

Abstract
Histone modifications and DNA methylation represent central dynamic and reversible processes that regulate gene expression and contribute to cellular phenotypes. These epigenetic marks have been shown to play fundamental roles in a diverse set of signaling and behavioral outcomes. Serotonin is a monoamine that regulates numerous physiological responses including those in the central nervous system. The cardinal signal transduction mechanisms via serotonin and its receptors are well established, but fundamental questions regarding complex interactions between the serotonin system and heritable epigenetic modifications that exert control on gene function remain a topic of intense research and debate. This review focuses on recent advances and contributions to our understanding of epigenetic mechanisms of serotonin receptor-dependent signaling, with focus on psychiatric disorders such as schizophrenia and depression.
Keywords: Schizophrenia, psychosis, antipsychotics, serotonin 5-HT2A receptor, hallucinogens, lysergic acid diethylamide (LSD), epigenetics, histone deacetylases (HDACs), G protein-coupled receptors (GPCRs)
Graphical abstract
The term “epigenetics” was coined by Conrad H. Waddington in the 1940s and alluded to the fundamental question of how a particular genome generates a highly complex organism containing innumerable cell types derived from a single fertilized egg.1 To that end, this field was mostly focused on processes related to embryonic development and cellular differentiation. Over the past decade, however, the definition of epigenetics has changed as the term now refers to the complete description of these potentially heritable changes across the genome that regulate gene transcription without modifying the underlying DNA sequence.2–7 Because these changes, which include primarily many types of histone and nucleotide modifications such as DNA methylation and DNA hydroxymethylation, are mitotically or meiotically heritable but environmentally modifiable, epigenetics has emerged as an important area of molecular biological studies focused on how environmental factors could mediate stable changes in brain function. In this review, we discuss recent advances in our understanding of epigenetic contribution to serotonin-dependent signaling, in particular the role of histone and DNA modifications in psychiatric disorders and their treatment, including schizophrenia8 and depression.9
OVERVIEW OF MECHANISMS OF EPIGENETIC REGULATION
The completion of the sequencing of the human genome is viewed as an important milestone.10,11 However, the primary sequence represents only the beginning of our understanding of how the genetic information is stored and, most importantly, read. In eukaryotic cells, in contrast to prokaryotes, the DNA is packaged in the form of a nucleoprotein complex called chromatin. The nucleosome is the basic repeating structural unit of chromatin, which contains 147 base pairs of DNA wrapped twice around an octamer of two copies of each core histone protein (H2A, H2B, H3, and H4). Histones are deeply evolutionarily conserved proteins with a globular domain and a flexible amino-terminal tail.5,12,13 By this design, biochemical modifications of these subunits can cause the histones to exist in one of two possible states: “open”, where the histone subunits are spaced apart and DNA that is wrapped around them is exposed to be transcribed, and “closed”, in which the histone subunits are packed tightly together and the DNA is not exposed and cannot be transcribed. Thus, the status of chromatin organization, and hence open or closed states of chromatin and DNA accessibility, depends on the so-called epigenetic modifications that fall into two main categories: DNA methylation and histone modifications.5,12,14–17 This epigenetic information has been shown to be fundamental during embryonic development and tissue-specific cellular differentiation.18,19 Recent studies also suggest that epigenetic modifications might constitute a new template for psychiatric interventions.20
DNA methylation is an epigenetic mark often associated with stable variations in gene expression that consists of the addition of a methyl group to the C5 position of cytosine at CpG dinucleotides. Most of the methylations occur within CpG islands, which are regions of DNA containing a high GC content (>55%).21 These GC-rich sequences colocalize with approximately 60% of all promoters and are largely free of DNA methylation. Only a small proportion of these CpG islands become methylated during development, particularly within genomic regions that are cell-type specific. Methylation of DNA is catalyzed by a family of DNA methyltransferases (DNMTs): DNMT1, DNMT2, DNMT3A, and DNMT3B. The regulatory factor DNMT3L stimulates the DNA methylation activity of DNMT3A and DNMT3B.22 During embryonic development, DNMTs are differentially expressed, and their spatiotemporal distribution has been suggested to play an important role in neurogenesis, neuronal maturation and memory formation.23–26 In mammals, patterns of DNA methylation are established during embryonic development by DNMT3A and DNMT3B, and maintained by a DNMT1-mediated copying mechanism when cells divide. DNA methylation is increased following learning26 and maintained to support long-term information storage in the cortex.27 In addition, demethylation of DNA appears to be involved in long-term changes of neuronal responsiveness.28 These and other findings suggest that methylation of cytosines in CpG sites influences normal brain physiology and neuropsychiatric disorders.
Although dynamic variation in the accumulation of 5-methylcytosine (5-mC) has emerged as a key factor in the regulation of brain function, 5-mC is not the only covalent modification of DNA in eukaryotes. Thus, it has been demonstrated that CpG dinucleotides can be successively oxidized and converted to 5-hydroxymethylcytosine (5-hmC), 5-formylcytosine (5-fC), and 5-carboxylcytosine (5-caC) by the TET family of DNA dioxygenases. An unusual DNA nucleotide, 5-hmC was detected while comparing the levels of 5-mC in cerebellar Purkinje neurons.29 In parallel, another group identified the ten-eleven translocase (TET) proteins that hydroxylate 5-mC to form 5-hmC.30 There is now considerable interest in this field because hydroxylation of 5-mC is likely the first step in the mechanism through which DNA methylation is reversed. These newly revealed DNA base modifications immediately drew broad attention from the research community and have been extensively reviewed. Understanding the dynamics of these modifications in living biological systems could lead to novel treatments for a number of psychiatric conditions.31,32
Covalent modifications at the N-terminal tail of histones correlate with open or closed states of chromatin depending on the type of modification. Thus, acetylation of histone H3 (H3ac) and acetylation of histone H4 (H4ac) are modifications that create a more open chromatin architecture.33 Histone methylation, in contrast, correlates with either transcriptional activation, such as methylation of lysine 4 on histone H3 (H3K4me) and methylation of lysine 36 on histone H3 (H3K36me), or repression, such as methylation of lysine 9 on histone H3 (H3K9me) and methylation of lysine 27 on histone H3 (H3K27me), depending on the histone and amino acid residue being methylated. Histone acetylation is catalyzed by histone acetyltransferases (HATs), and this modification can be reversed by the enzymatic action of histone deacetylases (HDACs). According to neighbor joining methods and Bayesian analysis,34 members of the HDAC family fall into four different phylogenetic classes: class I (HDAC1, 2, 3, and 8), class II (HDAC4, 5, 6, 7, 9, and 10), class III (SIR2 family of NAD+-dependent HDACs), and class IV (HDAC11). Class I and II (Zn-dependent) and class III (NAD+-dependent) show different distribution and are expressed among distinct cell types, including neurons, oligodendrocytes, and astrocytes.35 Most HDACs were found to be expressed primarily in neurons, whereas HDAC2–5 and HDAC11 were present in oligodendrocytes (suggesting a potential role in myelination) and only HDAC1, 3, and 5 were expressed in choroid plexus.35 The unusual expression of HDAC11 in hippocampus suggests a potential role in cognition and memory, whereas the high level of HDAC11 expression selectively in the Purkinje cell layer indicates a role in locomotor activity.35 These and other recent findings discussed below suggest that HDAC inhibitors might emerge as a new target for drug discovery in molecular psychiatry.36
ROLE OF EPIGENETIC MECHANISMS IN SEROTONIN-DEPENDENT SIGNALING
Abnormalities in serotonin (5-hydroxytryptamine; 5-HT)-dependent signaling during critical periods of neurodevelopment have been implicated in psychiatric disorders such as schizophrenia, depression, and autism.37,38 Atypical antipsychotic drugs, such as clozapine, olanzapine, and risperidone, present a high affinity as antagonists/inverse agonists for serotonin receptors, including 5-HT2A and 5-HT1A, and a much lower affinity for dopamine D2 receptors.39,40 It has also been demonstrated that clozapine is a partial agonist at cloned human 5-HT1A receptors.41 Based on their pharmacological properties, antidepressant drugs are classified into six major groups: tricyclic antidepressants (e.g., amitriptyline, imipramine, desipramine, and amoxapine), selective serotonin reuptake inhibitors (SSRIs; e.g., fluoxetine, paroxetine, and citalopram), selective serotonin and norepinephrine reuptake inhibitors (SSNRIs; e.g., venlafaxine), selective norepinephrine reuptake inhibitors (SNRIs; e.g., reboxetine), monoamine oxidase (MAO) inhibitors (e.g., isocarboxazid), and atypical antidepressants (e.g., mirtazapine and mianserin). Tricyclic antidepressants are classified into three subgroups based on their central ring of five links and are characterized by their inhibition of serotonin and norepinephrine uptake, as well as their properties to block the function of several monoaminergic receptors. Atypical antidepressants, such as mirtazapine and its structurally related mianserin, also block the function of 5-HT2A receptors. Recent findings suggest epigenetic mechanisms that affect both the serotonin system and phenotypes induced by treatment with antipsychotic and antidepressant drugs.
