

ABSTRACT
Cerebral Cavernous Malformation (CCM) is a major cerebrovascular disease of proven genetic origin affecting 0.3–0.5% of the general population. It is characterized by abnormally enlarged and leaky capillaries, which predispose to seizures, focal neurological deficits and intracerebral hemorrhage. Causative loss-of-function mutations have been identified in 3 genes, KRIT1 (CCM1), CCM2 and PDCD10 (CCM3). While providing new options for the development of pharmacological therapies, recent advances in knowledge of the functions of these genes have clearly indicated that they exert pleiotropic effects on several biological pathways.
Recently, we found that defective autophagy is a common feature of loss-of-function mutations of the 3 known CCM genes, and underlies major phenotypic signatures of CCM disease, including endothelial-to-mesenchymal transition and enhanced ROS production, suggesting a unifying pathogenetic mechanism and reconciling the distinct therapeutic approaches proposed so far.
In this invited review, we discuss autophagy as a possible unifying mechanism in CCM disease pathogenesis, and new perspectives and avenues of research for disease prevention and treatment, including novel potential drug repurposing and combination strategies, and identification of genetic risk factors as basis for development of personalized medicine approaches.
KEYWORDS: Autophagy, CCM genes, cerebral cavernous malformation (CCM), cerebrovascular diseases, endothelial-to-mesenchymal transition (EndMT), intracerebral hemorrhage (ICH), oxidative stress, reactive oxygen species (ROS)
Introduction
Cerebral cavernous malformation (CCM), also known as cavernous angioma or cavernoma, is a major vascular dysplasia, occurring mainly within the central nervous system and affecting 0.3–0.5% of the human population.1-3 CCM lesions consist of closely clustered, abnormally dilated and leaky capillaries, which can be single or multiple (up to hundreds) and may remain clinically silent or result in clinical symptoms of various type and severity at any age, including recurrent headaches, focal neurological deficits, seizures, stroke and intracerebral hemorrhage (ICH). Generally, only approximately 30% of people with CCM lesions eventually will develop clinical symptoms.1-3
This cerebrovascular disease is of proven genetic origin (OMIM 116860), arising sporadically or being inherited as autosomal dominant condition with incomplete penetrance and highly variable expressivity even among members of the same family, including wide differences in lesion number, size and susceptibility to ICH, suggesting that multiple factors can contribute to CCM disease pathogenesis.4 Genetic studies have so far identified causative mutations in 3 disease genes, KRIT1 (Krev interaction trapped 1, also known as CCM1), CCM2 and PDCD10 (programmed cell death 10, also known as CCM3), which account for about 50%, 20% and 10% of the CCM cases, respectively. The remaining 20% of cases have been attributed to mutations of a fourth as yet unidentified CCM gene.5 Notably, the hereditary form of the illness is often associated with multiple cavernous angiomas, whereas the sporadic form typically presents as a solitary lesion.
Despite significant progress and breakthroughs in the understanding of CCM disease pathogenesis over the last decade, with the potential for greatly advancing the development of therapeutic strategies for prevention and treatment, no direct therapeutic approaches for CCM disease exist so far, besides surgical removal of accessible lesions in patients with recurrent hemorrhage or intractable seizures. In particular, novel pharmacological strategies are required for preventing the most severe disease phenotype in susceptible individuals, including the development of numerous and large symptomatic lesions and ICH.
Comprehensive analysis of the 3 known CCM genes in mutation carriers has suggested that their functions need to be severely impaired for pathogenesis,5 whereas several studies in cellular and animal models have revealed a major role for these genes in the maintenance of endothelial cell-cell junction stability and blood-brain barrier (BBB) integrity.6-11 Nevertheless, endothelium-specific conditional knockout of CCM genes in mice resulted in a spatially and temporally restricted development of CCM lesions, indicating that loss of CCM genes is not sufficient to cause the disease, and suggesting that additional triggers occurring locally at the blood-brain interface, including microenvironmental stress factors, crucially contribute to CCM disease pathogenesis.4
In recent years, it has become clear that CCM genes play an important role in controlling signaling pathways involved in cell responses to oxidative stress, pointing to a novel pathogenic mechanism whereby the function of these genes may be relevant in preventing vascular dysfunctions triggered by oxidative stress events.4,12-14 In particular, original findings demonstrated that KRIT1 is involved in the maintenance of intracellular ROS homeostasis through the modulation of master regulators of cellular responses to oxidative stress, including FoxO1 and SOD2, which prevent accumulation of mitochondrial-derived superoxide anions, whereas KRIT1 loss-of-function is associated with ROS production and increased cell susceptibility to oxidative stress-mediated molecular and cellular dysfunctions.13 Moreover, subsequent findings showed that KRIT1 may exert a protective role against oxidative stress by limiting pro-oxidant and pro-inflammatory pathways and mechanisms, including JNK/c-Jun-dependent redox pathways.14 Accordingly, recent evidence in animal models has suggested that oxidative stress may play an even more critical role in CCM disease than previously described due to systemic effects.12 Furthermore, there is also evidence that CCM disease phenotypes can be reversed by ROS scavenging with antioxidant compounds.12,14,15
While these and other great advances in knowledge of the biological functions of CCM proteins have led to an explosion of disease-relevant molecular information,4,16 they have also clearly indicated that loss-of-function of CCM proteins has potentially pleiotropic effects on several biological pathways, thus bringing new research challenges.
