


Abstract
RNA–RNA interactions are fast emerging as a major functional component in many newly discovered non-coding RNAs. Basepairing is believed to be a major contributor to the stability of these intermolecular interactions, much like intramolecular basepairs formed in RNA secondary structure. As such, using algorithms similar to those for predicting RNA secondary structure, computational methods have been recently developed for the prediction of RNA–RNA interactions.
We provide the first comprehensive comparison comprising 14 methods that predict general intermolecular basepairs. To evaluate these, we compile an extensive data set of 54 experimentally confirmed fungal snoRNA–rRNA interactions and 102 bacterial sRNA–mRNA interactions. We test the performance accuracy of all methods, evaluating the effects of tool settings, sequence length, and multiple sequence alignment usage and quality.
Our results show that—unlike for RNA secondary structure prediction—the overall best performing tools are non-comparative energy-based tools utilizing accessibility information that predict short interactions on this data set. Furthermore, we find that maintaining high accuracy across biologically different data sets and increasing input lengths remains a huge challenge, causing implications for de novo transcriptome-wide searches. Finally, we make our interaction data set publicly available for future development and benchmarking efforts.
INTRODUCTION
A large percentage of the mammalian genome is transcribed into non-coding RNA (ncRNA) (1). As these ncRNAs may play important regulatory roles in the cell, efforts have been made to functionally annotate these transcripts (2,3). Previous research on ncRNAs such as sRNA (4) and miRNA (5) have shown that the identification of RNA–RNA interactions (RRI) between candidate ncRNAs and their targets is a key step to understanding the role of the RNA. Identifying and validating these interactions experimentally, however, can be slow and costly. To aid the identification of RNA–RNA interactions, a range of in silico methods have been proposed (6).
The prediction of RRIs can be viewed as a direct extension of RNA secondary structure prediction, employing similar theories and algorithms. In both settings, solutions are obtained by determining the set of Watson–Crick and wobble basepairs that correspond to the functionally relevant structure/interaction. Specifically, given two RNA sequences consisting of nucleotides adenine (A), cytosine (C), guanine (G) and uracil (U), determine the optimal set of intermolecular hydrogen bond basepairs between the two sequences.
More complex versions of the problem exist, such as those solving the joint structure, consisting of both the intramolecular basepairs within a single sequence in addition to the intermolecular basepairs. There is also the highly related RNA–RNA target prediction problem, where given a single query RNA sequence, and a set of potential target RNA sequences, find the correct pairing target for the query RNA. Tools solving the basic RRI prediction problem are much more common than those tackling these variations, and correct prediction of the complex variations often rely on correctly predicting the RRI problem first. As such, we will focus on the basic intermolecular RNA–RNA interaction problem given two sequences. In contrast to many previous evaluations, we impose no restriction on the specific type or length of the input RNA, aiming to evaluate RRIs tools in a general de novo scenario.
Algorithm strategies
We compare the predictive performance of 14 published computational methods (11 distinct program binaries) designed to predict interacting basepairs given two input RNA sequences. To better understand and compare these, we subdivide the RRI prediction algorithms based on their strategies into four types similar to those in other works (4,7).
The first type concerns itself only with intermolecular basepairs, both during computation and also for the final predicted result. Such algorithms are typically the fastest, having no need to predict intramolecular basepairs that could interfere and restrict certain intermolecular interactions. Ignoring restrictions and interferences, however, is exactly why these tools may incorrectly predict certain interactions where the existing RNA secondary structure needs to be taken into account. Algorithmically, these types of tools usually derive the set of interacting basepairs that maximize a certain value, commonly the stability of the entire interaction complex as quantified by the overall Gibbs free energy (ΔG) of stacking basepairs. We refer to these as ‘interaction-only’ methods, RNAduplex (8), RNAplex-c (9), RIsearch (10) and GUUGle (11) fall into this category. GUUGle is unique amongst these tools, being the only one that does not compute Gibbs free energies to score optimal interactions, but instead returns all ungapped interactions above a user-specific length, which we include as an absolute baseline for predictive performance.
The second type of method predicts only intermolecular basepairs, but factors in intramolecular interactions during computation, addressing the weakness of the first type. These algorithms utilize the McCaskill partition function algorithm (12,13) on the single input sequences to predict the pairing likelihood of nucleotides at each position. Thus, the stability of the intermolecular interaction at a specific position is now affected by both the predicted stability of the stacking basepairs, and also how likely the position will be made inaccessible by existing intramolecular basepairs. We refer to these as ‘accessibility-based’ methods which comprise RNAup (14), IntaRNA (15) and RNAplex-a (16).
The third type considers both inter- and intramolecular basepairs with restrictions during both computation and results, outputting in a joint structure. The most basic of these are termed ‘concatenation-based’ algorithms, literally concatenating the two input sequences and running it through classical RNA secondary structure prediction algorithms such as Mfold (17) and RNAfold (18). The main shortcoming of these methods stems from the classical RNA secondary structure algorithm's inability to predict unnested basepairs or pseudoknots, which translates to the inability to predict interactions that form on interior loops in the joint structure. Pairfold (19) and RNAcofold (20) fall into this category.
The fourth and final type is less well-defined and encompasses all non-concatenation methods that solve the joint structure, with little to no restrictions on interactions. The removal of restrictions often comes at the great expense of runtime performance, so these tools are typically restricted to relatively short input sequences. In this class, we have the program RactIP (21), made tractable for use on longer sequences by utilizing the technique of integer programming to optimize for runtime performance.
In addition to falling into one of the four categories, tools may optionally take multiple sequence alignments as input for each of the two input sequences. Based on successful RNA secondary structure prediction tools like Pfold (22) and RNAalifold (23), the addition of well-aligned and sufficiently divergent homologs provides additional information when predicting evolutionarily conserved basepairs. In theory, basepairs that are fully conserved or undergo compensatory mutations to retain the basepaired structure (i.e. covariation) are likely to be more functionally important than unconserved basepairs. RNAaliduplex (8) is the multiple sequence alignment version of RNAduplex (8), classified as the first interaction-only type. The interaction-only and accessibility-based version of RNAplex can optionally take multiple sequence alignments as input, which we will denote as RNAplex-cA and RNAplex-aA, respectively. PETcofold (7) belongs to the final complex joint structure category, given two multiple sequence alignment.
MATERIALS AND METHODS
Interaction prediction programs
Mentioned above, the programs used are summarized in Table 1, with more algorithmic details and exact settings described in the Supplementary Materials. On the table, in addition to splitting the tools into the four categories according to their strategy and usage of conservation, we also summarize whether they can output suboptimal results (instead of a single minimum free energy result) and the style of output they give.
Table 1. RNA–RNA interaction tools evaluated with categories and features. Strategy indicates the broad strategy of the algorithm in terms of prediction and output, described in the Introduction. Suboptimal indicates whether the tool can return suboptimal results in addition to the minimum free energy result. Conservation indicates whether it takes alignments as input. Interaction Length roughly describes the style of helices output, short helices typically not surpassing a dozen or so basepairs, with long helices reaching up to several times of that in total basepair count. Local interaction are a single interaction with gaps and bulges typically no longer than a few basepairs, while global predictions may span the entire sequence, containing multiple instances of local interactions, separated by long regions lacking intermolecular basepairs.
| Tool | Strategy | Suboptimal | Conservation | Interaction Length | Reference |
|---|---|---|---|---|---|
| GUUGle | Interaction only | Yes | No | Short Local | Gerlach & Giegerich (2006) (11) |
| RNAplex-c | Interaction only | Yes | No | Short Local | Tafer et al. (2008) (9) |
| RIsearch | Interaction only | Yes | No | Short Local | Wenzel et al. (2012) (10) |
| RNAduplex | Interaction only | Yes | No | Long Local | Lorenz et al. (2011) (8) |
| RNAplex-cA | Interaction only | Yes | Yes | Short Local | Tafer et al. (2011) (16) |
| RNAaliduplex | Interaction only | Yes | Yes | Long Local | Lorenz et al. (2011) (8) |
| Pairfold | Concatenation | No | No | Short Global | Andronescu et al. (2005) (19) |
| RNAcofold | Concatenation | No | No | Short Global | Bernhart et al. (2006) (20) |
| IntaRNA | Accessibility | Yes | No | Short Local | Busch et al. (2008) (15) |
| RNAplex-a | Accessibility | Yes | No | Short Local | Tafer et al. (2011) (16) |
| RNAup | Accessibility | No | No | Short Local | Mückstein et al. (2006) (14) |
| RNAplex-aA | Accessibility | Yes | Yes | Short Local | Tafer et al. (2011) (16) |
| RactIP | Complex joint | No | No | Long Global | Kato et al. (2010) (21) |
| PETcofold | Complex joint | No | Yes | Short Global | Seemann et al. (2011) (7) |
Related works and tools
Note that we focus on tools predicting general non-biology-specific RNA–RNA interactions, thus excluding many tools that utilize specific features of known classes of interactions. For example, the large collection of tools focused on predicting miRNA interactions and targets. While the general ideas of hybridization stability and accessibility apply to both the general and miRNA cases, modern miRNA prediction increasingly rely on miRNA-specific features that make their tools unsuited for predicting interactions outside of those for miRNAs. Reviews (5) and evaluations (24,25) of miRNA tools have been covered extensively by other works.