Transient inhibition of serotonin transporter (5-HTT) during early stages in development with the SSRI fluoxetine produces abnormal emotional behaviors in mice, such as a reduction in the total distance traveled in an open field and a decrease in the total number of arm entries in the elevated plus-maze.42 This behavioral phenotype was mimicked in mice genetically deficient in 5-HTT expression.42 Additionally, blockade of 5-HTT during postnatal days P2–P11 in mice leads to dendritic hypotrophy and reduced excitability of infralimbic cortical pyramidal neurons, whereas prelimbic cortical pyramidal neurons, which normally inhibit fear extinction, show an increased excitability.43 In contrast, it has also been demonstrated that administration of the SSRI escitalopram during postnatal days P5–P21 reduces anxiety-related behavior in adolescent mice,44 whereas 5-HTT knockout mice exhibited increased anxiety-related behaviors in both elevated plus maze and open field test.44 Because exploratory behavior was reduced in 5-HTT knockout mice, and early exposure to fluoxetine produced some but not all features associated with constitutive 5-HTT deficiency,44 these findings suggest changes in adult behavior induced by developmental exposure to antidepressants that are not recapitulated in 5-HTT knockout mice. Additional work will be necessary to identify the molecular and neurodevelopmental mechanisms that affect emotional behavior by early life blockade of the 5-HT transporter.
Using bisulfite sequencing to compare methylation of 20 CpG sites close to the transcription start site of the promoter region of the serotonin transporter gene (SLC6A4), recent findings in two cohorts (saliva-derived DNA from 80 young adults, and blood-derived DNA from 96 adolescents) suggest that methylation of the SLC6A4 promoter predicts amygdala reactivity using blood oxygen level-dependent (BOLD) functional magnetic resonance imaging (fMRI).45 Together, these results suggest that epigenetic modulation of 5-HTT (SLC6A4) affects behaviorally and clinically relevant brain functions. Insights gained from these studies will be key to identifying novel targets for diagnosis and therapy.
The serotonin 5-HT6 receptor, whose canonical signaling pathway consists of activation of heterotrimeric Gs proteins and the cyclic adenosine monophosphate (cAMP) pathway, is suspected to modulate neuronal development. Recent findings also demonstrate an alternative 5-HT6 receptor-dependent signaling pathway that participates in the control of neuronal migration.46 It has been reported that the carboxyl terminus of the 5-HT6 receptor recruits a population of signaling proteins,47 such as cyclin-dependent kinase 5 (Cdk5), which is implicated in various neuropsychiatric disorders including schizophrenia, major depression, and Alzheimer’s disease.48,49 This 5-HT6/Cdk5-dependent signaling pathway affected neuronal migration, neurite growth, and dendritic structure through a mechanism that required phosphorylation of the Cdk5 substrate histone H1.47 Additionally, these structural effects were independent of 5-HT6 receptor coupling to Gs.47 This effect on neurite growth of primary neurons was also reduced by mutating Ser350 into alanine in the carboxyl terminus of the 5-HT6 receptor.47 Although these findings suggest that Cdk5 and 5-HT6 are both implicated in the epigenetic regulation of neurite growth and neuronal migration, events that may be perturbed in developmental disorders, further work will be necessary to determine the precise epigenetic role of histone H1 as Cdk5 substrate in neuronal differentiation.
Long-term memories last a lifetime, whereas the RNA or protein markers that may underlie these memory traces are replaced with new functional copies on the order of hours or days.50 An attractive hypothesis to explain how memories remain stable despite the constant molecular turnover is related to epigenetic mechanisms that may affect the intrinsic properties of neurons in a long-term fashion. An example of this model is the epigenetic control of serotonin-dependent modulation of synaptic plasticity. Using the Aplysia central nervous system as a model, researchers found that serotonin induces methylation of a conserved CpG island in the promoter region of the CREB2 gene, leading to enhanced long-term synaptic facilitation.51 Interestingly, this epigenetic hypothesis is further supported by recent findings suggesting that induction of long-term memory (LTM) by serotonin in Aplysia requires epigenetic changes.52 LTM has been linked with functional strengthening of existing synapses and other processes including de novo synaptogenesis.50 A manipulation that can erase LTM permanently is inhibition of the constitutively active catalytic fragment of the atypical protein kinase Cζ (PKM).53 It has been found that LTM can persist following reconsolidation blockade and inhibition of PKM,52 indicating that consolidated memories may be far more refractory to modification or elimination than generally supposed. If these findings are confirmed in mammals, it would challenge the idea that the synapse is a cellular site for long-term memory storage.
Most antidepressants have a delayed onset of therapeutic efficacy.54 Specifically, SSRIs and tricyclic antidepressants often require several weeks of administration to reach full clinical efficacy. Using chronic social defeat stress as a model of depression in mice, researchers demonstrated that defeat stress induces lasting down-regulation of BDNF transcripts together with increased repressive histone methylation at their respective promoters in the hippocampus.55 Importantly, chronic treatment with the antidepressant imipramine, which inhibits serotonin uptake, induces a selective down-regulation of HDAC5.55 In addition, it was demonstrated that herpes simplex virus (HSV)-mediated overexpression of HDAC5 in the hippocampus prevented the antidepressant-like behavioral effects of chronic treatment with imipramine.55 More recent findings further support this view based on the demonstration that chronic treatment with the antidepressant fluoxetine induced a transient increase in BDNF expression in the adult visual cortex, an effect that occurred in association with increased H3ac at BDNF promoter regions and down-regulation of Hdac5.56 These experiments highlight an important role for the serotonergic system in processes of histone remodeling related to depression.
Most of the studies focused on the roles of HDACs in the CNS have been centered on the canonical function of these enzymes (i.e., deacetylation of histone tails). However, recent studies demonstrate that histones represent a small fraction of the total acetylome affected by HDAC-dependent function.57 Almost any kind of threat to homeostasis or stress will cause plasma glucocorticoid levels to rise, which has traditionally been linked to the physiological function of enhancing the organism’s resistance to stress. Using rodent models of post-traumatic stress disorder (PTSD), it has been suggested that cytoplasmic HDAC6 modulates acetylation of a key component of the glucocorticoid chaperone complex in the brain: Hsp90.58 According to findings based on transgenic Pet1-Cre mice crossed to HDAC6loxP/loxP mice in which a portion of the Hdac6 gene (exons 8–9) is floxed, it has been reported that deletion of HDAC6 exclusively in serotonin neurons reduces the anxiogenic-like effects of the glucocorticoid hormone cortico-sterone.58 The role of HDAC6 in emotional behavior via deacetylation of non-histone proteins, including α-tubulin, Hsp90, and cortactin, has been supported by the behavioral changes observed in HDAC6-KO mice, such as hyperactivity, less anxiety, and antidepressant-like behavior.59
Psychoactive drugs such as dissociatives (phencyclidine [PCP] and ketamine) and hallucinogens (lysergic acid diethylamide [LSD], psilocybin, and mescaline) induce alterations in perception, cognition, sensorimotor gating, and social skills that exhibit certain similarities with the endogenous psychosis of schizophrenia patients.60–64 These drugs, when injected in rodents, also elicit their own specific set of behaviors, such as deficits in working memory, deficits in sensorimotor gating, modulation of locomotion, and a particular side-to-side head movement known as the head-twitch response.60,65,66 Due to the psychotomimetic action of these compounds, LSD and other hallucinogens have been used repeatedly in the preclinical search for more effective antipsychotic compounds.65 In the 1990s, considerable interest was focused on the possible connection between the serotonergic and glutamatergic systems in schizophrenia.66,67 Milestone findings revealed that the mGlu2/3 receptor agonist LY354740 reversed the effects of PCP using paradigms such as working memory, deficits in sensorimotor gating, hyper-locomotor activity, and cortical glutamate efflux in an animal model.68
Follow-up studies by other groups demonstrated that LY354740 antagonizes the induction of excitatory postsynaptic potential and currents (EPSPs and EPSCs) via activation of the serotonin 5-HT2A receptor.69 It was also shown that LY354740 suppresses the head-twitch behavior induced by the hallucinogen (±)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane hydrochloride (DOI).70 Based on these findings, there was an enormous increase in the number of publications describing the potential use of mGlu2/3 agonists as antipsychotic-like drugs in rodents, as compounds such as LY354740, LY379268, and LY404039 were found to repress the cellular, electrophysiological, and behavioral effects induced by hallucinogenic 5-HT2A agonists.71 It has also been demonstrated that the antipsychotic-like effects of these mGlu2/3 receptor agonists are mediated via mGlu2-dependent and not mGlu3-dependent signaling.72–74
In light of these animal studies, it was shown that the mGlu2/3 receptor agonist LY404039 (active compound of LY2140023) improved both positive and negative symptoms of schizophrenia compared with placebo treatment.75 Importantly, however, follow-up studies provided convincing evidence that LY2140023 does not separate from placebo, whereas atypical antipsychotics such as risperidone and olanzapine were efficacious in the treatment of positive symptoms.76–79 Thus, improvement in PANSS total score (Positive and Negative Syndrome Scale) was significantly greater in the standard of care (SOC: olanzapine, risperidone, or aripiprazole) group.76–79 These findings preceded the press release by Eli Lilly and Company with the decision to stop clinical trials with LY2140023 for the treatment of schizophrenia. Recent preclinical and clinical findings, however, may provide an epigenetic model of why some of the schizophrenia patients do not respond to treatment with the mGlu2/3 receptor agonist LY404039 (Figure 1).