Defective autophagy is a key feature of cerebral cavernous malformations
Autophagy is a form of quality control inside the cell consisting in the removal of protein aggregates and excess or damaged organelles,17 including dysfunctional ROS-generating mitochondria, through their encapsulation by a double-membrane structure known as the autophagosome.18-20
Recently, using integrated research approaches involving the CCM_Italia multidisciplinary research network, we discovered a causal relationship between impaired autophagy and key phenotypic signatures of CCM disease.21 Specifically, using both cellular and animal models of CCM disease and surgical samples of human CCM lesions, we found that defective autophagy is a common feature of loss-of-function mutations of the 3 known CCM genes, and underlies major phenotypic signatures of CCM disease, including endothelial-to-mesenchymal transition (EndMT) and enhanced ROS production, suggesting a major role in CCM pathogenesis. Moreover, we demonstrated that defective autophagy caused by down-regulation of CCM genes is linked to the up-regulation of the mTOR (mammalian Target Of Rapamycin) kinase and mTOR-ULK1 regulatory pathway, and showed that pharmacological inhibition of mTOR and consequent activation of autophagy rescued major molecular and cellular disease phenotypes, including ROS accumulation and EndMT, suggesting novel mechanistic targets for therapeutic intervention.21
Taken together, these data point to a pivotal role for defective autophagy in CCM disease pathogenesis, and suggest a common mechanism for the efficacy of various potential therapeutic compounds proposed so far, thus providing a novel framework for the development of new pharmacological strategies to prevent or alleviate adverse clinical outcomes of CCM lesions.
From pleiotropic effects toward unifying mechanisms
So far, multiple molecules and molecular mechanisms have been involved in CCM disease pathogenesis, which reflects the multiple functions attributed to the 3 known CCM proteins over the last decade. Indeed, besides their role in the modulation of redox-sensitive pathways and mechanisms described above, CCM proteins have been demonstrated to cooperate in promoting the formation and maintenance of VE-cadherin–based adherens junctions between endothelial cells.16 In particular, KRIT1/CCM1 has been demonstrated to act as a Rap1 effector that regulates VE-cadherin-mediated endothelial cell-cell junctions,7 β-catenin signaling,22 and endothelial polarity.23 Moreover, there is compelling evidence that CCM proteins inhibit the RhoA GTPase pathway to maintain vascular integrity and BBB stability, while their loss-of-function promotes the activation of the RhoA GTPase and its effector ROCK, which increase cellular contractility and destabilize endothelial cell-cell junctions, thereby decreasing barrier function and increasing vascular permeability.9-11 Furthermore, loss-of-function of CCM proteins has been shown to cause the activation of the TGF-β/BMP and β-catenin pathways, which underlies the induction of EndMT associated with the onset and progression of CCM disease.8,24
In addition, there is also evidence that CCM proteins ensure the quiescence of endothelial cells and inhibit angiogenic responses by either limiting accumulation of intracellular ROS and altered redox signaling,13,14,21 activating the Delta-Notch signaling,25-27 inhibiting the vascular endothelial growth factor (VEGF) 28 and MAP kinase signaling,29 or regulating the β1 integrin-Klf2-mediated mechanotransduction pathway.30
On the other hand, CCM proteins may also regulate the integrin-based focal adhesions that connect endothelial cells to the underlying extracellular matrix, and integrin-mediated signaling.30-32 Specifically, there is evidence that CCM proteins limit β1 integrin–dependent endothelial cell adhesion and contractility, and fibronectin remodeling by stabilizing ICAP-1, an inhibitor of β1 integrin,31 and may control endothelial β1 integrin-dependent mechanotransduction in response to shear stress.30,32
CCM proteins therefore act as “nodes” that tune and orchestrate the crosstalk between integrins and cadherins, which coordinately regulates cell–extracellular matrix and cell-cell interactions and actin cytoskeleton dynamics involved in the maintenance of vascular integrity and barrier function, thereby promoting vascular maturation/stabilization and inhibiting vascular permeability. Consistently, the small GTPase Rap1, a major KRIT1/CCM1 molecular interactor,7 has been previously reported to play a pivotal role in the signaling crosstalk between cadherins and integrins,33,34 whereas emerging evidence indicates that ROS and redox signaling, also linked to KRIT1/CCM1 function,13,14 may set the talk.35 Furthermore, it has been also suggested that the induction of EndMT associated with CCM disease8 might be connected to and downstream of deregulation of redox signaling and c-Jun activity.4,14
Taken together, the accumulated evidence indicates that CCM proteins exert pleiotropic effects on numerous biological pathways, suggesting that a unifying mechanism should exist that explains these pleiotropic functions as well as the broad spectrum of phenotypic hallmarks linked to their loss-of-function and underlying CCM disease, including decreased endothelial cell-cell junction stability, altered cell-matrix adhesion and cytoskeleton dynamics, induction of EndMT, increased proliferative and angiogenic potential, disturbed redox signaling and enhanced cell sensitivity to oxidative stress.
