PMCID: PMCPmc3245175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic acids research. 2009;37(Web Server issue):W623–33. Epub 2009/06/06. 10.1093/nar/gkp456 ; PubMed Central PMCID: PMCPmc2703903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Database resources of the National Center for Biotechnology Information. Nucleic acids research. 2013;41(Database issue):D8–d20. Epub 2012/11/30. 10.1093/nar/gks1189 ; PubMed Central PMCID: PMCPmc3531099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landrum GRO-schwro.
- 38.Rakesh KS, Jagadish S, Vinayaka AC, Hemshekhar M, Paul M, Thushara RM, et al. A new ibuprofen derivative inhibits platelet aggregation and ROS mediated platelet apoptosis. PLoS One. 2014;9(9):e107182 Epub 2014/09/23. 10.1371/journal.pone.0107182 ; PubMed Central PMCID: PMCPmc4169656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davies TG, Verdonk ML, Graham B, Saalau-Bethell S, Hamlett CC, McHardy T, et al. A structural comparison of inhibitor binding to PKB, PKA and PKA-PKB chimera. Journal of molecular biology. 2007;367(3):882–94. Epub 2007/02/06. 10.1016/j.jmb.2007.01.004 . [DOI] [PubMed] [Google Scholar]
- 40.Molecular Operating Environment (MOE), Chemical Computing Group Inc., Montreal, Canada, 2014.
- 41.Labute P. Protonate3D: assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins. 2009;75(1):187–205. Epub 2008/09/25. 10.1002/prot.22234 ; PubMed Central PMCID: PMCPmc3056144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rangappa KS. New cholinesterase inhibitors: synthesis and structure–activity relationship studies of 1, 2‐benzisoxazole series and novel imidazolyl‐d2‐isoxazolines. Journal of physical organic chemistry. 2005;18(8):773–8. [Google Scholar]
- 43.Bratulescu G. Synthesis of 4, 5-Substituted Imidazoles by a Fast Condensation of 1, 2-Diketones and Urotropine in Heterogeneous Medium. Synthesis. 2009;(14):2319–20. [Google Scholar]
- 44.Kim SW, Kim S-M, Bae H, Nam D, Lee J-H, Lee S-G, et al. Embelin inhibits growth and induces apoptosis through the suppression of Akt/mTOR/S6K1 signaling cascades. The Prostate. 2013;73(3):296–305. 10.1002/pros.22574 [DOI] [PubMed] [Google Scholar]
- 45.Bharathkumar H, Mohan CD, Ananda H, Fuchs JE, Li F, Rangappa S, et al. Microwave-assisted synthesis, characterization and cytotoxic studies of novel estrogen receptor α ligands towards human breast cancer cells. Bioorganic & medicinal chemistry letters. 2015;25(8):1804–7. [DOI] [PubMed] [Google Scholar]
- 46.Subramaniam A, Shanmugam MK, Ong TH, Li F, Perumal E, Chen L, et al. Emodin inhibits growth and induces apoptosis in an orthotopic hepatocellular carcinoma model by blocking activation of STAT3. British Journal of Pharmacology. 2013;170(4):807–21. 10.1111/bph.12302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neelgundmath M, Dinesh KR, Mohan CD, Li F, Dai X, Siveen KS, et al. Novel synthetic coumarins that targets NF-κB in Hepatocellular carcinoma. Bioorganic & medicinal chemistry letters. 2015;25(4):893–7. [DOI] [PubMed] [Google Scholar]
- 48.Mohan CD, Bharathkumar H, Bulusu KC, Pandey V, Rangappa S, Fuchs JE, et al. Development of a novel azaspirane that targets the Janus kinase-signal transducer and activator of transcription (STAT) pathway in hepatocellular carcinoma in vitro and in vivo. The Journal of biological chemistry. 2014;289(49):34296–307. Epub 2014/10/17. 10.1074/jbc.M114.601104 ; PubMed Central PMCID: PMCPmc4256360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keerthy HK, Mohan CD, Sivaraman Siveen K, Fuchs JE, Rangappa S, Sundaram MS, et al. Novel synthetic biscoumarins target tumor necrosis factor-alpha in hepatocellular carcinoma in vitro and in vivo. The Journal of biological chemistry. 2014;289(46):31879–90. Epub 2014/09/19. 10.1074/jbc.M114.593855 ; PubMed Central PMCID: PMCPmc4231667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park KR, Nam D, Yun HM, Lee SG, Jang HJ, Sethi G, et al. beta-Caryophyllene oxide inhibits growth and induces apoptosis through the suppression of PI3K/AKT/mTOR/S6K1 pathways and ROS-mediated MAPKs activation. Cancer letters. 2011;312(2):178–88. Epub 2011/09/20. . [DOI] [PubMed] [Google Scholar]
- 51.Bharathkumar H, Paricharak S, Dinesh K, Siveen KS, Fuchs JE, Rangappa S, et al. Synthesis, biological evaluation and in silico and in vitro mode-of-action analysis of novel dihydropyrimidones targeting PPAR-γ. RSC Advances. 2014;4(85):45143–6. [Google Scholar]
- 52.Sun Y, Mao X, Fan C, Liu C, Guo A, Guan S, et al. CXCL12-CXCR4 axis promotes the natural selection of breast cancer cell metastasis. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35(8):7765–73. Epub 2014/05/09. 10.1007/s13277-014-1816-1 ; PubMed Central PMCID: PMCPmc4158177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391(6662):43–50. Epub 1998/01/09. 10.1038/34112 . [DOI] [PubMed] [Google Scholar]
- 54.Roopashree R, Mohan CD, Swaroop TR, Jagadish S, Raghava B, Balaji KS, et al. Novel synthetic bisbenzimidazole that targets angiogenesis in Ehrlich ascites carcinoma bearing mice. Bioorg Med Chem Lett. 2015;25(12):2589–93. Epub 2015/04/30. 10.1016/j.bmcl.2015.04.010 . [DOI] [PubMed] [Google Scholar]
- 55.Kim HM, Kim CS, Lee JH, Jang SJ, Hwang JJ, Ro S, et al. CG0009, a novel glycogen synthase kinase 3 inhibitor, induces cell death through cyclin D1 depletion in breast cancer cells. PLoS One. 2013;8(4):e60383 Epub 2013/04/09. 10.1371/journal.pone.0060383 ; PubMed Central PMCID: PMCPmc3614906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Siddharth S, Mohapatra P, Preet R, Das D, Satapathy SR, Choudhuri T, et al. Induction of apoptosis by 4-(3-(tert-butylamino)imidazo[1,2-alpha]pyridine-2-yl) benzoic acid in breast cancer cells via upregulation of PTEN. Oncology research. 2013;21(1):1–13. Epub 2013/12/18. 10.3727/096504013x13786659070190 . [DOI] [PubMed] [Google Scholar]
- 57.Lali FV, Hunt AE, Turner SJ, Foxwell BM. The pyridinyl imidazole inhibitor SB203580 blocks phosphoinositide-dependent protein kinase activity, protein kinase B phosphorylation, and retinoblastoma hyperphosphorylation in interleukin-2-stimulated T cells independently of p38 mitogen-activated protein kinase. The Journal of biological chemistry. 2000;275(10):7395–402. Epub 2000/03/04. . [DOI] [PubMed] [Google Scholar]
- 58.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–74. Epub 2007/07/03. 10.1016/j.cell.2007.06.009 ; PubMed Central PMCID: PMCPmc2756685. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC)
(DOC)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.