Figure 1.

(A, B) Schematic model of the effect of chronic atypical antipsychotic treatment on the epigenetic status of the mGlu2 (Grm2) gene. Activation of the 5-HT2A receptor by the endogenous neurotransmitter serotonin represses the promoter activity of the HDAC2 gene in mouse and in human frontal cortex (see also ref 83). Atypical antipsychotic drugs, such as clozapine and risperidone, reverse the 5-HT2A receptor-dependent repression of HDAC2, an effect that is associated with increased HDAC2 promoter activity and repressive histone modifications at the mGlu2 promoter. This epigenetic effect of chronic atypical antipsychotic treatment may limit the therapeutic effects of mGlu2/3 agonists such as LY2140023 (pomaglumetad, pro-drug of LY404039). (C) Decreased acetylation of histone H3 (H3ac) at the mGlu2 promoter in prefrontal cortex of schizophrenia patients treated with atypical antipsychotic drugs (see also ref 83).
In schizophrenia patients, many articles describe improvement hours or days immediately after intramuscular anti-psychotic drug administration.80 However, psychiatric disorders in general, and schizophrenia in particular, are often characterized by chronic drug administration (weeks, months, or even years of sustained drug treatment). The prolonged use of these drugs in schizophrenia treatment emphasized the importance of understanding the consequences of the use of these compounds long-term. In fact, a recent 20-year follow-up study demonstrates that long-lasting treatment with anti-psychotic medications does not eliminate or reduce the frequency of psychosis in schizophrenia patients.81 Thus, treated patients will usually enter a stabilization phase in which psychotic symptoms have been controlled, but patients remain at risk for relapse if treatment is interrupted.81 This demonstrates that the antipsychotics currently available are designed only to treat the symptoms of schizophrenia and not the underlying causes of the disease. Additionally, whereas these findings suggest that the therapeutic effects of antipsychotic drugs are acute, they also validate the importance of understanding the consequences of long-term antipsychotic drug treatment. For chronic treatment with typical and atypical antipsychotic drugs in mouse models, it has been demonstrated that chronic treatment with atypical antipsychotics markedly decreases the density of 5-HT2A receptors in frontal cortex.82–84 Considering that the density of 5-HT2A receptors is increased in post-mortem frontal cortex of antipsychotic-free schizophrenic subjects, and reduced to control levels in postmortem frontal cortex of antipsychotic-treated schizophrenic subjects,84,85 these findings suggest that down-regulation of 5-HT2A receptor density by chronic atypical antipsychotic treatment may be one of the molecular mechanisms involved in their therapeutic effects (see refs 86 and 87 for review articles discussing the level of expression of 5-HT2A receptor in the schizophrenia brain). However, it was also demonstrated that this down-regulation of 5-HT2A receptor density in mouse frontal cortex by chronic treatment with atypical antipsychotic drugs leads to down-regulation of the transcription of mGlu2, an effect that was associated with repressive histone modifications at the promoter region of the mGlu2 gene in mouse and human frontal cortex.83 This epigenetic change occurs in association with a 5-HT2A receptor-dependent up-regulation of HDAC2 and increased binding of HDAC2 to the mGlu2 promoter.83,88 Based on these findings (Figure 1), it was proposed that chronic treatment with atypical antipsychotic drugs induces a selective 5-HT2A receptor-dependent up-regulation of HDAC2 in frontal cortex of individuals with schizophrenia, which consequently induces repressive epigenetic marks at the mGlu2 promoter and thereby limits the therapeutic effects of mGlu2/3 agonists in these patients.83,88,89
Notably, this hypothesis has recently been substantiated by re-evaluation of previous clinical work. Thus, preliminary data suggest that previous exposure of schizophrenia patients to chronic treatment with atypical antipsychotic drugs, such as olanzapine and risperidone, prevents the therapeutic effects of LY404039, whereas this mGlu2/3 receptor agonist induces antipsychotic effects in patients previously treated with typical antipsychotics, such as haloperidol, that separate from placebo.90 In addition, a genetic association between SNPs in the 5-HT2A (Htr2a) gene and the response to LY404039 treatment has been demonstrated.91 Further preclinical and clinical investigation is definitely needed to validate the molecular mechanisms underlying these effects of typical versus atypical antipsychotic drug treatment on therapeutic responses to LY404039. However, based on previous findings,83 a potential approach to reverse or prevent the repressive epigenetic changes induced at the mGlu2 promoter in frontal cortex of schizophrenia patients chronically treated with atypical antipsychotics may be the use of adjunctive treatment with HDAC2 inhibitors. Thus, adjunctive treatment with suberoylanilide hydroxamic acid (SAHA), which is a selective inhibitor of class I and class II HDACs, prevented the repressive histone modifications induced at the mGlu2 promoter by chronic treatment with atypical antipsychotic drugs and improved their therapeutic-like effects in mouse models.83
ROLE OF PRENATAL ENVIRONMENT IN SEROTONIN-DEPENDENT SIGNALING
There is extensive evidence to suggest that genetics plays a significant role in the etiology of schizophrenia.92–94 The genetic determinants, however, are complex, as clearly elucidated from studies on twins. Monozygotic twins, whose DNA sequences are ~100% identical, have a concordance for schizophrenia of nearly 50%.95–98 Such results favor a significant contribution of genetic factors to the etiology of schizophrenia. At the same time, they argue for a significant role of environmental events in the development of this complex psychiatric disease. Epidemiological studies have indicated that maternal infection during pregnancy with a wide variety of agents, including viruses (influenza99,100 and rubella101), bacteria (bronchopheumonia102), and protozoa (Toxoplasma gondii103) increase the risk of schizophrenia in the adult offspring. Similarly, maternal adverse life events that occurred during pregnancy, such as war, famine, and death or illness in a first-degree relative, have been associated with schizophrenia in the adult offspring.104–106 A conservative hypothesis would be that the vulnerability to schizophrenia is multifactorial, caused by the interaction between genes and environmental factors. Nevertheless, recent genome-wide association studies focused on de novo mutations show that the 5-HT2A gene (Htr2a) might potentially be implicated in schizophrenia-related synaptic netwoks.93 Additionally, alterations in density of 5-HT2A receptor binding have been reported in frontal cortex of schizophrenia subjects, using both radioligand binding assays in post-mortem samples84,85,107 and positron emission tomography (PET) in schizophrenia patients.108,109 (See also refs 86 and 87 for review articles). The potential link between alterations in 5-HT2A receptor-dependent signaling and schizophrenia-related behaviors has been suggested recently by findings based upon rodent models of prenatal adverse life events.