Remarkably, most of the reported CCM protein functions and effects are directly or indirectly related to autophagy and its tight interconnection with redox homeostasis and signaling,21,36,37 suggesting that the modulation of autophagy may represent the underlying and unifying mechanism for CCM protein physiopathological functions. Consistently, autophagy is a converging point of multiple physiological and pathological pathways, and may exert pleiotropic effects on several molecular and cellular processes.18 Specifically, autophagy plays a pivotal role in various signaling pathways linked to CCM proteins, including the Sirt1/FoxO1,13,38,39 JNK/c-Jun,14,40 β-catenin,22,41 RhoGTPase/ROCK,42 and TGF-β 8,21 pathways. Furthermore, most phenotypic hallmarks of CCM disease can be linked to autophagic dysfunctions, including enhanced ROS production21,43 and EndMT,21 as well as altered cell adhesion and angiogenic potential,44-46 and enhanced endothelial cell sensitivity to oxidant-induced injury,39 suggesting that the multiple pathogenic mechanisms proposed so far for CCM disease may originate from impaired autophagy and its interplay with redox imbalance and oxidative stress.
On this basis and with new understanding of the interplay between autophagy and redox regulation in cell signaling and adaptive response to oxidative stress,36,37,43,47,48 it is therefore plausible that dysfunction of autophagy might represent a unifying mechanism that explains the broad disregulation of signal transduction induced by loss-of-function of CCM proteins and the consequent multiple effects on CCM disease pathogenesis.
Reconciling therapeutic approaches for CCM disease
To date there are not direct pharmacological therapies for CCM disease. However, the great progress in understanding CCM protein functions and disease mechanisms has opened promising therapeutic opportunities. Indeed, multiple therapeutic approaches have been proposed so far for CCM disease prevention and treatment, with the potentiality to be effective at least in limiting the disease severity, including the following:
Statins (e.g. Simvastatin) and Fasudil, which have been suggested to act by inhibiting Rho GTPase signaling10,49 and Rho kinase (ROCK) activity,9,50 respectively;
Sulindac sulfide and its analogs, which have been shown to inhibit the β-catenin and TGF-β pathways8,24;
Cholechalciferol (vitamin D3), known to exert an antioxidant activity among others,12,51 and distinct compounds with well-established antioxidant properties, such as N-acetylcysteine (NAC), a potent glutathione precursor,13,14 Tempol, a scavenger of superoxide anions,12 and Avenanthramide, a polyphenol from oats,15 which have been demonstrated to counteract disease phenotypes linked to oxidative stress, including actin stress fiber formation, adherens junction weakening, and endothelial barrier dysfunction;
mTOR inhibitors, such as Rapamycin (also known as Sirolimus) and Torin1, which act as inducers of autophagy.21
Intriguingly, all of the different compounds proposed as non-invasive drug treatment approaches for CCM disease may exert potential pleiotropic effects, acting as either antioxidants or autophagy inducers or both. Indeed, whereas there is evidence that the Rho GTPase pathway can be directly activated by ROS,52 both statins and fasudil are known to exert powerful antioxidant activities in endothelial cells, including the inhibition of superoxide production and the improvement of both ROS scavenging and NO bioavailability.53-55 Moreover, it has been demonstrated that both sulindac sulfone and sulindac sulfide display a powerful ROS scavenging activity, whose potency resulted even higher than that of endogenous antioxidants such as reduced glutathione (GSH).56 Furthermore, it is well established that autophagy inducers limit ROS accumulation and oxidative stress by stimulating the autophagic degradation of ROS-generating mitochondria.57
On the other hand, there is compelling evidence that most, if not all, of the potential therapeutic compounds listed above have been also shown to trigger autophagy by inhibiting the mTOR pathway, including statins,58,59 fasudil,60 sulindac derivatives,61,62 vitamin D3,63-65 and established antioxidant compounds, such as resveratrol.66
Taken together with these observations, our recent finding that defective autophagy plays a pivotal role in CCM disease pathogenesis 21 points to autophagy as a major unifying mechanism that accommodates the different molecular pathways and potential therapeutic compounds described so far, thus providing a novel framework for the development of new pharmacological strategies based on repurposed drugs and combined therapies for more effective prevention and treatment of the most severe forms of CCM disease.