Two notable related tools solving the interaction target prediction problem are RNAPredator (26), which utilizes RNAplex to predict the target partner of small bacterial sRNAs and CopraRNA (27), which uses IntaRNA to tackle the same sRNA target prediction problem. A recent assessment of target prediction tools for sRNA was done by Pain et al. (28), showing CopraRNA as the best tool currently available for the task.
Finally, there were methods that fit the criteria of our evaluation but were excluded due to our inability to obtain or run them due to availability or practical reasons. These include GRNAs (29) (unavailable publicly), InteRNA (30) (algorithmically impractical), rip (31) (unavailable publicly) and ripalign (32) (algorithmically impractical).
Multiple sequence alignment programs
A subset of our tools are conservation-based and require high quality multiple sequence alignment to perform optimally. These tools take multiple sequence alignments as input, with the objective of using evolutionary information in the alignments to improve the accuracy performance of the algorithm. The quality of the alignment is a large limiting factor to the performance of these tools, so we evaluate the performance of the tools as a function of the alignments’ minimum percent identity. Specifically, we start with the full unfiltered alignment, and then remove all sequences with a percent identity (relative to the reference species) lower than the minimum threshold, and run the resulting alignments with the algorithms selected. No filtering was done using minimum sequence count or total tree length.
Our initial selection of aligners was based on recent assessments of multiple sequence aligners (33,34), where MAFFT (in ‘accurate mode’ or L-INS-i) (35) and ProbConsRNA (36) were selected. While these tools have been shown to perform well at aligning homologues with conserved sequences, it is unknown if they can correctly align homologous conserved basepairing structure which may exhibit covariation and thus lose sequence conservation. In order to alleviate this, we also examine alignments from two structure-aware aligners LocARNA (37) and SPARSE (38). The latest version of MAFFT also included two strucuture-aware alignment modes Q-INS-i and X-INS-i, both of which we test. We conduct a very brief assessment of predictive performance on alignmnents created by the listed aligners, and made our final selection to use MAFFT Q-INS-i based on balance between accuracy and runtime performance, detailed in the Results section.
Data sets
The evaluation of any new computational tool requires the compilation of a set of experimentally verified results. While RNA secondary structure tools have long benefited from curated and compiled data sets such as RNA STRAND (39) and Rfam (40), RNA–RNA interaction tool evaluation have so far only relied on ad hoc and varying data sets. Generally, tools have aimed to obtain a set of biologically functional interactions, consisting mostly of miRNA, bacterial small RNAs (sRNA) and snoRNAs. In this paper, we aggregate the sRNA and snoRNA interaction pairs that have been used in various papers, and here present what we believe is one of the largest, freely accessible and digitized collections of such RNA–RNA interactions. In contrast to previous data sets, we attempt to alleviate the biological bias of focusing on a single type of data, gathering both snoRNA and sRNA. Additionally, we also eliminate any bias on input length, taking the full length of target rRNA and mRNA sequences. We make the data set available in tab-delimited format as part of the Supplementary Files, containing the full sequences and basepair information for future tool development and benchmarking efforts.
Bacterial sRNA
Small RNAs or sRNAs, are non-coding regulatory RNAs found in bacteria, shown to bind to the translational start sites of mRNAs, controlling the stability and translation of their targets. Forty to 400 nucleotides in length (41), sRNAs do not simply bind in zipper-like fashion to the mRNA across its entire length like the majority of miRNAs. Instead, sRNA–mRNA interactions vary significantly in stability and length, modulated by existing RNA secondary structures on both the sRNA and mRNA strands. Thus, the identification of the functionally relevant interaction serves as a challenging and relevant problem in RNA–RNA interaction prediction.
Functionally relevant sRNA–mRNA pairs are obtained from previously published experimental works, mostly derived from biochemical mapping experiments in Escherichia coli and Salmonella enterica. In earlier works, a set of 18 interactions collected for IntaRNA(15) was used by several works (10,42). An expanded set tripling the interaction count was used for analysis of sRNA target binding regions (43,44), stated to be equivalent to sRNATarBase (45) published in parallel. Finally, the most recent and comprehensive set of over 100 interactions was compiled to evaluate the partner prediction problem in CopraRNA (27), a direct expansion of the aforementioned set.
Our sRNA data set is a curated and digitized version of the interactions presented by CopraRNA, recovered from the Supplementary Material files and at times graphical figures in cited experimental publications to obtain the exact basepairing. The end product is a set of 109 sRNA–mRNA interactions (64 E. coli, 45 S. enterica) from 18 sRNAs against 82 mRNA targets. sRNA lengths range from 72 to 237 nucleotides, with a mean length of 123 nt. The majority of interactions involve only one interaction site, but some pairs involve interactions at two disjoint sites. For these interactions (OxyS-fhlA and RprA-csgD in E. coli, GcvB-cycA, GcvB-tppB and MicF-lpxR in S. enterica), each continuous segment is counted as one unique interaction for performance evaluation, resulting in these pairs having two solutions each. While it is technically possible to combine both sites into a long interaction containing a lengthy unpaired region in the middle, splitting it allows for both better predictive performance for the tools evaluated and also allows us to limit the maximum interaction length.
We used RefSeq (46) genomes for E.coli str. K-12 substr. MG1655 (NC_000913.3) and S.enterica subsp. enterica serovar Typhimurium str. LT2 (NC_003197) as our reference sequence. Sequences for sRNA and mRNA targets were extracted from genomes using gene names and associated GFF annotation files, along with 300 bases upstream of the translation start site. Once sequences were extracted, interactions from Supplementary Materials and manuscript figures were mapped onto the sequences and all interactions were confirmed to correspond to valid basepairs. The output data are stored in a computer-parsable CSV file, each line containing the full sRNA and mRNA sequences, along with the exact binding location and the basepair formation, available as Supplementary Files.
Fungal snoRNA
Small nucleolar RNAs or snoRNAs are non-coding RNAs found in eukaryote and archaea, shown to stably bind to rRNAs, guiding essential chemical modifications at specific positions (47). These RNAs are generally classified into C/D box snoRNAs that guide methylation and H/ACA snoRNAs that guide pseudouridylation. We focus on C/D box snoRNAs out of necessity, as H/ACA snoRNA interactions heavily depend on correctly folding intramolecular hairpins, making them great for evaluating joint structures, but falling outside the scope of this work. For those interested in H/ACA predictions, we refer readers to the recent work RNAsnoop (48), which includes a small comparison between three H/ACA prediction tools. A single C/D box snoRNA typically has one or two binding sites, ranging from 10 to 21 nt in length, forming highly complementary interactions with its target rRNA site. Despite the highly complementary interactions, the possible existence of multiple binding sites on the snoRNA, and the length of the rRNA targets (up to thousands of nucleotides) presents a very different problem than that posed by the sRNA set.
We chose to use yeast snoRNA–rRNA interactions from Saccharomyces cerevisiae, due to the completeness of the data set and annotations available. Our data set consists of 52 C/D box interactions obtained from Methylation Guide snoRNA Database (49), with additional interactions from the UMASS Amherst Yeast snoRNA Database (50). The 52 interactions are made by 43 unique snoRNAs and two rRNA targets. SnoRNA lengths range from 78 to 255 nucleotides with a mean length of 104 nt, the full rRNA sequences are the 1800 nt 18S rRNA and 3396 nt 25S rRNA.