A relevant line of research has recently been focused on the importance of maternal infection during pregnancy as a risk factor for schizophrenia.110 Using the human influenza virus A/NWS/33CHINI, it has been shown that maternal influenza infection induces behavioral changes in the adult offspring that model schizophrenia-related symptoms, such as deficits in PPI in the acoustic startle response and alterations in exploratory behavior and social interaction.111 Notably, more recent findings demonstrate that maternal infection with the mouse-adapted influenza A/WSN/33 (H1N1) virus causes up-regulation of 5-HT2A binding and 5-HT2A mRNA expression in frontal cortex in the adult offspring.112 Additionally, the behavioral response to the hallucinogenic 5-HT2A agonist DOI was shown to be affected by maternal influenza viral infection.112 Thus, adult mice born to influenza virus-infected mothers show increased head-twitch behavior and dysregulated locomotor activity in response to DOI.112 Additional findings obtained by independent groups testing the effects of several environmental factors, such as variable stress during pregnancy,113 maternal immune activation during pregnancy,114 Roman Low- and High-Avoidance (RLA-I versus RHA-I) rat strains,115 transgenic mice with a knock-in of a tryptophan hydroxylase 2 (Tph2) R439H mutation,116 and repeated administration of methamphetamine,117 validate that adverse environmental effects induce up-regulation of 5-HT2A receptor in frontal cortex and dysregulation of 5-HT2A receptor-dependent signaling and behavioral function (Figure 2). Although together with previous findings in post-mortem human brain of schizophrenic subjects and in schizophrenia patients (see above), these results suggest that up-regulation of 5-HT2A receptor density might be involved in schizophrenia-related phenotypes, further investigation will be necessary to understand the molecular and epigenetic mechanisms responsible for the effects of prenatal insults on 5-HT2A receptor density and its function. The fundamental role of gene–environment interactions in the function of the 5-HT2A receptor is further supported by an association between changes in placental methylation of the 5-HT2A gene and deficits in infant neurobehavioral outcomes.118 Together with the schematic model presented in Figure 1 (see above), these data also suggest that whereas up-regulation of 5-HT2A receptor density in frontal cortex of untreated schizophrenic subjects might predispose to psychosis, down-regulation of frontal cortex 5-HT2A receptor density by chronic treatment with atypical antipsychotic drugs may be one of the mechanisms underlying their therapeutic effects.
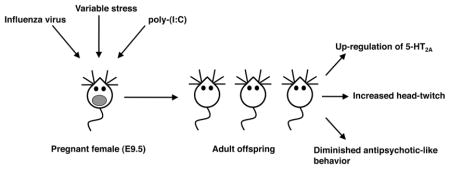
Figure 2.

(A, B) Increased 5-HT2A receptor density as defined by [3H]ketanserin binding saturation curves in post-mortem frontal cortex of schizophrenic subjects and individually matched controls. Note that 5-HT2A receptor density is increased in untreated schizophrenic subjects (A) and unaffected in antipsychotic-treated schizophrenic subjects (B) compared with controls (see also refs 84 and 85). (C) In mouse models, prenatal insults during pregnancy such as maternal influenza viral infection, maternal variable and unpredictable stress, and maternal immune activation by injection of poly(I:C) induce alterations in expression and behavior function of the 5-HT2A receptor in the adult offspring. These prenatal insults also decrease mGlu2-dependent antipsychotic-like behavioral effects of the mGlu2/3 agonist LY379268. (D) Increased 5-HT2A receptor binding in frontal cortex of adult mice born to influenza virus-infected mothers (see also refs 112 and 113). (E) Increased head-twitch behavior induced by the hallucinogenic 5-HT2A receptor agonist DOI in adult mice born to influenza virus-infected mothers (see also refs 112 and 113).
Additional evidence suggests that environmental factors affect serotonin-dependent function in mouse and humans. For example, using immunohistochemical approaches, it was reported that maternal stress in rats induces a decrease 5-HT1A-like immunoreactivity in the hippocampus of the adult offspring.119 These findings correlate with the increased DNA methylation in the promoter region of the 5-HT1A (Htr1a) gene observed in schizophrenia and bipolar disorder.120 Combining the paradigms of maternal stress with heterozygous 5-HTT deficient mice (5-HTT+/−), genome-wide hippocampal gene expression profiling suggests that numerous genes and related pathways are differentially affected by prenatal stress and the 5-HTT genotype.121 Among these, MAPK and neurotrophin signaling pathways were shown to be dysregulated by both the 5-HTT+/− genotype and prenatal stress exposure.121 Additionally, exposure of 5-HTT+/− mice to prenatal stress induced behavioral deficits related to cognition.121
Post-mortem human brain studies have revealed decreased forebrain expression of glutamic acid decarboxylase (GAD1), which correlates with increased DNA methylation of the GAD1 promoter.122 It is known that maternal behavior affects epigenetic programming in the offspring.123–125 When the effects of maternal care on GAD1 promoter activity were examined in the hippocampus of rats, it was demonstrated that mice born to high licking/grooming mothers showed enhanced hippocampal GAD1 expression, which was accompanied by decreased DNA methylation at the GAD1 promoter, compared with mice born to low licking/grooming mothers.126 Epigenetic assays revealed enhanced H3K9ac binding to the GAD1 promoter in the hippocampus of mice born to high licking/grooming mothers.126 Additionally, treatment of hippocampal neuronal cultures with 5-HT significantly increased GAD1 mRNA levels.126 Together, these data suggest that serotonin-dependent signaling may serve as a tool to epigenetically affect the function of GAD1 and consequently the GABAergic system, which is linked to the pathophysiology of schizophrenia.
FUTURE DIRECTIONS
This review has summarized the increasing array of findings that support a role of serotonin-dependent epigenetic regulation of physiological and behavioral responses. We have focused our attention on two main aspects related to epigenetic mechanisms of serotonin signaling in whole animal models of psychiatric disorders such as schizophrenia and depression: the role of epigenetic mechanisms in serotonin receptor-dependent signaling and how environmental events during pregnancy induce stable adaptations and maladaptations of the serotonin system in the brain. The mechanisms of basic transcriptional and epigenetic mechanisms are varied and highly complex, and further studies are needed to understand what controls the epigenetic state of specific subsets of genes. An important question is related to how epigenetic changes are translated into transcriptional modulation. For example, no single epigenetic modification examined to date has been shown to be deterministic for a change in gene promoter activity. This suggests that numerous modifications that work in concert are required for transcriptional changes. Although epigenetic enzymes such as HDACs have emerged recently as attractive therapeutic targets, a major limitation is that manipulation of such targets will affect chromatin structure at hundreds or thousands of genes throughout the brain. Attractive approaches to succeed with these limitations are the use of synthetic zinc finger proteins (ZFPs) or sequence-specific transcription activator-like effectors (TALEs) coupled to an enzymatic moiety. These approaches would be designed to target a particular chromatin modification to a given gene within a region of the CNS. These goals represent perhaps one of the greatest challenges in serotonin research.