Conclusions and future perspectives
Intense investigation over the past two decades has led to the identification and characterization of 3 genes associated with CCM disease, showing that their loss-of-function mutations affect multiple important molecular mechanisms and signaling pathways, leading to a variety of disease phenotypes, suggesting potentially pleiotropic effects. It was then legitimate to ask the question as to whether, despite the many kinds of molecules and signaling pathways involved, there may be at least some unifying mechanisms underlying CCM disease pathogenesis. Taken together, our previous and recent findings confirmed that indeed a unifying mechanism might exist, consisting in the interplay between defective autophagy and redox imbalance, which might be integral to the development and progression of CCM lesions by sensitizing endothelial cells to local oxidative stress events (Fig. 1). Indeed, whereas our previously accumulated data pointed to a major role for altered redox signaling and enhanced cell sensitivity to oxidative stress in CCM disease pathogenesis,4,12,13,35 our recent findings demonstrated that defective autophagy is also involved and plays a pivotal role,21 suggesting that a combined therapy approach based on both antioxidants and autophagy inducers might represent a novel option for the pharmacological treatment of CCM disease.
Figure 1.
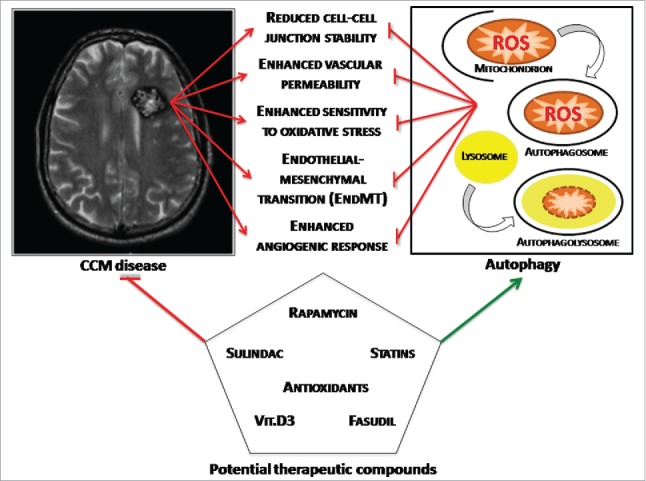
Toward a unifying mechanism for CCM disease pathogenesis. The interplay between defective autophagy and redox imbalance underlies development and progression of CCM lesions by sensitizing endothelial cells to local oxidative stress events, representing a major unifying mechanism that accommodates the different molecular pathways and potential therapeutic compounds described so far.
Further studies aimed at better characterizing the fine-tuned interplay between autophagy and oxidative stress in CCM disease pathogenesis might provide additional therapeutic options for preventing and reversing adverse clinical outcomes of CCM lesions.
In addition, the identification of putative genetic susceptibility factors that might influence the clinical severity of CCM disease, including single nucleotide polymorphisms (SNPs) in genes linked to autophagy modulation and interindividual variability in susceptibility to oxidative stress,4,67 should provide useful insights into the development of novel targeted strategies for personalized medicine approaches tailored to high-risk individuals.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors wish to gratefully acknowledge the Italian Research Network for Cerebral Cavernous Malformation (CCM Italia, http://www.ccmitalia.unito.it) and the Associazione Italiana Angiomi Cavernosi (AIAC, http://www.ccmitalia.unito.it/aiac) for supporting the study with clinical information and biological materials. The authors kindly thank Santina Barbaro for helpful discussion.
Funding
This work was supported by grants from Telethon to Saverio Francesco Retta (GGP15219/Coordinator) and Paolo Pinton (GGP15219/B). Eliana Trapani is supported by a PhD Fellowship from the Telethon Foundation. Luca Goitre is supported by a PostDoc Fellowship from the University of Torino.