We use the Saccharomyces cerevisiae S288c genome from the Saccharomyces Genome Database (51), and extract snoRNA and rRNA sequences by gene name using the associated GFF genome annotation file. Interaction data from the Methylation Guide snoRNA Database (49) was reformatted and mapped onto the sequences, confirming the correctness of the basepairs. The final output is a computer-parsable CSV file with each line representing an interaction, containing the full snoRNA and rRNA sequence, as well as the exact interaction sites and basepairing formation, available as Supplementary Files.
Multiple sequence alignments
Whereas minimum free energy (MFE) methods only require a single sequence from each of the target and query RNAs, conservation-based methods require multiple sequence alignments (MSA) of homologues.
For the sRNA database, we obtained the list of 244 completed Enterobacteriaceae genomes from KEGG (52) and complete genomes listed from RefSeq. We used GotohScan (53) to find homologs for input reference sequences, and generated FASTA files of unaligned hits from the output. FASTA files were then aligned with MAFFT (35) in structurally-aware Q-INS-i mode with default settings. Finally, using percent identity (% ID) to the references species, we kept the top hit for any species that had more than one homolog hit. The above process resulted in MSA of sRNA and mRNA sequences, which were then used to make pairs of alignments containing the same species. For each interaction, the corresponding sRNA and mRNA alignments were taken, and the intersection of species was kept. The number of species after these intersections range from 42 to 216, with a mean species count of 170. Pipeline scripting was done in Perl and R, with FASTA sequence and RNA structure manipulation done using the R4RNA package (54).
For the snoRNA database, we obtained the entirety of the RefSeq release 68 Fungi sequences Nov 2014, containing just under 3000 species. We followed the same procedure as described, also using MAFFT Q-INS-i on the full length rRNAs despite significantly longer runtimes. Species count for finalized alignments range from 5 to 44 with a mean count of 23.
Performance measures
We use the True Positive Rate (TPR, also called sensitivity) and Positive Predictive Value (PPV, also called selectivity (55)) to measure predictive performance. We only consider intermolecular basepairs, ignoring all intramolecular predictions. Given a set of predicted basepairs and a set of experimentally validated basepairs, each predicted basepair is either a True Positive (TP) if it also appears in the known set else it is a False Positive (FP). All basepairs in the known set that are not predicted as TP are False Negatives (FN), i.e. the prediction algorithm incorrectly predicts the basepair as non-pairing. Hence:
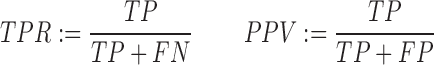 |
True negatives basepairs (TN) have traditionally been of little practical use for RNA secondary structure prediction evaluation, with the same applying in this work. Regardless, we describe its computation as it is required in some of our statistical analysis. We compute the number of TN basepairs as the ‘total’ number of possible basepairs minus the number of TP basepairs as defined above. We estimate the total number of possible basepairs as 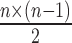 , where n is the length of the concatenated sequence (8). This value is typically several magnitudes larger than the other values, and typically makes the specificity measure (also known as the true negative rate:
, where n is the length of the concatenated sequence (8). This value is typically several magnitudes larger than the other values, and typically makes the specificity measure (also known as the true negative rate: 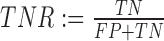 ) largely meaningless, as it is effectively 1 for all tools evaluated. For this reason, we use TPR and PPV, which are independent of the TN count.
) largely meaningless, as it is effectively 1 for all tools evaluated. For this reason, we use TPR and PPV, which are independent of the TN count.
Finally, we also use Matthews Correlation Coefficient (MCC) (56) as a rough summary of both TPR and PPV as defined as:
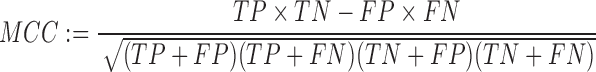 |
MCC ranges from 1 for predictions with maximum TPR and PPV, to −1 for very poor predictions, although in practice, the physical constraints of RNA basepairs result in a range between 0 and 1 for non-random predictions, and has been shown to be an approximation of the geometric mean of TPR and PPV (57).
Considerations were made to subtract compatible basepairs (55) from the False Positive count, but doing so was found to be too lenient for tools which took a shotgun approach to predicting interactions.
RESULTS
Minimum free energy results on sRNA data set
We run the 10 energy-based tools against 109 sRNA–mRNA pairs, using the full length sRNA against truncated mRNA targets. Knowing the biology of sRNAs, it is not unreasonable for users to focus on a region around the translation start site (TSS). Previous studies have used similar windows, such as −150 and +100 bps relative to the TSS (44). With all interactions falling within −130 and +104 bps, we begin with a conservative window of −150 and +150 bps. We run the analysis under two sets of options, the first being the most basic use case scenario of all default settings, followed by runs using optimal recommended settings when available.
Results for 109 pairs run on 10 tools are visualized as a heatmap in Supplementary Figure S1 created using ggplot (58) in R, with default options on left, optimal options in middle and differences shown on the right. Hierarchical clustering of the results run with optimal settings clarifies an otherwise confusing set of results, and provides some immediate insight into tool similarity. As expected, results for accessibility-based IntaRNA and RNAplex-a, interaction-only RIsearch and RNAplex-c, and concatenation-based Pairfold and RNAcofold show highly similar performance profiles (Supplementary Figure S2, left). RNAduplex and RactIP also cluster together, which was not apparent from their algorithmic strategies.
The mean of performance results is shown on Supplementary Table S1 for the sRNA data set, seen for results run with default options, optimal options and the difference between the two runs where applicable. Setting correct options for RIsearch and RNAplex-c are essential for obtaining competitive results, whereas all other tools gain only a small increase in performance (Supplementary Figure S1 middle). For these two tools, the optimal setting involves setting the per nucleotide extension penalty to 0.3 kcal mol−1–the average duplex energy between two random RNA bases (9). For the two accessibility-based tools, optimal settings involve restricting the length of interactions to 60 nt, just larger than the longest interaction in the data set.
According to mean MCC (Supplementary Table S1), the best performing tool on the data set is IntaRNA (0.62) followed closely by RNAplex-a (0.58). The simple inclusion of accessibility information may not completely explain this advantage over other tools, given that RNAup too uses accessibility, yet only achieves a mean MCC of 0.39. Tools that perform poorly according to MCC, such as RNAduplex, appear to be severely penalized for predicting a large number of interacting basepairs resulting in a poor PPV.
Suboptimal interaction results on sRNA data set
While all the energy-based tools used produce a single minimum free energy secondary structure by default, a majority of tools also allows the prediction of suboptimal results. In practice, this allows both for an increased sensitivity and also the ability to correctly predict pairs of interacting sequences with multiple binding sites. We determine the increases obtained by turning on suboptimal results for tools that have this option.
For this test, we use the same sRNA data set and optimal options as used previously, with the exception of the new suboptimal results option enabled, set to allow for all suboptimal structures within reason to be returned. Specifically, when given the choice to set an energy threshold, we have the tools return all interactions with a predicted ΔG stability of ≤0 kcal/mol. This is high enough to include the minimum free energy structures and any suboptimal results that would be of practical interest, and low enough to keep output file sizes under control. It should be emphasized that this energy cutoff is not the one used for performance evaluation as follows. The definitions defined for MFE performance evaluation persist, but the predicted basepairs are now the union of intermolecular basepairs (i.e. no duplicates) from all suboptimal structures below a specific energy threshold. This specific energy threshold is unique to each tool for each interaction prediction, and is chosen to maximize the MCC value for that specific run. While knowing the energy threshold that maximizes the MCC up-front is impossible in the typical use case where the interactions are not known beforehand, we aim to derive the theoretical maximum of including suboptimal results, and thereby make potential recommendations on how to set such a threshold for future de novo runs.
Results for suboptimal results are visualized in Figure 1 (middle), retaining the same column and row ordering for easy comparison with the optimal MFE results in Figure 1 (left). Visually, the tools with the largest changes are RNAduplex, RIsearch and RNAplex-c, differences highlighted in Figure 1 (right). IntaRNA and RNAplex-a have relatively smaller gains to performance. Summarized in Supplementary Table S2, we see that the three former tools roughly double TPR, but see little change to PPV rates. This increase in TPR likely stems from the increased total number of predicted basepairs, three to four times the number of bases for these three tools.