Acknowledgments
The preparation of this review has been supported at least in part by the National Institutes of Health (Grant R01MH084894). T.H. was recipient of Minority Scholars Award Scholarship from the A.M.A. Foundation.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Sun H, Kennedy PJ, Nestler EJ. Epigenetics of the depressed brain: Role of histone acetylation and methylation. Neuropsychopharmacology. 2013;38:124–137. doi: 10.1038/npp.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502:480–488. doi: 10.1038/nature12751. [DOI] [PubMed] [Google Scholar]
- 3.de Laat W, Duboule D. Topology of mammalian developmental enhancers and their regulatory landscapes. Nature. 2013;502:499–506. doi: 10.1038/nature12753. [DOI] [PubMed] [Google Scholar]
- 4.Apostolou E, Hochedlinger K. Chromatin dynamics during cellular reprogramming. Nature. 2013;502:462–471. doi: 10.1038/nature12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–974. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502:489–498. doi: 10.1038/nature12752. [DOI] [PubMed] [Google Scholar]
- 7.Dupont C, Armant DR, Brenner CA. Epigenetics: Definition, mechanisms and clinical perspective. Semin Reprod Med. 2009;27:351–357. doi: 10.1055/s-0029-1237423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jakovcevski M, Akbarian S. Epigenetic mechanisms in neurological disease. Nat Med. 2012;18:1194–1204. doi: 10.1038/nm.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nestler EJ. Epigenetic mechanisms of depression. JAMA Psychiatry. 2014;71:454–456. doi: 10.1001/jamapsychiatry.2013.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, Szustakowki J, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 11.McPherson JD, Marra M, Hillier L, Waterston RH, Chinwalla A, Wallis J, Sekhon M, Wylie K, Mardis ER, Wilson RK, Fulton R, Kucaba TA, Wagner-McPherson C, Barbazuk WB, Gregory SG, Humphray SJ, French L, Evans RS, Bethel G, Whittaker A, Holden JL, McCann OT, Dunham A, Soderlund C, Scott CE, Bentley DR, Schuler G, Chen HC, Jang W, Green ED, Idol JR, Maduro VV, Montgomery KT, Lee E, Miller A, Emerling S, Kucherlapati R, Gibbs R, Scherer S, Gorrell JH, Sodergren E, Clerc-Blankenburg K, Tabor P, Naylor S, Garcia D, de Jong PJ, Catanese JJ, Nowak N, Osoegawa K, Qin S, Rowen L, Madan A, Dors M, Hood L, Trask B, Friedman C, Massa H, Cheung VG, Kirsch IR, Reid T, Yonescu R, Weissenbach J, Bruls T, Heilig R, Branscomb E, Olsen A, Doggett N, Cheng JF, Hawkins T, Myers RM, Shang J, Ramirez L, Schmutz J, Velasquez O, Dixon K, Stone NE, Cox DR, Haussler D, Kent WJ, Furey T, Rogic S, Kennedy S, Jones S, Rosenthal A, Wen G, Schilhabel M, Gloeckner G, Nyakatura G, Siebert R, Schlegelberger B, Korenberg J, Chen XN, Fujiyama A, Hattori M, Toyoda A, Yada T, Park HS, Sakaki Y, Shimizu N, Asakawa S, Kawasaki K, Sasaki T, Shintani A, Shimizu A, Shibuya K, Kudoh J, Minoshima S, Ramser J, Seranski P, Hoff C, Poustka A, Reinhardt R, Lehrach H. A physical map of the human genome. Nature. 2001;409:934–941. doi: 10.1038/35057157. [DOI] [PubMed] [Google Scholar]
- 12.Dulac C. Brain function and chromatin plasticity. Nature. 2010;465:728–735. doi: 10.1038/nature09231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 14.Day JJ, Sweatt JD. Epigenetic mechanisms in cognition. Neuron. 2011;70:813–829. doi: 10.1016/j.neuron.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol. 2007;14:1008–1016. doi: 10.1038/nsmb1337. [DOI] [PubMed] [Google Scholar]
- 16.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 17.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 18.Ptak C, Petronis A. Epigenetics and complex disease: From etiology to new therapeutics. Annu Rev Pharmacol Toxicol. 2008;48:257–276. doi: 10.1146/annurev.pharmtox.48.113006.094731. [DOI] [PubMed] [Google Scholar]
- 19.Orkin SH, Hochedlinger K. Chromatin connections to pluripotency and cellular reprogramming. Cell. 2011;145:835–850. doi: 10.1016/j.cell.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maze I, Shen L, Zhang B, Garcia BA, Shao N, Mitchell A, Sun H, Akbarian S, Allis CD, Nestler EJ. Analytical tools and current challenges in the modern era of neuroepigenomics. Nat Neurosci. 2014;17:1476–1490. doi: 10.1038/nn.3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antequera F. Structure, function and evolution of CpG island promoters. Cell Mol Life Sci. 2003;60:1647–1658. doi: 10.1007/s00018-003-3088-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suetake I, Shinozaki F, Miyagawa J, Takeshima H, Tajima S. DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J Biol Chem. 2004;279:27816–27823. doi: 10.1074/jbc.M400181200. [DOI] [PubMed] [Google Scholar]
- 23.Watanabe D, Uchiyama K, Hanaoka K. Transition of mouse de novo methyltransferases expression from Dnmt3b to Dnmt3a during neural progenitor cell development. Neuroscience. 2006;142:727–737. doi: 10.1016/j.neuroscience.2006.07.053. [DOI] [PubMed] [Google Scholar]
- 24.Kadriu B, Guidotti A, Chen Y, Grayson DR. DNA methyltransferases1 (DNMT1) and 3a (DNMT3a) colocalize with GAD67-positive neurons in the GAD67-GFP mouse brain. J Comp Neurol. 2012;520:1951–1964. doi: 10.1002/cne.23020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, Fan G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010;13:423–430. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 27.Miller CA, Gavin CF, White JA, Parrish RR, Honasoge A, Yancey CR, Rivera IM, Rubio MD, Rumbaugh G, Sweatt JD. Cortical DNA methylation maintains remote memory. Nat Neurosci. 2010;13:664–666. doi: 10.1038/nn.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen SX, Cherry A, Tari PK, Podgorski K, Kwong YK, Haas K. The transcription factor MEF2 directs developmental visually driven functional and structural metaplasticity. Cell. 2012;151:41–55. doi: 10.1016/j.cell.2012.08.028. [DOI] [PubMed] [Google Scholar]
- 29.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethyl-cytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song CX, Yi C, He C. Mapping recently identified nucleotide variants in the genome and transcriptome. Nat Biotechnol. 2012;30:1107–1116. doi: 10.1038/nbt.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev Genet. 2012;13:7–13. doi: 10.1038/nrg3080. [DOI] [PubMed] [Google Scholar]
- 33.Graff J, Tsai LH. Histone acetylation: Molecular mnemonics on the chromatin. Nat Rev Neurosci. 2013;14:97–111. doi: 10.1038/nrn3427. [DOI] [PubMed] [Google Scholar]
- 34.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 35.Broide RS, Redwine JM, Aftahi N, Young W, Bloom FE, Winrow CJ. Distribution of histone deacetylases 1–11 in the rat brain. J Mol Neurosci. 2007;31:47–58. doi: 10.1007/BF02686117. [DOI] [PubMed] [Google Scholar]
- 36.Penney J, Tsai LH. Histone deacetylases in memory and cognition. Sci Signal. 2014;7:re12. doi: 10.1126/scisignal.aaa0069. [DOI] [PubMed] [Google Scholar]
- 37.Ansorge MS, Hen R, Gingrich JA. Neurodevelopmental origins of depressive disorders. Curr Opin Pharmacol. 2007;7:8–17. doi: 10.1016/j.coph.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 38.Weisstaub NV, Zhou M, Lira A, Lambe E, Gonzalez-Maeso J, Hornung JP, Sibille E, Underwood M, Itohara S, Dauer WT, Ansorge MS, Morelli E, Mann JJ, Toth M, Aghajanian G, Sealfon SC, Hen R, Gingrich JA. Cortical 5-HT2A receptor signaling modulates anxiety-like behaviors in mice. Science. 2006;313:536–540. doi: 10.1126/science.1123432. [DOI] [PubMed] [Google Scholar]
- 39.Miyamoto S, Miyake N, Jarskog LF, Fleischhacker WW, Lieberman JA. Pharmacological treatment of schizophrenia: a critical review of the pharmacology and clinical effects of current and future therapeutic agents. Mol Psychiatry. 2012;17:1206–1227. doi: 10.1038/mp.2012.47. [DOI] [PubMed] [Google Scholar]