References
- [1].Batra S, Lin D, Recinos PF, Zhang J, Rigamonti D. Cavernous malformations: natural history, diagnosis and treatment. Nat Rev Neurol 2009; 5:659-70; PMID:19953116; http://dx.doi.org/ 10.1038/nrneurol.2009.177 [DOI] [PubMed] [Google Scholar]
- [2].Fontanella M. Cerebral Cavernous Malformations, ed. Minerva Medica 2015; pp. 1-140. ISBN-13 978-88-7711-842-4. http://www.minervamedica.it/it/volumi/specialita-mediche/neurochirurgia/scheda.php?cod=L10023 [Google Scholar]
- [3].Rigamonti D. Cavernous Malformations of the nervous system. Cambridge University Press, 2011; http://dx.doi.org/ 10.1017/CBO9781139003636 [DOI] [Google Scholar]
- [4].Trapani E, Retta SF. Cerebral cavernous malformation (CCM) disease: from monogenic forms to genetic susceptibility factors. J Neurosurg Sci 2015; 59:201-9; PMID:25896717 [PubMed] [Google Scholar]
- [5].Riant F, Bergametti F, Ayrignac X, Boulday G, Tournier-Lasserve E. Recent insights into cerebral cavernous malformations: the molecular genetics of CCM. Febs Journal 2010; 277:1070-5; PMID:20096038; http://dx.doi.org/ 10.1111/j.1742-4658.2009.07535.x [DOI] [PubMed] [Google Scholar]
- [6].Corr M, Lerman I, Keubel JM, Ronacher L, Misra R, Lund F, Sarelius IH, Glading AJ. Decreased Krev Interaction-Trapped 1 Expression Leads to Increased Vascular Permeability and Modifies Inflammatory Responses In Vivo. Arterioscl Throm Vas 2012; 32:2702-+; http://dx.doi.org/ 10.1161/ATVBAHA.112.300115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Glading A, Han J, Stockton RA, Ginsberg MH. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell-cell junctions. J Cell Biol 2007; 179:247-54; PMID:17954608; http://dx.doi.org/ 10.1083/jcb.200705175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M, Ferrarini L, Orsenigo F, Papa E, Boulday G, et al.. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 2013; 498:492-+; PMID:23748444; http://dx.doi.org/ 10.1038/nature12207 [DOI] [PubMed] [Google Scholar]
- [9].Stockton RA, Shenkar R, Awad IA, Ginsberg MH. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J Exp Med 2010; 207:881-96; PMID:20308363; http://dx.doi.org/ 10.1084/jem.20091258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Whitehead KJ, Chan AC, Navankasattusas S, Koh W, London NR, Ling J, Mayo AH, Drakos SG, Jones CA, Zhu W, et al.. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat Med 2009; 15:177-84; PMID:19151728; http://dx.doi.org/ 10.1038/nm.1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zheng XJ, Xu C, Di Lorenzo A, Kleaveland B, Zou ZY, Seiler C, Chen M, Cheng L, Xiao JP, He J, et al.. CCM3 signaling through sterile 20-like kinases plays an essential role during zebrafish cardiovascular development and cerebral cavernous malformations. Journal of Clinical Investigation 2010; 120:2795-804; PMID:20592472; http://dx.doi.org/ 10.1172/JCI39679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gibson CC, Zhu W, Davis CT, Bowman-Kirigin JA, Chan AC, Ling J, Walker AE, Goitre L, Delle Monache S, Retta SF, et al.. Strategy for identifying repurposed drugs for the treatment of cerebral cavernous malformation. Circulation 2015; 131:289-99; PMID:25486933; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.114.010403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Goitre L, Balzac F, Degani S, Degan P, Marchi S, Pinton P, Retta SF. KRIT1 regulates the homeostasis of intracellular reactive oxygen species. PLoS One 2010; 5:e11786; PMID:20668652; http://dx.doi.org/ 10.1371/journal.pone.0011786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Goitre L, De Luca E, Braggion S, Trapani E, Guglielmotto M, Biasi F, Forni M, Moglia A, Trabalzini L, Retta SF. KRIT1 loss of function causes a ROS-dependent upregulation of c-Jun. Free Radic Biol Med 2014; 68:134-47; PMID:24291398; http://dx.doi.org/ 10.1016/j.freeradbiomed.2013.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Moglia A, Goitre L, Gianoglio S, Baldini E, Trapani E, Genre A, Scattina A, Dondo G, Trabalzini L, Beekwilder J, Retta SF. Evaluation of the bioactive properties of avenanthramide analogs produced in recombinant yeast. Biofactors, 2015; 41(1):15-27. PMID:2563935124481819 [DOI] [PubMed] [Google Scholar]
- [16].Draheim KM, Fisher OS, Boggon TJ, Calderwood DA. Cerebral cavernous malformation proteins at a glance. J Cell Sci 2014; 127:701-7; PMID:24481819; http://dx.doi.org/ 10.1242/jcs.138388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell 2014; 159:1263-76; PMID:25480292; http://dx.doi.org/ 10.1016/j.cell.2014.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013; 368:651-62; PMID:23406030; http://dx.doi.org/ 10.1056/NEJMra1205406 [DOI] [PubMed] [Google Scholar]