Figure 1.

Predictive accuracy measured by Matthews Correlation Coefficient (MCC) for energy-based interaction prediction tools on the sRNA–mRNA data set. Minimum free energy (MFE) (left) versus suboptimal (middle) results shown with differences (right), pairs and tools clustered hierarchically according to MFE results to group like results (Supplementary Figure S1). Suboptimal results only available in IntaRNA, RIsearch, RNAduplex and both versions of RNAplex.
Measured by MCC, IntaRNA (0.69) and RNAplex-a (0.69) remain the two top performing tools, while RNAplex-c (0.52) and RIsearch (0.50) jump ahead to take third and fourth spot. Surprisingly, IntaRNA has minimal change to the number of predicted basepairs even with the suboptimal, meaning that the validated interactions are often its MFE prediction already. GUUGle, which simply returns all valid ungapped interactions, does surprisingly well with an MCC of 0.44 if we simply take all predictions greater than 9 basepairs (mean 9.90). It obtains a TPR equal to RNAplex-a, but suffers from inadequate PPV, suggesting that Gibbs free energy serves as a good means to determine functional interactions from random ones. It is noted that while there is a shift in performance, the clustering of tools remains similar to before with only MFE results for this data set (Supplementary Figure S2, right).
In addition to a clustering of tools, the results of each tool also cluster to some extent. In Supplementary Figures S11 and S12, we see MCC performance distributions of the energy-based tools on sRNA data returning MFE and suboptimal results (when applicable), respectively. We see a large number of results at 0 MCC when MFE results are returned, since predictions are either a hit or miss. With suboptimal options enabled, this peak at 0 mostly disappears for tools with the option. On this Supplementary Figure S3, we show the TPR and PPV for each tool on each prediction, with a two-dimensional density plot showing the rough clustering of results for each tool. Ignoring the large majority of points that end up with no predictions, we see a large concentration of accessibility-based predictions with a PPV of 1 and a range of TPR values. In contrast, tools without accessibility have a strong concentration of results with a TPR of 1, but a range of PPV values.
The Gibbs free energy (ΔG) in Supplementary Table S2 denotes the energy threshold used, below which bases are considered to be positively predicted. In practice, these could serve as guidelines for thresholds. The variance in energies between tools, however, makes setting clear guidelines difficult. The Rank denotes the average number of suboptimal results kept for each interaction to obtain the performances seen (i.e. MFE results effectively have Rank of 1). A lower rank doesn't necessarily mean worse performance, since it might simply reflect a tool that successfully predicts the entire interaction via small separate interactions.
Effect of increasing target sequence size on sRNA data set
Using optimal options and suboptimal results where applicable, we test the accuracy performance of energy-based tools on the sRNA data set, but this time increasing the length of the target mRNA sequence. Previously, we had used the full-length sRNA sequence against a 300-nt window around the translation start site, 150 nt upstream, 150 nt downstream. Here, we keep the 150 nt upstream the same, but gradually increase the length of the coding sequence (CDS) downstream by increments of 100 nt, until we are 1150 nt upstream, having target sequences up to 1300 nt, roughly the length of the average bacterial gene.
The resulting performance as measured by MCC is seen in Supplementary Figure S5, showing a monotonically decreasing trend for all tools. All tools have difficulties maintaining PPV, resulting in an overall decrease in MCC as the search space increases. This is alleviated somewhat by tools that produce suboptimal results, as they are able to maintain a high TPR rate, which is often untrue for tools that only return MFE results. The rate of decrease varies between tools, with a few such as IntaRNA and RNAplex-a having relatively linear trends, while RIsearch and RNAplex-c having fast asymptotic trends. Extrapolating, it is likely that increasing the length even further will result in a further decrease in performance for all tools, which would have worrying implications for full-transcriptome searches.
MFE energy-based results on snoRNA data set
In order to examine whether the performances observed in the sRNA data set are generalizable to interactions of other types, we evaluate the performance of all tools on our second data set, consisting of 52 C/D snoRNA–rRNA interactions. We proceed straight to evaluating the performance using optimal settings, adjusting the maximal interaction length to 25, just large enough to capture all known interactions in the data set. We start by running MFE results following by suboptimal results.
We run the energy-based tools on two versions of the data set using the same 52 snoRNA and rRNA pairs. We first determine the performance in an ideal scenario, knowing the general binding region of the snoRNA on the rRNA sequence, having the full length snoRNA interact with a 300 nt subsequence of the target rRNA centered around the center of the binding site. We then test a more realistic de novo scenario, interacting the full snoRNA against the entirety of the target rRNA.
MFE results on the short and long data set are seen on are seen in Supplementary Table S7 and Supplementary Figure S10, which repeat our observation that increasing the search space results in a significant drop in performance for all tools. In contrast to suboptimal results, the drop in MCC is caused by decreasing performance in both TPR and PPV. In Supplementary Figures S13 and S14, we see MCC performance distributions of the energy-based tools on snoRNA data returning MFE and suboptimal results (when applicable).
Suboptimal energy-based results on snoRNA data set
On Supplementary Table S8, we see a numerical summary of the effects of enabling the suboptimal results option when available. The MCC gains obtained for enabling suboptimal results on this snoRNA data set (0.03–0.08) are much smaller compared to the effects seen with sRNA (Supplementary Table S2, 0.07–0.19). This is due to the MFE results often being the known interaction, making the additional predictions gained from suboptimal results mostly unnecessary. As seen in the TPR and PPV columns, enabling suboptimal options results in a trade-off, increasing TPR at the expense of PPV, ultimate resulting in a higher MCC similar to the sRNA data set.
The results of using the short ideal window versus full rRNA are shown in Supplementary Figure S6 and summarized in Supplementary Table S3, with average results for each tools across the data set and metrics. As seen in the sRNA data set, tools form pairs and cluster closely together S9 compared to Supplementary Figure S2.
For the short ideal case (Supplementary Table S3 (Short) rows), the average MCC performance is higher for all tools in comparison to the sRNA data set, likely due to the simpler and more uniform interactions in this data set. The simpler interactions are reflected in a much higher TPR rate for all tools with 6 out of the 10 energy-based tools achieving a TPR rate of ≥0.91, detecting almost all known interactions. With the exception of GUUGle, RIsearch and RNAplex-c which double their PPV rates from around 0.40 to 0.80, most tools only see relatively small improvements to PPV.
When we extend the target to full length rRNAs (Supplementary Table S3 (Long) rows), we see a significant decrease in performance for all tools as the number of positive predictions increase, the majority of them false. Based on MCC, IntaRNA, RNAup, RNAduplex, RactIP and RNAplex-a suffer a relatively smaller drop in MCC (−0.10 to −0.14), while the remaining tools suffer a larger decrease (−0.24 to −0.29). For the latter tools, interaction-only tools with suboptimal options (GUUGle, RIsearch and RNAplex-c) see little change in TPR, but experience significant decreases in PPV, explaining the MCC drop. Concatenation tools Pairfold and RNAcofold see significant drops in TPR, PPV and MCC performances.
Based on the Gibbs free energy cutoffs and the predicted number of basepairs (Supplementary Table S3 Bps columns), increasing the target search space does not significantly change the energy threshold, but the number of basepairs that pass this threshold increases. With the exception of RNAup, tools that do not compute suboptimal results do extremely poorly with the increased search space. Of the remaining tools that do compute suboptimal results, accessibility seems to be key to preventing huge losses in PPV, perhaps explaining why RNAup remains competitive.
Conservation-based predictions for sRNA data set
As seen in Supplementary Table S4 and Figure 2, the minimum percent identity (% ID) has a large effect on the MCC, TPR and PPV values of the four tools evaluated. We observe a monotonically increasing trend for TPR as the minimum percent identity increases, suggesting that the experimentally determined basepairs are not extremely well conserved, and are only detected when a majority of divergent homologues are filtered out. PPV values fluctuate depending on the tool, with RNAplex-c and RNAaliduplex seeing a slight decreasing trend. With the exception of PETcofold, there appears to be a clear trade-off between TPR and PPV, with TPR increasing while PPV decreases as the minimum percent identity of the alignment increases.