- 40.Meltzer HY. Update on typical and atypical antipsychotic drugs. Annu Rev Med. 2013;64:393–406. doi: 10.1146/annurev-med-050911-161504. [DOI] [PubMed] [Google Scholar]
- 41.Newman-Tancredi A, Chaput C, Verriele L, Millan MJ. Clozapine is a partial agonist at cloned, human serotonin 5-HT1A receptors. Neuropharmacology. 1996;35:119–121. doi: 10.1016/0028-3908(95)00170-0. [DOI] [PubMed] [Google Scholar]
- 42.Ansorge MS, Zhou M, Lira A, Hen R, Gingrich JA. Early-life blockade of the 5-HT transporter alters emotional behavior in adult mice. Science. 2004;306:879–881. doi: 10.1126/science.1101678. [DOI] [PubMed] [Google Scholar]
- 43.Rebello TJ, Yu Q, Goodfellow NM, Caffrey Cagliostro MK, Teissier A, Morelli E, Demireva EY, Chemiakine A, Rosoklija GB, Dwork AJ, Lambe EK, Gingrich JA, Ansorge MS. Postnatal day 2 to 11 constitutes a 5-HT-sensitive period impacting adult mPFC function. J Neurosci. 2014;34:12379–12393. doi: 10.1523/JNEUROSCI.1020-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Altieri SC, Yang H, O’Brien HJ, Redwine HM, Senturk D, Hensler JG, Andrews AM. Perinatal vs Genetic Programming of Serotonin States Associated with Anxiety. Neuro-psychopharmacology. 2014 doi: 10.1038/npp.2014.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nikolova YS, Koenen KC, Galea S, Wang CM, Seney ML, Sibille E, Williamson DE, Hariri AR. Beyond genotype: Serotonin transporter epigenetic modification predicts human brain function. Nat Neurosci. 2014;17:1153–1155. doi: 10.1038/nn.3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Riccio O, Potter G, Walzer C, Vallet P, Szabo G, Vutskits L, Kiss JZ, Dayer AG. Excess of serotonin affects embryonic interneuron migration through activation of the serotonin receptor 6. Mol Psychiatry. 2009;14:280–290. doi: 10.1038/mp.2008.89. [DOI] [PubMed] [Google Scholar]
- 47.Duhr F, Deleris P, Raynaud F, Seveno M, Morisset-Lopez S, Mannoury la Cour C, Millan MJ, Bockaert J, Marin P, Chaumont-Dubel S. Cdk5 induces constitutive activation of 5-HT6 receptors to promote neurite growth. Nat Chem Biol. 2014;10:590–597. doi: 10.1038/nchembio.1547. [DOI] [PubMed] [Google Scholar]
- 48.Graff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, Nieland TJ, Fass DM, Kao PF, Kahn M, Su SC, Samiei A, Joseph N, Haggarty SJ, Delalle I, Tsai LH. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483:222–226. doi: 10.1038/nature10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramos-Miguel A, Meana JJ, Garcia-Sevilla JA. Cyclin-dependent kinase-5 and p35/p25 activators in schizophrenia and major depression prefrontal cortex: basal contents and effects of psychotropic medications. Int J Neuropsychopharmacol. 2013;16:683–689. doi: 10.1017/S1461145712000879. [DOI] [PubMed] [Google Scholar]
- 50.Kandel ER, Dudai Y, Mayford MR. The molecular and systems biology of memory. Cell. 2014;157:163–186. doi: 10.1016/j.cell.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 51.Rajasethupathy P, Antonov I, Sheridan R, Frey S, Sander C, Tuschl T, Kandel ER. A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell. 2012;149:693–707. doi: 10.1016/j.cell.2012.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen S, Cai D, Pearce K, Sun PY, Roberts AC, Glanzman DL. Reinstatement of long-term memory following erasure of its behavioral and synaptic expression in Aplysia. Elife. 2014;3:e03896. doi: 10.7554/eLife.03896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sacktor TC. How does PKMzeta maintain long-term memory? Nat Rev Neurosci. 2011;12:9–15. doi: 10.1038/nrn2949. [DOI] [PubMed] [Google Scholar]
- 54.Gingrich JA, Hen R. Dissecting the role of the serotonin system in neuropsychiatric disorders using knockout mice. Psychopharmacology (Berlin) 2001;155:1–10. doi: 10.1007/s002130000573. [DOI] [PubMed] [Google Scholar]
- 55.Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- 56.Maya Vetencourt JF, Tiraboschi E, Spolidoro M, Castren E, Maffei L. Serotonin triggers a transient epigenetic mechanism that reinstates adult visual cortex plasticity in rats. Eur J Neurosci. 2011;33:49–57. doi: 10.1111/j.1460-9568.2010.07488.x. [DOI] [PubMed] [Google Scholar]
- 57.Spange S, Wagner T, Heinzel T, Kramer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. 2009;41:185–198. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 58.Espallergues J, Teegarden SL, Veerakumar A, Boulden J, Challis C, Jochems J, Chan M, Petersen T, Deneris E, Matthias P, Hahn CG, Lucki I, Beck SG, Berton O. HDAC6 regulates glucocorticoid receptor signaling in serotonin pathways with critical impact on stress resilience. J Neurosci. 2012;32:4400–4416. doi: 10.1523/JNEUROSCI.5634-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fukada M, Hanai A, Nakayama A, Suzuki T, Miyata N, Rodriguiz RM, Wetsel WC, Yao TP, Kawaguchi Y. Loss of deacetylation activity of Hdac6 affects emotional behavior in mice. PloS ONE. 2012;7:e30924. doi: 10.1371/journal.pone.0030924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hanks JB, Gonzalez-Maeso J. Animal models of serotonergic psychedelics. ACS Chem Neurosci. 2013;4:33–42. doi: 10.1021/cn300138m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moreno JL, Gonzalez-Maeso J. Preclinical models of antipsychotic drug action. Int J Neuropsychopharmacol. 2013;16:2131–2144. doi: 10.1017/S1461145713000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schmid Y, Enzler F, Gasser P, Grouzmann E, Preller KH, Vollenweider FX, Brenneisen R, Muller F, Borgwardt S, Liechti ME. Acute Effects of Lysergic Acid Diethylamide in Healthy Subjects. Biol Psychiatry. 2014 doi: 10.1016/j.biop-sych.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 63.Vollenweider FX, Vollenweider-Scherpenhuyzen MF, Babler A, Vogel H, Hell D. Psilocybin induces schizophrenia-like psychosis in humans via a serotonin-2 agonist action. Neuroreport. 1998;9:3897–3902. doi: 10.1097/00001756-199812010-00024. [DOI] [PubMed] [Google Scholar]
- 64.Kometer M, Schmidt A, Jancke L, Vollenweider FX. Activation of serotonin 2A receptors underlies the psilocybin-induced effects on alpha oscillations, N170 visual-evoked potentials, and visual hallucinations. J Neurosci. 2013;33:10544–10551. doi: 10.1523/JNEUROSCI.3007-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Colpaert FC. Discovering risperidone: The LSD model of psychopathology. Nat Rev Drug Discovery. 2003;2:315–320. doi: 10.1038/nrd1062. [DOI] [PubMed] [Google Scholar]
- 66.Carlsson A, Waters N, Holm-Waters S, Tedroff J, Nilsson M, Carlsson ML. Interactions between monoamines, glutamate, and GABA in schizophrenia: New evidence. Annu Rev Pharmacol Toxicol. 2001;41:237–260. doi: 10.1146/annurev.pharmtox.41.1.237. [DOI] [PubMed] [Google Scholar]
- 67.Aghajanian GK, Marek GJ. Serotonin model of schizophrenia: Emerging role of glutamate mechanisms. Brain Res Rev. 2000;31:302–312. doi: 10.1016/s0165-0173(99)00046-6. [DOI] [PubMed] [Google Scholar]
- 68.Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281:1349–1352. doi: 10.1126/science.281.5381.1349. [DOI] [PubMed] [Google Scholar]
- 69.Marek GJ, Wright RA, Schoepp DD, Monn JA, Aghajanian GK. Physiological antagonism between 5-hydroxytryptamine(2A) and group II metabotropic glutamate receptors in prefrontal cortex. J Pharmacol Exp Ther. 2000;292:76–87. [PubMed] [Google Scholar]
- 70.Gewirtz JC, Marek GJ. Behavioral evidence for interactions between a hallucinogenic drug and group II metabotropic glutamate receptors. Neuropsychopharmacology. 2000;23:569–576. doi: 10.1016/S0893-133X(00)00136-6. [DOI] [PubMed] [Google Scholar]