- [19].Sica V, Galluzzi L, Bravo-San Pedro JM, Izzo V, Maiuri MC, Kroemer G. Organelle-Specific Initiation of Autophagy. Mol Cell 2015; 59:522-39; PMID:26295960; http://dx.doi.org/ 10.1016/j.molcel.2015.07.021 [DOI] [PubMed] [Google Scholar]
- [20].Rimessi A, Bonora M, Marchi S, Patergnani S, Marobbio CM, Lasorsa FM, Pinton P. Perturbed mitochondrial Ca2+ signals as causes or consequences of mitophagy induction. Autophagy 2013; 9:1677-86; PMID:24121707; http://dx.doi.org/ 10.4161/auto.24795 [DOI] [PubMed] [Google Scholar]
- [21].Marchi S, Corricelli M, Trapani E, Bravi L, Pittaro A, Delle Monache S, Ferroni L, Patergnani S, Missiroli S, Goitre L, et al.. Defective autophagy is a key feature of cerebral cavernous malformations. EMBO Mol Med 2015; 7(11):1403-17; PMID:26417067; http://dx.doi.org/ 10.15252/emmm.201505316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Glading AJ, Ginsberg MH. Rap1 and its effector KRIT1/CCM1 regulate beta-catenin signaling. Dis Model Mech 2010; 3:73-83; PMID:20007487; http://dx.doi.org/ 10.1242/dmm.003293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lampugnani MG, Orsenigo F, Rudini N, Maddaluno L, Boulday G, Chapon F, Dejana E. CCM1 regulates vascular-lumen organization by inducing endothelial polarity. J Cell Sci 2010; 123:1073-80; PMID:20332120; http://dx.doi.org/ 10.1242/jcs.059329 [DOI] [PubMed] [Google Scholar]
- [24].Bravi L, Rudini N, Cuttano R, Giampietro C, Maddaluno L, Ferrarini L, Adams RH, Corada M, Boulday G, Tournier-Lasserve E, et al.. Sulindac metabolites decrease cerebrovascular malformations in CCM3-knockout mice. P Natl Acad Sci USA 2015; 112:8421-6; http://dx.doi.org/ 10.1073/pnas.1501352112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wustehube J, Bartol A, Liebler SS, Brutsch R, Zhu Y, Felbor U, Sure U, Augustin HG, Fischer A. Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling. Proc Natl Acad Sci U S A 2010; 107:12640-5; PMID:20616044; http://dx.doi.org/ 10.1073/pnas.1000132107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].You C, Sandalcioglu IE, Dammann P, Felbor U, Sure U, Zhu Y. Loss of CCM3 impairs DLL4-Notch signalling: implication in endothelial angiogenesis and in inherited cerebral cavernous malformations. J Cell Mol Med 2013; 17:407-18; PMID:23388056; http://dx.doi.org/ 10.1111/jcmm.12022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Schulz GB, Wieland E, Wustehube-Lausch J, Boulday G, Moll I, Tournier-Lasserve E, Fischer A. Cerebral Cavernous Malformation-1 Protein Controls DLL4-Notch3 Signaling Between the Endothelium and Pericytes. Stroke 2015; 46:1337-43; PMID:25791711; http://dx.doi.org/ 10.1161/STROKEAHA.114.007512 [DOI] [PubMed] [Google Scholar]
- [28].DiStefano PV, Kuebel JM, Sarelius IH, Glading AJ. KRIT1 protein depletion modifies endothelial cell behavior via increased vascular endothelial growth factor (VEGF) signaling. J Biol Chem 2014; 289:33054-65; PMID:25320085; http://dx.doi.org/ 10.1074/jbc.M114.582304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cullere X, Plovie E, Bennett PM, MacRae CA, Mayadas TN. The cerebral cavernous malformation proteins CCM2L and CCM2 prevent the activation of the MAP kinase MEKK3. Proc Natl Acad Sci U S A 2015; 112(46):14284-9; PMID:26540726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Renz M, Otten C, Faurobert E, Rudolph F, Zhu Y, Boulday G, Duchene J, Mickoleit M, Dietrich AC, Ramspacher C, et al.. Regulation of beta1 integrin-Klf2-mediated angiogenesis by CCM proteins. Dev Cell 2015; 32:181-90; PMID:25625207; http://dx.doi.org/ 10.1016/j.devcel.2014.12.016 [DOI] [PubMed] [Google Scholar]
- [31].Faurobert E, Rome C, Lisowska J, Manet-Dupe S, Boulday G, Malbouyres M, Balland M, Bouin AP, Keramidas M, Bouvard D, et al.. CCM1-ICAP-1 complex controls beta1 integrin-dependent endothelial contractility and fibronectin remodeling. J Cell Biol 2013; 202:545-61; PMID:23918940; http://dx.doi.org/ 10.1083/jcb.201303044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Macek Jilkova Z, Lisowska J, Manet S, Verdier C, Deplano V, Geindreau C, Faurobert E, Albiges-Rizo C, Duperray A. CCM proteins control endothelial beta1 integrin dependent response to shear stress. Biol Open 2014; 3:1228-35; PMID:25432514; http://dx.doi.org/ 10.1242/bio.201410132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Balzac F, Avolio M, Degani S, Kaverina I, Torti M, Silengo L, Small JV, Retta SF. E-cadherin endocytosis regulates the activity of Rap1: a traffic light GTPase at the crossroads between cadherin and integrin function. J Cell Sci 2005; 118:4765-83; PMID:16219685; http://dx.doi.org/ 10.1242/jcs.02584 [DOI] [PubMed] [Google Scholar]