Figure 2.

Performance on conservation-based tools using alignments for the sRNA data set, alignment sequences filtered by minimum percent identity.
Maximal MCC performances for tools are obtained at 70% ID for RNAaliduplex (0.31), 80% ID for PETcofold (0.43), 90% ID for RNAplex-cA (0.56) and 100% ID for RNAplex-aA (0.69). For the three tools that have direct energy-based counterparts, these MCC values are greater or equal to the performance values seen in Supplementary Table S2, with an increase in performance of 0.01 RNAaliduplex, 0.04 RNAplex-cA and 0.00 RNAplex-aA. Take note that optimal conservation-based MCC values were obtained at different minimum percent identity thresholds, with the RNAplex-aA threshold of 100% effectively being the energy-based methods as no divergent information was present in the alignment.
Conservation-based predictions for snoRNA data set
We test the effects of multiple sequence alignment inputs on the snoRNA data set with truncated rRNA targets, again filtering alignments by minimum percent identity. As seen in Supplementary Figure S4 and Table S5, this time we see that increasing percent identity actually results in a drop in MCC performance for three of the four MSA-based tools. Again, while we see a trade-off between increasing TPR and decreasing PPV as the minimum percent identity increases, the gains in TPR are quite minor, with the loss of PPV fairly significant as the minimum percent identity increases. These results seem to suggest that in this data set, the benefits of an increase in PPV outweigh the penalities of decreased sensitivity, leading to an overall MCC that is superior to energy-based tools.
Maximal MCC performances for tools are obtained at 75% ID for RNAaliduplex (0.67), 80% ID for PETcofold (0.58), 80% ID for RNAplex-cA (0.83) and 80% ID for RNAplex-aA (0.82). For the three tools that have direct energy-based counterparts, these MCC values are greater or roughly equal to the performance values seen in Supplementary Table S3, with an increase in performance of 0.29 RNAaliduplex, 0.09 RNAplex-cA and −0.01 RNAplex-aA. In contrast to the sRNA results, the increase in performance can be directly attributed to the conservation information, with an alignment of identical sequences (i.e. 100% minimum ID) resulting in inferior results.
Surprisingly, when comparing the two versions of RNAplex, the simpler RNAplex-cA that does not take in accessibility profiles does better than its accessibility-based comparative counterpart. With an optimal MCC value of 0.93, its accuracy is superior to all energy-based methods on the short snoRNA–rRNA data set as seen on Supplementary Table S3 (MCC (Short) row).
We follow up this evaluation by testing the same conservation-based tools on the snoRNA–rRNA data set, but use the full-length rRNAs instead of the windowed binding sites. Simulating a de novo use case, we use no minimum percent identity filter, obtaining results seen in Supplementary Table S6. As in the case of energy-based algorithms on Supplementary Table S3 (MCC (Long) row), we see a drop in overall performance as measured by MCC. However, the usage of conservation information maintains a relatively higher PPV value. Most evident in RNAplex-cA, we observe a TPR rate of 0.94 and PPV of 0.81, resulting in an MCC measure of 0.85, eclipsing all other tools conservation or energy-based on the untruncated snoRNA–rRNA data set.
Effect of different aligners on predictive performance
Using the snoRNA–rRNA data set, we run sequence-based aligners ProbConsRNA (36) and MAFFT (L-INS-i mode) (35), and structurally-aware aligners LocARNA (37), SPARSE (38) and two modes of MAFFT (Q-IN-i and X-INS-i). For each aligner, we give as input the full-length, unfiltered, unaligned snoRNA and rRNA alignemnts as inputs. After obtaining alignments, we trim the target rRNA to a window surrounding the known interaction site and ensure each pair of alignments has the same species. Finally, we progessively filter the alignment according to minimum percent identity every 10%. We then run conservation-based interactions on the resulting alignments and evaluate performance in terms of MCC, TPR and PPV.
All tools successfully aligned snoRNA within a reasonable amount of time but only ProbConsRNA and two modes of MAFFT (L-INS-i and Q-INS-i) successfully completed the two rRNA alignments (18S and 25S) within a week of continuous runtime. For the tools that failed their rRNA alignments, MAFFT Q-INS-i rRNA alignments were used. This test was to be repeated on the sRNA–mRNA data set, but multiple tools were unable to complete the mRNA alignments within a week of runtime.
Accuracy performance of the four conservation-based tools on the different alignments are measured in MCC (Supplementary Figure S18), TPR (Supplementary Figure S19) and PPV (Supplementary Figure S20). According to MCC, no tool seems clearly superior to all the others, while SPARSE and ProbConsRNA clearly produce inferior results on specific tools. The three MAFFT modes perform extremely similarly. Considering both performance accuracy and runtime speed, the choice of MAFFT Q-INS-i is arguably the best choice given the selection of alignment algorithms.
Combining energy-based and conservation-based results
A common way to increase TPR or PPV is to combine results of multiple tools. Due to the number of tools we have, the potential number of combinations is unrealistic to fully explore. However, we take some time to test this technique on the three energy-based tools with conservation-based counterparts.
In Supplementary Table S3, we show the predictive performance of RNAduplex, RNAplex-c and RNAplex-a paired with comparative counterparts RNAaliduplex, RNAplex-cA and RNAplex-aA. For each pair of tools, we show the performance of the MFE algorithm, the MSA algorithm (MFE + alignment input), the union of results and the intersection of results.
As expected, the union of results increases the TPR to values greater than the MSA or MFE results individually, but results in a large decrease in PPV. The intersection increases the PPV to values greater than the MSA or MFE results individually, and results in a decrease of TPR. Based of MCC results, taking the union or intersection can be highly beneficial at times, resulting in values greater than MFE or MSA results. However, the results are inconsistent and it is hard to recommend a consistent setting for specific tools.
However, if TPR or PPV are of particular interest in predicting basepair interactions, these results suggest that taking the union or intersection of results is not a bad approach.
Basepair covariation in data sets and background
To gain some understanding into the different effects that alignments had on the sRNA versus snoRNA data sets, we computed basepair covaration score and basepair conservation for the known inter- and intramolecular basepairs in the data sets. Supplementary Figure S17 shows the binned distribution of basepairs according to their covariation scores (where −2 is unconserved, 0 is perfectly conserved and 2 is covarying with compensatory mutations (59)) and conservation (1 is perfect conservation of nucleotide, 0 is no conservation). Specifically, we take the multiple sequence alignment as previously described for the data set, and compute the two scores for every known intermolecular basepair between the snoRNA and rRNA, sRNA and mRNA, and the intramolecular basepairs between rRNA and rRNA derived from the solved structures from the Comparative RNA Website (60). We also repeat the same scoring with helices absent from the known structure predicted on the same rRNA alignment to obtain a background, and the known rRNA helices projected onto a shuffled (with Multiperm (61)) rRNA alignment for a fully random control. We filter all alignments used to 80% minimum percent identity in both data sets and controls, which is where most tools have been shown to perform best across both data sets.
The results from the plots are summarized in Table 2. From these plots, we clearly see that the majority of basepairs show strong conservation (1) and no covariation (0). Overall, we see a much stronger positive covariation score in intramolecular rRNA–rRNA results, with the intermolecular basepairs in snoRNA–rRNA and sRNA–rRNA showing less covariation. The intermolecular interactions show surprisingly similar covariation and conservation scores, both with slightly negative covariation scores and extremely high conservation scores.