- 71.Moreno JL, Sealfon SC, Gonzalez-Maeso J. Group II metabotropic glutamate receptors and schizophrenia. Cell Mol Life Sci. 2009;66:3777–3785. doi: 10.1007/s00018-009-0130-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fell MJ, Svensson KA, Johnson BG, Schoepp DD. Evidence for the role of metabotropic glutamate (mGlu)2 not mGlu3 receptors in the preclinical antipsychotic pharmacology of the mGlu2/3 receptor agonist (−)-(1R,4S,5S,6S)-4-amino-2-sulfonylbicyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY404039) J Pharmacol Exp Ther. 2008;326:209–217. doi: 10.1124/jpet.108.136861. [DOI] [PubMed] [Google Scholar]
- 73.Spooren WP, Gasparini F, van der Putten H, Koller M, Nakanishi S, Kuhn R. Lack of effect of LY314582 (a group 2 metabotropic glutamate receptor agonist) on phencyclidine-induced locomotor activity in metabotropic glutamate receptor 2 knockout mice. Eur J Pharmacol. 2000;397:R1–2. doi: 10.1016/s0014-2999(00)00269-7. [DOI] [PubMed] [Google Scholar]
- 74.Woolley ML, Pemberton DJ, Bate S, Corti C, Jones DN. The mGlu2 but not the mGlu3 receptor mediates the actions of the mGluR2/3 agonist, LY379268, in mouse models predictive of antipsychotic activity. Psychopharmacology (Berlin) 2008;196:431–440. doi: 10.1007/s00213-007-0974-x. [DOI] [PubMed] [Google Scholar]
- 75.Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, Avedisova AS, Bardenstein LM, Gurovich IY, Morozova MA, Mosolov SN, Neznanov NG, Reznik AM, Smulevich AB, Tochilov VA, Johnson BG, Monn JA, Schoepp DD. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- 76.Adams DH, Kinon BJ, Baygani S, Millen BA, Velona I, Kollack-Walker S, Walling DP. A long-term, phase 2, multicenter, randomized, open-label, comparative safety study of pomaglumetad methionil (LY2140023 monohydrate) versus atypical antipsychotic standard of care in patients with schizophrenia. BMC Psychiatry. 2013;13:143. doi: 10.1186/1471-244X-13-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kinon BJ, Zhang L, Millen BA, Osuntokun OO, Williams JE, Kollack-Walker S, Jackson K, Kryzhanovskaya L, Jarkova N. A multicenter, inpatient, phase 2, double-blind, placebo-controlled dose-ranging study of LY2140023 monohydrate in patients with DSM-IV schizophrenia. J Clin Psychopharmacol. 2011;31:349–355. doi: 10.1097/JCP.0b013e318218dcd5. [DOI] [PubMed] [Google Scholar]
- 78.Stauffer VL, Millen BA, Andersen S, Kinon BJ, Lagrandeur L, Lindenmayer JP, Gomez JC. Pomaglumetad methionil: No significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophr Res. 2013;150:434–441. doi: 10.1016/j.schres.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 79.Adams DH, Zhang L, Millen BA, Kinon BJ, Gomez JC. Pomaglumetad Methionil (LY2140023 Monohydrate) and Aripiprazole in Patients with Schizophrenia: A Phase 3, Multicenter, Double-Blind Comparison. Schizophr Res Treat. 2014;2014:758212. doi: 10.1155/2014/758212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Agid O, Kapur S, Arenovich T, Zipursky RB. Delayed-onset hypothesis of antipsychotic action: A hypothesis tested and rejected. Arch Gen Psychiatry. 2003;60:1228–1235. doi: 10.1001/archpsyc.60.12.1228. [DOI] [PubMed] [Google Scholar]
- 81.Harrow M, Jobe TH, Faull RN. Does treatment of schizophrenia with antipsychotic medications eliminate or reduce psychosis? A 20-year multi-follow-up study. Psychol Med. 2014;44:3007–3016. doi: 10.1017/S0033291714000610. [DOI] [PubMed] [Google Scholar]
- 82.Moreno JL, Holloway T, Umali A, Rayannavar V, Sealfon SC, Gonzalez-Maeso J. Persistent effects of chronic clozapine on the cellular and behavioral responses to LSD in mice. Psychopharmacology (Berlin) 2013;225:217–226. doi: 10.1007/s00213-012-2809-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kurita M, Holloway T, Garcia-Bea A, Kozlenkov A, Friedman AK, Moreno JL, Heshmati M, Golden SA, Kennedy PJ, Takahashi N, Dietz DM, Mocci G, Gabilondo AM, Hanks J, Umali A, Callado LF, Gallitano AL, Neve RL, Shen L, Buxbaum JD, Han MH, Nestler EJ, Meana JJ, Russo SJ, Gonzalez-Maeso J. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat Neurosci. 2012;15:1245–1254. doi: 10.1038/nn.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gonzalez-Maeso J, Ang RL, Yuen T, Chan P, Weisstaub NV, Lopez-Gimenez JF, Zhou M, Okawa Y, Callado LF, Milligan G, Gingrich JA, Filizola M, Meana JJ, Sealfon SC. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature. 2008;452:93–97. doi: 10.1038/nature06612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Muguruza C, Moreno JL, Umali A, Callado LF, Meana JJ, Gonzalez-Maeso J. Dysregulated 5-HT(2A) receptor binding in postmortem frontal cortex of schizophrenic subjects. Eur Neuropsychopharmacol. 2013;23:852–864. doi: 10.1016/j.euroneuro.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gonzalez-Maeso J, Sealfon SC. Psychedelics and schizophrenia. Trends Neurosci. 2009;32:225–232. doi: 10.1016/j.tins.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 87.Dean B. Interpreting the significance of decreased cortical serotonin 2A receptors in schizophrenia. Prog Neuro-Psychopharmacol Biol Psychiatry. 2009;33:1583–1584. doi: 10.1016/j.pnpbp.2009.08.006. Author reply: Deng, C., Kang, K., Wang, Q, and Huang, X.-F. (2009) Serotonin 2A receptor and its association with the pathology of schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 33, 1585–1586. [DOI] [PubMed] [Google Scholar]
- 88.Kurita M, Moreno JL, Holloway T, Kozlenkov A, Mocci G, Garcia-Bea A, Hanks JB, Neve R, Nestler EJ, Russo SJ, Gonzalez-Maeso J. Repressive Epigenetic Changes at the mGlu2 Promoter in Frontal Cortex of 5-HT2A Knockout Mice. Mol Pharmacol. 2013;83:1166–1175. doi: 10.1124/mol.112.084582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kurita M, Holloway T, Gonzalez-Maeso J. HDAC2 as a new target to improve schizophrenia treatment. Expert Rev Neurother. 2013;13:1–3. doi: 10.1586/ern.12.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stauffer VL. The development of pomaglumetad methionil as a glutamate-based pharmacotherapy for schizophrenia: Lessons learned. Curr Neuropharmacol. 2014;12(Suppl 1):57. [Google Scholar]
- 91.Liu W, Downing AC, Munsie LM, Chen P, Reed MR, Ruble CL, Landschulz KT, Kinon BJ, Nisenbaum LK. Pharmacogenetic analysis of the mGlu2/3 agonist LY2140023 monohydrate in the treatment of schizophrenia. Pharmacogenomics J. 2010;12:246–254. doi: 10.1038/tpj.2010.90. [DOI] [PubMed] [Google Scholar]
- 92.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, Carrera N, Humphreys I, Johnson JS, Roussos P, Barker DD, Banks E, Milanova V, Grant SG, Hannon E, Rose SA, Chambert K, Mahajan M, Scolnick EM, Moran JL, Kirov G, Palotie A, McCarroll SA, Holmans P, Sklar P, Owen MJ, Purcell SM, O’Donovan MC. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O’Dushlaine C, Chambert K, Bergen SE, Kahler A, Duncan L, Stahl E, Genovese G, Fernandez E, Collins MO, Komiyama NH, Choudhary JS, Magnusson PK, Banks E, Shakir K, Garimella K, Fennell T, DePristo M, Grant SG, Haggarty SJ, Gabriel S, Scolnick EM, Lander ES, Hultman CM, Sullivan PF, McCarroll SA, Sklar P. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cardno AG, Gottesman II. Twin studies of schizophrenia: From bow-and-arrow concordances to star wars Mx and functional genomics. Am J Med Genet. 2000;97:12–17. [PubMed] [Google Scholar]
- 96.Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci. 2002;25:409–432. doi: 10.1146/annurev.neuro.25.112701.142754. [DOI] [PubMed] [Google Scholar]