- [34].Retta SF, Balzac F, Avolio M. Rap1: a turnabout for the crosstalk between cadherins and integrins. Eur J Cell Biol 2006; 85:283-93; PMID:16546572; http://dx.doi.org/ 10.1016/j.ejcb.2005.09.007 [DOI] [PubMed] [Google Scholar]
- [35].Goitre L, Pergolizzi B, Ferro E, Trabalzini L, Retta SF. Molecular Crosstalk between Integrins and Cadherins: Do Reactive Oxygen Species Set the Talk? J Signal Transduct 2012; 2012:807682; PMID:22203898; http://dx.doi.org/ 10.1155/2012/807682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol 2007; 17:422-7; PMID:17804237; http://dx.doi.org/ 10.1016/j.tcb.2007.07.009 [DOI] [PubMed] [Google Scholar]
- [37].Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death and Differentiation 2015; 22:377-88; PMID:25257172; http://dx.doi.org/ 10.1038/cdd.2014.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhao Y, Yang J, Liao WJ, Liu XY, Zhang H, Wang S, Wang DL, Feng JN, Yu L, Zhu WG. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol 2010; 12:665-U88; PMID:20543840; http://dx.doi.org/ 10.1038/ncb2069 [DOI] [PubMed] [Google Scholar]
- [39].Liu J, Bi X, Chen T, Zhang Q, Wang SX, Chiu JJ, Liu GS, Zhang Y, Bu P, Jiang F. Shear stress regulates endothelial cell autophagy via redox regulation and Sirt1 expression. Cell Death Dis 2015; 6:e1827; PMID:26181207; http://dx.doi.org/ 10.1038/cddis.2015.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yogev O, Shaulian E. Jun proteins inhibit autophagy and induce cell death. Autophagy 2010; 6:566-7; PMID:20404571; http://dx.doi.org/ 10.4161/auto.6.4.11950 [DOI] [PubMed] [Google Scholar]
- [41].Lin R, Feng J, Dong S, Pan R, Zhuang H, Ding Z. Regulation of autophagy of prostate cancer cells by beta-catenin signaling. Cell Physiol Biochem 2015; 35:926-32; PMID:25633614; http://dx.doi.org/ 10.1159/000369749 [DOI] [PubMed] [Google Scholar]
- [42].Mleczak A, Millar S, Tooze SA, Olson MF, Chan EY. Regulation of autophagosome formation by Rho kinase. Cell Signal 2013; 25:1-11; PMID:22975682; http://dx.doi.org/ 10.1016/j.cellsig.2012.09.010 [DOI] [PubMed] [Google Scholar]
- [43].Huang J, Lam GY, Brumell JH. Autophagy Signaling Through Reactive Oxygen Species. Antioxid Redox Sign 2011; 14:2215-31; http://dx.doi.org/ 10.1089/ars.2010.3554 [DOI] [PubMed] [Google Scholar]
- [44].Tuloup-Minguez V, Hamai A, Greffard A, Nicolas V, Codogno P, Botti J. Autophagy modulates cell migration and beta1 integrin membrane recycling. Cell Cycle 2013; 12:3317-28; PMID:24036548; http://dx.doi.org/ 10.4161/cc.26298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kumar S, Guru SK, Pathania AS, Kumar A, Bhushan S, Malik F. Autophagy triggered by magnolol derivative negatively regulates angiogenesis. Cell Death Dis 2013; 4:e889; PMID:24176847; http://dx.doi.org/ 10.1038/cddis.2013.399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kim KW, Paul P, Qiao J, Lee S, Chung DH. Enhanced autophagy blocks angiogenesis via degradation of gastrin-releasing peptide in neuroblastoma cells. Autophagy 2013; 9:1579-90; PMID:24108003; http://dx.doi.org/ 10.4161/auto.25987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 2011; 36:30-8; PMID:20728362; http://dx.doi.org/ 10.1016/j.tibs.2010.07.007 [DOI] [PubMed] [Google Scholar]
- [48].Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J 2012; 441:523-40; PMID:22187934; http://dx.doi.org/ 10.1042/BJ20111451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Li DY, Whitehead KJ. Evaluating Strategies for the Treatment of Cerebral Cavernous Malformations. Stroke 2010; 41:S92-S4; PMID:20876517; http://dx.doi.org/ 10.1161/STROKEAHA.110.594929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].McDonald DA, Shi CB, Shenkar R, Stockton RA, Liu FF, Ginsberg MH, Marchuk DA, Awad IA. Fasudil Decreases Lesion Burden in a Murine Model of Cerebral Cavernous Malformation Disease. Stroke 2012; 43:571-4; PMID:22034008; http://dx.doi.org/ 10.1161/STROKEAHA.111.625467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wiseman H. Vitamin D is a membrane antioxidant. Ability to inhibit iron-dependent lipid peroxidation in liposomes compared to cholesterol, ergosterol and tamoxifen and relevance to anticancer action. FEBS Lett 1993; 326:285-8; PMID:8325381; http://dx.doi.org/ 10.1016/0014-5793(93)81809-E [DOI] [PubMed] [Google Scholar]