Table 2. Covariation and conservation mean ± standard deviation scores for known and control inter- and intramolecular, along with the percentage of conserved (and canonical), covarying (both double and single-sided) and invalid (non-canonical, gapped) basepairs in the same alignments.
| Data set | Covariation | Conservation | % Basepairs covarying | % Basepairs conserved | % Basepairs invalid | Basepairs |
|---|---|---|---|---|---|---|
| rRNA–rRNA | 0.06 ± 0.34 | 0.87 ± 0.19 | 0.07 ± 0.04 | 0.84 ± 0.02 | 0.10 ± 0.01 | 1504 |
| snoRNA–rRNA | −0.03 ± 0.13 | 0.98 ± 0.06 | 0.01 ± 0.01 | 0.97 ± 0.03 | 0.02 ± 0.03 | 664 |
| sRNA–mRNA | −0.03 ± 0.14 | 0.97 ± 0.08 | 0.01 ± 0.02 | 0.96 ± 0.06 | 0.03 ± 0.04 | 1879 |
| Non-functional rRNA–rRNA | −0.17 ± 0.29 | 0.83 ± 0.18 | 0.06 ± 0.01 | 0.69 ± 0.23 | 0.24 ± 0.24 | 1500 |
| Shuffled rRNA–rRNA | −0.46 ± 0.40 | 0.72 ± 0.24 | 0.05 ± 0.00 | 0.22 ± 0.00 | 0.73 ± 0.00 | 1504 |
DISCUSSION
Settings and overfitting
As shown in Supplementary Table S1, the effect of differing settings is significant for multiple tools on various data sets. Given no prior information and guidance, determining optimal settings is a non-trivial task. Even with recommended settings from authors, we noticed that settings that were optimal for one data set cannot be assumed to work well with another. While the tools evaluated can technically be applied to any RNA sequence, the need for biologically-specific settings adds an extra layer of challenge for users.
In theory, it would be possible to perform an exhaustive search through the multi-variable setting space to find the setting values that maximize performance accuracy for each tool on each data set. It is debatable, however, whether such data set-specific settings would still perform well on other data sets and de novo user data as such parameters would potentially be extremely overfit and largely meaningless on other data sets and research settings.
We showed that enabling the suboptimal options for tools that support it consistently increases the overall predictive performance (Supplementary Table S2). As touched upon previously, however, in practice the user would then have to deal with a ranked list of results instead of the single output. We have shown that the number of ranked results to use for optimal performance and the optimal Gibbs energy threshold cutoff varies greatly depending on both the tool and data set in question, making specific suggestions difficult.
Performance effects of conservation
According to benchmark evaluations in RNA secondary structure prediction, compensatory mutations in multiple sequence alignments can greatly aid the accurate prediction of basepairs (55).
For RNA–RNA interactions prediction, the inclusion of conservation information by giving alignments seems to bring mixed results depending on the tool and data set. For interaction-only tools like RNAplex-c, the addition of conservation information increases the specificity, resulting in an overall MCC performance increase. When used in conjunction with accessibility-based methods (e.g. RNAplex-a), additional alignment information does not seem to significantly increase the performance, and may even decrease performance due to alignments of questionable quality. Out of all the tools evaluated, the highest MCC performance (0.93) was achieved by RNAplex-cA on the snoRNA data set with a relatively divergent input alignment (65% ID), showing that in the ideal case, conservation information can provide the best predictions. The number of variables that need to be correctly determined for this optimal result (i.e. alignment settings, percent identity threshold, settings, suboptimal results to keep), could make it impractical in a de novo setting.
Previous studies have observed that sRNA binding sites exhibit a high sequence conservation but low basepair conservation, further stating that covariation can only help a subset of interactions (44). However, other studies also on sRNA have shown that under ideal circumstances (43) and sophisticated alignment methods (16), conservation can serve as a beneficial feature.
In addition to issues with the alignments due to the biology of sequence, the tools used to obtain homologues and align them can greatly effect the predictions, as shown through our usage of six different alignment methods. Choosing a proper aligner is a non-trivial task, with a need to consider computational restraints, RRI prediction tool and whether an algorithmically more complex algorithm is actually worth a potential increase in performance. While our alignments could undoubtedly be improved with expert knowledge and manual curation, our work hopefully shows the issues with a high-throughput de novo RNA–RNA interaction screen.
The fact that different tools react differently to different data sets and minimum percent identity settings in this work, and also compared to what is known from RNA secondary structure studies is perplexing. Possible avenues of explanation include things attributable to the user, such as alignment input and tool settings of which there are already a non-trivial amount to control. Additionally, it is known that many of the evolutionary models and scores employed in these intermolecular basepairs prediction algorithms were trained on intramolecular basepairs (7), for which we have suggested may be under different selective pressures and evolve differently. For all conservation-based algorithms used, they combine thermodynamic and evolutionary components, with tunable weighs for each component trained on specific data sets. These weights undoubtedly play a role in how tools react to changes in alignment quality, but require a non-trivial amount of work to optimize for specific data sets.
Taken together, the benefits of conservation information highly depend on the type of interaction (and possibly even the specific transcript pair) in question, complicated by the significant effect that homolog and alignment quality have on the results. Even under ideal circumstances, however, previous work has shown that accessibility aids correct prediction more than conservation (43) and that using both results in only a slightly higher performance. In practice, the effort and curation required to generate high quality alignments required is non-trivial making conservation-based potentially less appealing than energy-based methods.
It is of particular note that nearly all the tools evaluated that utilize conservation information use an extremely simplified evolutionary model, namely the covariation score. While such a score is computationally fast to compute, it fails to account for many of the finer details of basepair evolution. For example, the covariation score does not normalize the observed conservation against the background conservation of the surrounding region, nor is it aware of any phylogeny which may serve to strengthen or weaken the importance of an observed evolutionary event. By utilizing a more complex model, such as Felsenstein's evolutionary model (62) used in Pfold (22) (and by extension PETcofold (7)), it is theoretically possible to capture background evolutionary rates and weigh different types of mutations (e.g. transversions and transitions (63)) while being informed by the alignment phylogeny.
Target size and interaction search space
Consistently observed in all tools across all data sets tested, increasing the length of the input sequences leads a decrease in predictive performance. For tools that enable multiple suboptimal results, there is little change to TPR, but difficulties in maintaining a high PPV results in an overall low MCC value. For tools that only produce a MFE result, both TPR and PPV suffer.
The inability of these tools to scale properly as input size increases is most problematic when applying these tools to predict potential interactions on a transcriptome-wide scale. These observations agree with existing interaction target prediction tools for sRNAs such as CopraRNA and RNApredator shown to have PPV values of 44% and 28%, and TPR values of 23% and 32%, respectively (27,28).
While we demonstrate on the untruncated snoRNA data set that conservation may be a strong feature to include for an increased PPV rate, the difficulties discussed likely affect its usefulness in practice. CopraRNA uses homolog information in its computations, and it is uncertain whether it is the limitations of the algorithm, the alignment quality or the biology that limit its performance.
Runtime and memory performance
We show CPU runtimes and physical memory usages for the energy-based tools outputting suboptimal results (where applicable) when running on the sRNA–mRNA data set with increasing long mRNA sequences in Supplementary Figures S15 and S16, respectively.
With the exception of RNAup, all tools ran in a few seconds to under a minute. Notably, GUUGle, RIsearch and RNAduplex returned results effectively immediately up to the maximal input length of 1150 basepairs. IntaRNA, RNAplex-c, RNAplex-a and RNAcofold saw roughly linearly increasing runtimes up to roughly 10 s. Pairfold and RactIP had polynomial runtimes with the longest jobs finishing in under a minute. RNAup had runtimes several times larger than all other tools, with longer jobs taking several minutes.
For physical memory, most tools could comfortably run under a hundred or so megabytes, with RNAup being the exception taking several times more. GUUGle, RIsearch and RNAduplex again used negligible amounts of memory. Interestingly, both versions of RNAplex showed a constant memory usage, and it is unclear whether this is a consequence of the scanning-like algorithm, or a pre-allocation of memory whose limit we have yet to encounter. RNAcofold, Pairfold, RactIP and IntaRNA use increasing amounts of memory relative to each other, each with increasing memory usage as a function of input length.
Extrapolating from these performances, it is likely that several tools would see little problem when applied to larger genome-wide searches, while others would need to be modified or perhaps used in a secondary pass in a larger pipeline.
CONCLUSION
RNA–RNA interaction prediction has increasingly become a field of intense interest, driven by advances in sequencing technology, uncovering a vast number of novel non-coding RNAs. The potential of using RNA–RNA interactions in identifying ncRNA targets may help us determine its networks and functions in the cell.
Fast and accurate full genome computational RNA interaction target searches are a sought after goal, which we believe starts with a strong foundation of being able to accurately predict interactions sites given two transcripts. In this work, we have conducted the most comprehensive assessment of general RNA–RNA interaction prediction tools to date. For this, we have compiled a comprehensive benchmark data set, consisting of two biologically different types of functional and experimentally confirmed RRIs: bacterial sRNA–mRNA interactions that regulate translation, and yeast snoRNA–rRNA interactions that guide nucleotide modifications. Instead of artificially truncating input sequences around their known interaction sites, we provide the full query and target transcript sequence to simulate a realistic setting for de novo discoveries. We make our data set publicly available for future research and development of new tools.