- 97.Gottesman II. Schizophrenia Genesis: The Origins of Madness. W.H. Freeman and Company; New York: 1991. [Google Scholar]
- 98.McGuffin P, Asherson P, Owen M, Farmer A. The strength of the genetic effect. Is there room for an environmental influence in the aetiology of schizophrenia? Br J Psychiatry. 1994;164:593–599. doi: 10.1192/bjp.164.5.593. [DOI] [PubMed] [Google Scholar]
- 99.Menninger KA. Psychoses associated with influenza. J Am Med Assoc. 1919;72:235–241. [Google Scholar]
- 100.Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, Babulas VP, Susser ES. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch Gen Psychiatry. 2004;61:774–780. doi: 10.1001/archpsyc.61.8.774. [DOI] [PubMed] [Google Scholar]
- 101.Brown AS, Cohen P, Harkavy-Friedman J, Babulas V, Malaspina D, Gorman JM, Susser ES. A.E. Bennett Research Award. Prenatal rubella, premorbid abnormalities, and adult schizophrenia. Biol Psychiatry. 2001;49:473–486. doi: 10.1016/s0006-3223(01)01068-x. [DOI] [PubMed] [Google Scholar]
- 102.Sorensen HJ, Mortensen EL, Reinisch JM, Mednick SA. Association between prenatal exposure to bacterial infection and risk of schizophrenia. Schizophr Bull. 2009;35:631–637. doi: 10.1093/schbul/sbn121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brown AS, Schaefer CA, Quesenberry CP, Jr, Liu L, Babulas VP, Susser ES. Maternal exposure to toxoplasmosis and risk of schizophrenia in adult offspring. Am J Psychiatry. 2005;162:767–773. doi: 10.1176/appi.ajp.162.4.767. [DOI] [PubMed] [Google Scholar]
- 104.Khashan AS, Abel KM, McNamee R, Pedersen MG, Webb RT, Baker PN, Kenny LC, Mortensen PB. Higher risk of offspring schizophrenia following antenatal maternal exposure to severe adverse life events. Arch Gen Psychiatry. 2008;65:146–152. doi: 10.1001/archgenpsychiatry.2007.20. [DOI] [PubMed] [Google Scholar]
- 105.Malaspina D, Corcoran C, Kleinhaus KR, Perrin MC, Fennig S, Nahon D, Friedlander Y, Harlap S. Acute maternal stress in pregnancy and schizophrenia in offspring: A cohort prospective study. BMC Psychiatry. 2008;8:71. doi: 10.1186/1471-244X-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.van Os J, Selten JP. Prenatal exposure to maternal stress and subsequent schizophrenia. The May 1940 invasion of The Netherlands. Br J Psychiatry. 1998;172:324–326. doi: 10.1192/bjp.172.4.324. [DOI] [PubMed] [Google Scholar]
- 107.Gurevich EV, Joyce JN. Alterations in the cortical serotonergic system in schizophrenia: a postmortem study. Biol Psychiatry. 1997;42:529–545. doi: 10.1016/S0006-3223(97)00321-1. [DOI] [PubMed] [Google Scholar]
- 108.Rasmussen H, Erritzoe D, Andersen R, Ebdrup BH, Aggernaes B, Oranje B, Kalbitzer J, Madsen J, Pinborg LH, Baare W, Svarer C, Lublin H, Knudsen GM, Glenthoj B. Decreased frontal serotonin2A receptor binding in anti-psychotic-naive patients with first-episode schizophrenia. Arch Gen Psychiatry. 2010;67:9–16. doi: 10.1001/archgenpsychiatry.2009.176. [DOI] [PubMed] [Google Scholar]
- 109.Erritzoe D, Rasmussen H, Kristiansen KT, Frokjaer VG, Haugbol S, Pinborg L, Baare W, Svarer C, Madsen J, Lublin H, Knudsen GM, Glenthoj BY. Cortical and Subcortical 5-HT(2A) Receptor Binding in Neuroleptic-Naive First-Episode Schizophrenic Patients. Neuropsychopharmacology. 2008;33:2435–2441. doi: 10.1038/sj.npp.1301656. [DOI] [PubMed] [Google Scholar]
- 110.Meyer U, Feldon J, Fatemi SH. In-vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci Biobehav Rev. 2009;33:1061–1079. doi: 10.1016/j.neubiorev.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 111.Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moreno JL, Kurita M, Holloway T, Lopez J, Cadagan R, Martinez-Sobrido L, Garcia-Sastre A, Gonzalez-Maeso J. Maternal Influenza Viral Infection Causes Schizophrenia-Like Alterations of 5-HT2A and mGlu2 Receptors in the Adult Offspring. J Neurosci. 2011;31:1863–1872. doi: 10.1523/JNEUROSCI.4230-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Holloway T, Moreno JL, Umali A, Rayannavar V, Hodes GE, Russo SJ, Gonzalez-Maeso J. Prenatal Stress Induces Schizophrenia-Like Alterations of Serotonin 2A and Metabotropic Glutamate 2 Receptors in the Adult Offspring: Role of Maternal Immune System. J Neurosci. 2013;33:1088–1098. doi: 10.1523/JNEUROSCI.2331-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Malkova NV, Gallagher JJ, Yu CZ, Jacobs RE, Patterson PH. Manganese-enhanced magnetic resonance imaging reveals increased DOI-induced brain activity in a mouse model of schizophrenia. Proc Natl Acad Sci U S A. 2014;111:E2492–2500. doi: 10.1073/pnas.1323287111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Klein AB, Ultved L, Adamsen D, Santini MA, Tobena A, Fernandez-Teruel A, Flores P, Moreno M, Cardona D, Knudsen GM, Aznar S, Mikkelsen JD. 5-HT(2A) and mGlu2 receptor binding levels are related to differences in impulsive behavior in the Roman Low- (RLA) and High- (RHA) avoidance rat strains. Neuroscience. 2014;263:36–45. doi: 10.1016/j.neuroscience.2013.12.063. [DOI] [PubMed] [Google Scholar]
- 116.Jorgensen CV, Jacobsen JP, Caron MG, Klein AB, Knudsen GM, Mikkelsen JD. Cerebral 5-HT2A receptor binding, but not mGluR2, is increased in tryptophan hydroxylase 2 decrease-of-function mice. Neurosci Lett. 2013;555:118–122. doi: 10.1016/j.neulet.2013.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chiu HY, Chan MH, Lee MY, Chen ST, Zhan ZY, Chen HH. Long-lasting alterations in 5-HT2A receptor after a binge regimen of methamphetamine in mice. Int J Neuropsychopharmacol. 2014:1–12. doi: 10.1017/S1461145714000455. [DOI] [PubMed] [Google Scholar]
- 118.Paquette AG, Lesseur C, Armstrong DA, Koestler DC, Appleton AA, Lester BM, Marsit CJ. Placental HTR2A methylation is associated with infant neurobehavioral outcomes. Epigenetics. 2013;8:796–801. doi: 10.4161/epi.25358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Van den Hove DL, Lauder JM, Scheepens A, Prickaerts J, Blanco CE, Steinbusch HW. Prenatal stress in the rat alters 5-HT1A receptor binding in the ventral hippocampus. Brain Res. 2006;1090:29–34. doi: 10.1016/j.brainres.2006.03.057. [DOI] [PubMed] [Google Scholar]
- 120.Carrard A, Salzmann A, Malafosse A, Karege F. Increased DNA methylation status of the serotonin receptor 5HTR1A gene promoter in schizophrenia and bipolar disorder. J Affective Disord. 2011;132:450–453. doi: 10.1016/j.jad.2011.03.018. [DOI] [PubMed] [Google Scholar]
- 121.van den Hove DL, Jakob SB, Schraut KG, Kenis G, Schmitt AG, Kneitz S, Scholz CJ, Wiescholleck V, Ortega G, Prickaerts J, Steinbusch H, Lesch KP. Differential effects of prenatal stress in 5-Htt deficient mice: towards molecular mechanisms of gene x environment interactions. PloS ONE. 2011;6:e22715. doi: 10.1371/journal.pone.0022715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Huang HS, Akbarian S. GAD1 mRNA expression and DNA methylation in prefrontal cortex of subjects with schizophrenia. PloS ONE. 2007;2:e809. doi: 10.1371/journal.pone.0000809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 124.Weaver IC, Meaney MJ, Szyf M. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc Natl Acad Sci U S A. 2006;103:3480–3485. doi: 10.1073/pnas.0507526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonte B, Szyf M, Turecki G, Meaney MJ. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12:342–348. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang TY, Hellstrom IC, Bagot RC, Wen X, Diorio J, Meaney MJ. Maternal care and DNA methylation of a glutamic acid decarboxylase 1 promoter in rat hippocampus. J Neurosci. 2010;30:13130–13137. doi: 10.1523/JNEUROSCI.1039-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]