- [52].Aghajanian A, Wittchen ES, Campbell SL, Burridge K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. Plos One 2009; 4:e8045; PMID:19956681; http://dx.doi.org/ 10.1371/journal.pone.0008045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Adam O, Laufs U. Antioxidative effects of statins. Arch Toxicol 2008; 82:885-92; PMID:18670762; http://dx.doi.org/ 10.1007/s00204-008-0344-4 [DOI] [PubMed] [Google Scholar]
- [54].Kuhlmann CR, Gerigk M, Bender B, Closhen D, Lessmann V, Luhmann HJ. Fluvastatin prevents glutamate-induced blood-brain-barrier disruption in vitro. Life Sci 2008; 82:1281-7; PMID:18534629; http://dx.doi.org/ 10.1016/j.lfs.2008.04.017 [DOI] [PubMed] [Google Scholar]
- [55].Ma Z, Zhang J, Ji E, Cao G, Li G, Chu L. Rho kinase inhibition by fasudil exerts antioxidant effects in hypercholesterolemic rats. Clin Exp Pharmacol Physiol 2011; 38:688-94; PMID:21711379; http://dx.doi.org/ 10.1111/j.1440-1681.2011.05561.x [DOI] [PubMed] [Google Scholar]
- [56].Costa D, Gomes A, Reis S, Lima JL, Fernandes E. Hydrogen peroxide scavenging activity by non-steroidal anti-inflammatory drugs. Life Sci 2005; 76:2841-8; PMID:15808884; http://dx.doi.org/ 10.1016/j.lfs.2004.10.052 [DOI] [PubMed] [Google Scholar]
- [57].Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico-Petruzzelli M, et al.. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ 2015; 22:58-73; PMID:25236395; http://dx.doi.org/ 10.1038/cdd.2014.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhang J, Yang Z, Xie L, Xu L, Xu D, Liu X. Statins, autophagy and cancer metastasis. Int J Biochem Cell Biol 2013; 45:745-52; PMID:23147595; http://dx.doi.org/ 10.1016/j.biocel.2012.11.001 [DOI] [PubMed] [Google Scholar]
- [59].Wei YM, Li X, Xu M, Abais JM, Chen Y, Riebling CR, Boini KM, Li PL, Zhang Y. Enhancement of Autophagy by Simvastatin through Inhibition of Rac1-mTOR Signaling Pathway in Coronary Arterial Myocytes. Cell Physiol Biochem 2013; 31:925-37; PMID:23817226; http://dx.doi.org/ 10.1159/000350111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Iorio F, Isacchi A, di Bernardo D, Brunetti-Pierri N. Identification of small molecules enhancing autophagic function from drug network analysis. Autophagy 2010; 6:1204-5; PMID:20930556; http://dx.doi.org/ 10.4161/auto.6.8.13551 [DOI] [PubMed] [Google Scholar]
- [61].Gurpinar E, Grizzle WE, Shacka JJ, Mader BJ, Li N, Piazza NA, Russo S, Keeton AB, Piazza GA. A Novel Sulindac Derivative Inhibits Lung Adenocarcinoma Cell Growth through Suppression of Akt/mTOR Signaling and Induction of Autophagy. Molecular Cancer Therapeutics 2013; 12:663-74; PMID:23443799; http://dx.doi.org/ 10.1158/1535-7163.MCT-12-0785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chiou SK, Hoa N, Hodges A. Sulindac sulfide induces autophagic death in gastric epithelial cells via survivin down-regulation: a mechanism of NSAIDs-induced gastric injury. Biochem Pharmacol 2011; 81:1317-23; PMID:21458423; http://dx.doi.org/ 10.1016/j.bcp.2011.03.019 [DOI] [PubMed] [Google Scholar]
- [63].Lisse TS, Hewison M. Vitamin D: a new player in the world of mTOR signaling. Cell Cycle 2011; 10:1888-9; PMID:21558808; http://dx.doi.org/ 10.4161/cc.10.12.15620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wu S, Sun J. Vitamin D, vitamin D receptor, and macroautophagy in inflammation and infection. Discov Med 2011; 11:325-35; PMID:21524386 [PMC free article] [PubMed] [Google Scholar]
- [65].Kim TH, Choi SJ, Lee YH, Song GG, Ji JD. Combined therapeutic application of mTOR inhibitor and vitamin D(3) for inflammatory bone destruction of rheumatoid arthritis. Med Hypotheses 2012; 79:757-60; PMID:22967804; http://dx.doi.org/ 10.1016/j.mehy.2012.08.022 [DOI] [PubMed] [Google Scholar]
- [66].Liu ML, Wilk SA, Wang AP, Zhou LJ, Wang RH, Ogawa W, Deng CX, Dong LQ, Liu F. Resveratrol Inhibits mTOR Signaling by Promoting the Interaction between mTOR and DEPTOR. J Biol Chem 2010; 285:36387-94; PMID:20851890; http://dx.doi.org/ 10.1074/jbc.M110.169284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Choquet H, Trapani E, Goitre L, Trabalzini L, Akers A, Fontanella M, Hart BL, Morrison LA, Pawlikowska L, Kim H, Retta SF. Cytochrome P450 and Matrix Metalloproteinase Genetic Modifiers of Disease Severity in Cerebral Cavernous Malformation type 1. Free Radic Biol Med. 2016; 92:100-109; PMID:26795600; http://dx.doi.org/ 10.1016/j.freeradbiomed.2016.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]