Evaluating all tools against all interactions in our data set, we test not only the predictive accuracy of tools, but also the effects of various common settings seen in multiple tools. Of the four increasingly complex prediction strategies we grouped our tools into, those that did accessibility-based predictions generally fared the best, with IntaRNA consistently performing well across all data sets, RNAplex-a performing closely on many occasions and RNAup being an exception to this observation.
The effects of adding evolutionary conservation information to predictions is highly mixed, ranging from detrimental effects on the sRNA data set to impressive gains in performance on the untruncated snoRNA data set. We further observe that the addition of conservation information to accessibility information often results in an overall decrease in performance. This is unexpected given that the best methods for predicting RNA secondary structure work in a comparative way (by harnessing information on evolutionary conservation of base-pairs) and should also be seen as a warning as many of the current methods for predicting general RNA–RNA interaction deliberately employ similar ideas.
With the field's goal of applying RNA–RNA interaction prediction to full-genome searches of RNA targets, we conduct a controlled experiment by increasing the target sequence length. As expected, we observed large drops in prediction accuracy, resulting in implications for large-scale searches.
The comparatively new field of general RNA–RNA interaction prediction thus needs a range of novel ideas compared to RNA secondary structure prediction to address the challenges shown by our benchmark tests.
Current prediction accuracies that pale in comparison to general RNA secondary structure prediction algorithms. It may be that we have reached the theoretical limits that a generalized non-biology-specific prediction algorithm can achieve, and that further performance gains can only be achieved by developing tools specific to a biological class of interactions. Various existing tools for miRNA prediction already pursue this, taking advantage of binding motifs and highly specific interactions lengths. There are also tools such as PLEXY (64), which can accurately predict C/D snoRNA binding sites by using nucleotide sequence motifs to narrow down the window of prediction. Alternatively, recent advancements have been made in high-throughput RNA structure and interaction probing (65). These corresponding enzymatic probes and pairing constraints produced have the potential to greatly assist predictions, replacing pure in silico accessibility profiles with experimental binding evidence. Regardless, we hope that our assessment and the accompanying data set will help improve the current state-of-the-art in RNA–RNA interaction prediction.
Supplementary Material
Acknowledgments
This work was supported by grants to I.M.M. from the Natural Sciences and Engineering Research Council (NSERC) of Canada and from the Canada Foundation for Innovation. D.L. is funded by Canadian Institutes of Health Research/Michael Smith Foundation for Health Research Strategic Training Program in Bioinformatics at the University of British Columbia and the NSERC Postgraduate Scholarship.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: Natural Sciences and Engineering Research Council (NSERC) of Canada.
Conflict of interest statement. None declared.
REFERENCES
- 1.Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F., et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amaral P.P., Dinger M.E., Mercer T.R., Mattick J.S. The eukaryotic genome as an RNA machine. Science. 2008;319:1787–1789. doi: 10.1126/science.1155472. [DOI] [PubMed] [Google Scholar]
- 3.Mercer T.R., Mattick J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013;20:300–307. doi: 10.1038/nsmb.2480. [DOI] [PubMed] [Google Scholar]
- 4.Backofen R., Hess W.R. Computational prediction of sRNAs and their targets in bacteriA. RNA Biol. 2010;7:33–42. doi: 10.4161/rna.7.1.10655. [DOI] [PubMed] [Google Scholar]
- 5.Peterson S.M., Thompson J.A., Ufkin M.L., Sathyanarayana P., Liaw L., Congdon C.B. Common features of microRNA target prediction tools. Front. Genet. 2014;5:1–10. doi: 10.3389/fgene.2014.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meyer I.M. Predicting novel RNA-RNA interactions. Curr. Opin. Struc. Biol. 2008;18:387–393. doi: 10.1016/j.sbi.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Seemann S.E., Richter A.S., Gesell T., Backofen R., Gorodkin J. PETcofold: predicting conserved interactions and structures of two multiple alignments of RNA sequences. Bioinformatics. 2011;27:211–219. doi: 10.1093/bioinformatics/btq634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lorenz R., Bernhart S.H., Höner Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tafer H., Hofacker I.L. RNAplex: a fast tool for RNA-RNA interaction search. Bioinformatics. 2008;24:2657–2663. doi: 10.1093/bioinformatics/btn193. [DOI] [PubMed] [Google Scholar]
- 10.Wenzel A., Akbasli E., Gorodkin J. RIsearch: fast RNA-RNA interaction search using a simplified nearest-neighbor energy model. Bioinformatics. 2012;28:2738–2746. doi: 10.1093/bioinformatics/bts519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerlach W., Giegerich R. GUUGle: a utility for fast exact matching under RNA complementary rules including G-U base pairing. Bioinformatics. 2006;22:762–764. doi: 10.1093/bioinformatics/btk041. [DOI] [PubMed] [Google Scholar]
- 12.McCaskill J.S. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers. 1990;29:1105–1119. doi: 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- 13.Bernhart S.H., Mückstein U., Hofacker I.L. RNA Accessibility in cubic time. Algorithms Mol. Biol. 2011;6:3. doi: 10.1186/1748-7188-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mückstein U., Tafer H., Hackermüller J., Bernhart S.H., Stadler P.F., Hofacker I.L. Thermodynamics of RNA-RNA binding. Bioinformatics. 2006;22:1177–1182. doi: 10.1093/bioinformatics/btl024. [DOI] [PubMed] [Google Scholar]
- 15.Busch A., Richter A.S., Backofen R. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics. 2008;24:2849–2856. doi: 10.1093/bioinformatics/btn544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tafer H., Amman F., Eggenhofer F., Stadler P.F., Hofacker I.L. Fast accessibility-based prediction of RNA-RNA interactions. Bioinformatics. 2011;27:1934–1940. doi: 10.1093/bioinformatics/btr281. [DOI] [PubMed] [Google Scholar]
- 17.Zuker M., Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981;9:133–148. doi: 10.1093/nar/9.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hofacker I.L., Fontana W., Stadler P.F., Bonhoeffer L.S., Tacker M., Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh. Chem. 1994;125:167–188. [Google Scholar]
- 19.Andronescu M., Zhang Z.C., Condon A. Secondary structure prediction of interacting RNA molecules. J. Mol. Biol. 2005;345:987–1001. doi: 10.1016/j.jmb.2004.10.082. [DOI] [PubMed] [Google Scholar]
- 20.Bernhart S.H., Tafer H., Mückstein U., Flamm C., Stadler P.F., Hofacker I.L. Partition function and base pairing probabilities of RNA heterodimers. Algorithms Mol. Biol. 2006;1:3. doi: 10.1186/1748-7188-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kato Y., Sato K., Hamada M., Watanabe Y., Asai K., Akutsu T. RactIP: fast and accurate prediction of RNA-RNA interaction using integer programming. Bioinformatics. 2010;26:i460–i466. doi: 10.1093/bioinformatics/btq372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knudsen B. Pfold: RNA secondary structure prediction using stochastic context-free grammars. Nucleic Acids Res. 2003;31:3423–3428. doi: 10.1093/nar/gkg614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernhart S.H., Hofacker I.L., Will S., Gruber A.R., Stadler P.F. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics. 2008;9:474. doi: 10.1186/1471-2105-9-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baek D., Villén J., Shin C., Camargo F.D., Gygi S.P., Bartel D.P. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alexiou P., Maragkakis M., Papadopoulos G.L., Reczko M., Hatzigeorgiou A.G. Lost in translation: An assessment and perspective for computational microrna target identification. Bioinformatics. 2009;25:3049–3055. doi: 10.1093/bioinformatics/btp565. [DOI] [PubMed] [Google Scholar]
- 26.Eggenhofer F., Tafer H., Stadler P.F., Hofacker I.L. RNApredator: fast accessibility-based prediction of sRNA targets. Nucleic Acids Res. 2011;39:W149–W154. doi: 10.1093/nar/gkr467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wright P.R., Richter A.S., Papenfort K., Mann M., Vogel J., Hess W.R., Backofen R., Georg J. Comparative genomics boosts target prediction for bacterial small RNAs. Proc. Natl. Acad. Sci. U.S.A. 2013;110:E3487–E3496. doi: 10.1073/pnas.1303248110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pain A., Ott A., Amine H., Rochat T., Bouloc P., Gautheret D. An assessment of bacterial small RNA target prediction programs. RNA Biol. 2015;12:509–513. doi: 10.1080/15476286.2015.1020269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montaseri S., Zare-Mirakabad F., Moghadam-Charkari N. RNA-RNA interaction prediction using genetic algorithm. Algorithms Mol. Biol. 2014;9:17. doi: 10.1186/1748-7188-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salari R., Mathias M., Will S. Time and space efficient RNA-RNA interaction prediction via sparse folding. Lect. Notes Comput. Sci. 2010;6044:473–490. [Google Scholar]
- 31.Huang F.W.D., Qin J., Reidys C.M., Stadler P.F. Partition function and base pairing probabilities for RNA-RNA interaction prediction. Bioinformatics. 2009;25:2646–2654. doi: 10.1093/bioinformatics/btp481. [DOI] [PubMed] [Google Scholar]
- 32.Li A.X., Marz M., Qin J., Reidys C.M. RNA-RNA interaction prediction based on multiple sequence alignments. Bioinformatics. 2011;27:456–463. doi: 10.1093/bioinformatics/btq659. [DOI] [PubMed] [Google Scholar]
- 33.Pervez M.T., Babar M., Nadeem A., Aslam M., Awan A., Aslam N., Hussain T., Naveed N., Qadri S., Waheed U., et al. Evaluating the accuracy and efficiency of multiple sequence alignment methods. Evol. Bioinform. 2014;10:205–217. doi: 10.4137/EBO.S19199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pais F.S.M., Ruy P.D.C., Oliveira G., Coimbra R.S. Assessing the efficiency of multiple sequence alignment programs. Algorithm. Mol. Biol. 2014;9:4. doi: 10.1186/1748-7188-9-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Do C.B. ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome. Res. 2005;15:330–340. doi: 10.1101/gr.2821705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Will S., Reiche K., Hofacker I.L., Stadler P.F., Backofen R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 2007;3:e65. doi: 10.1371/journal.pcbi.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Will S., Otto C., Miladi M., Möhl M., Backofen R. SPARSE: quadratic time simultaneous alignment and folding of RNAs without sequence-based heuristics. Bioinformatics. 2015;31:2489–2496. doi: 10.1093/bioinformatics/btv185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andronescu M., Bereg V., Hoos H.H., Condon A. RNA STRAND: the RNA secondary structure and statistical analysis database. BMC Bioinformatics. 2008;9:340. doi: 10.1186/1471-2105-9-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burge S.W., Daub J., Eberhardt R., Tate J., Barquist L., Nawrocki E.P., Eddy S.R., Gardner P.P., Bateman A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013;41:D226–D232. doi: 10.1093/nar/gks1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Storz G., Opdyke J.A., Zhang A. Controlling mRNA stability and translation with small, noncoding RNAs. Curr. Opin. Microbiol. 2004;7:140–144. doi: 10.1016/j.mib.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 42.Chitsaz H., Salari R., Sahinalp S.C., Backofen R. A partition function algorithm for interacting nucleic acid strands. Bioinformatics. 2009;25:i365–i373. doi: 10.1093/bioinformatics/btp212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peer A., Margalit H. Accessibility and evolutionary conservation mark bacterial small-rna target-binding regions. J. Bacteriol. 2011;193:1690–1701. doi: 10.1128/JB.01419-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richter A.S., Backofen R. Accessibility and conservation: general features of bacterial small RNA-mRNA interactions. RNA Biol. 2012;9:954–965. doi: 10.4161/rna.20294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao Y., Wu J., Liu Q., Zhao Y., Ying X., Cha L., Wang L., Li W. sRNATarBase: a comprehensive database of bacterial sRNA targets verified by experiments. RNA. 2010;16:2051–2057. doi: 10.1261/rna.2193110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tatusova T., Ciufo S., Federhen S., Fedorov B., McVeigh R., O'Neill K., Tolstoy I., Zaslavsky L. Update on RefSeq microbial genomes resources. Nucleic Acids Res. 2014;43:D599–D605. doi: 10.1093/nar/gku1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bachellerie J.P., Cavaillé J., Hüttenhofer A. The expanding snoRNA world. Biochimie. 2002;84:775–790. doi: 10.1016/s0300-9084(02)01402-5. [DOI] [PubMed] [Google Scholar]
- 48.Tafer H., Kehr S., Hertel J., Hofacker I.L., Stadler P.F. RNAsnoop: efficient target prediction for H/ACA snoRNAs. Bioinformatics. 2010;26:610–616. doi: 10.1093/bioinformatics/btp680. [DOI] [PubMed] [Google Scholar]
- 49.Lowe T.M., Eddy S.R. A computational screen for methylation guide snoRNAs in yeast. Science. 1999;283:1168–1171. doi: 10.1126/science.283.5405.1168. [DOI] [PubMed] [Google Scholar]
- 50.Piekna-Przybylska D., Decatur W.A., Fournier M.J. New bioinformatic tools for analysis of nucleotide modifications in eukaryotic rRNA. RNA. 2007;13:305–312. doi: 10.1261/rna.373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cherry J.M., Hong E.L., Amundsen C., Balakrishnan R., Binkley G., Chan E.T., Christie K.R., Costanzo M.C., Dwight S.S., Engel S.R., et al. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 2012;40:D700–D705. doi: 10.1093/nar/gkr1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kanehisa M., Goto S., Sato Y., Furumichi M., Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hertel J., de Jong D., Marz M., Rose D., Tafer H., Tanzer A., Schierwater B., Stadler P.F. Non-coding RNA annotation of the genome of Trichoplax adhaerens. Nucleic Acids Res. 2009;37:1602–1615. doi: 10.1093/nar/gkn1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lai D., Proctor J.R., Zhu J.Y.A., Meyer I.M. R-CHIE: a web server and R package for visualizing RNA secondary structures. Nucleic Acids Res. 2012;40:e95. doi: 10.1093/nar/gks241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gardner P.P., Giegerich R. A comprehensive comparison of comparative RNA structure prediction approaches. BMC Bioinformatics. 2004;5:140. doi: 10.1186/1471-2105-5-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matthews B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta. 1975;405:442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- 57.Gorodkin J., Stricklin S.L., Stormo G.D. Discovering common stem-loop motifs in unaligned RNA sequences. Nucleic Acids Res. 2001;29:2135–2144. doi: 10.1093/nar/29.10.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wickham H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011;3:180–185. [Google Scholar]
- 59.Hofacker I.L., Fekete M., Stadler P.F. Secondary structure prediction for aligned RNA sequences. J. Mol. Biol. 2002;319:1059–1066. doi: 10.1016/S0022-2836(02)00308-X. [DOI] [PubMed] [Google Scholar]
- 60.Cannone J.J., Subramanian S., Schnare M.N., Collett J.R., D'Souza L.M., Du Y., Feng B., Lin N., Madabusi L.V., Müller K.M., et al. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3:2. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anandam P., Torarinsson E., Ruzzo W.L. Multiperm: shuffling multiple sequence alignments while approximately preserving dinucleotide frequencies. Bioinformatics. 2009;25:668–669. doi: 10.1093/bioinformatics/btp006. [DOI] [PubMed] [Google Scholar]
- 62.Felsenstein J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- 63.Knies J.L., Dang K.K., Vision T.J., Hoffman N.G., Swanstrom R., Burch C.L. Compensatory evolution in RNA secondary structures increases substitution rate variation among sites. Mol. Biol. Evol. 2008;25:1778–1787. doi: 10.1093/molbev/msn130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kehr S., Bartschat S., Stadler P.F., Tafer H. PLEXY: efficient target prediction for box C/D snoRNAs. Bioinformatics. 2011;27:279–280. doi: 10.1093/bioinformatics/btq642. [DOI] [PubMed] [Google Scholar]
- 65.Helwak A., Tollervey D. Mapping the miRNA interactome by cross-linking ligation and sequencing of hybrids (CLASH) Nat. Protoc. 2014;9:711–728. doi: 10.1038/nprot.2014.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.