


Abstract
G-quadruplexes (G4) are nucleic acid conformations of guanine-rich sequences, in which guanines are arranged in the square-planar G-tetrads, stacked on one another. G4 motifs form in vivo and are implicated in regulation of such processes as gene expression and chromosome maintenance. The structure and stability of various G4 topologies were determined experimentally; however, the driving forces for their formation are not fully understood at the molecular level. Here, we used all-atom molecular dynamics to probe the microscopic origin of the G4 motif stability. By computing the free energy profiles governing the dissociation of the 3′-terminal G-tetrad in the telomeric parallel-stranded G4, we examined the thermodynamic and kinetic stability of a single G-tetrad, as a common structural unit of G4 DNA. Our results indicate that the energetics of guanine association alone does not explain the overall stability of the G-tetrad and that interactions involving sugar–phosphate backbone, in particular, the constrained minimization of the phosphate–phosphate repulsion energy, are crucial in providing the observed enthalpic stabilization. This enthalpic gain is largely compensated by the unfavorable entropy change due to guanine association and optimization of the backbone topology.
INTRODUCTION
Certain guanine-rich DNA (and RNA) sequences have long been known to fold into intra- or inter-molecular four-stranded secondary structures called G-quadruplexes (G4) (1,2). Recently, G4 have been also detected in ciliate and mammalian cells (3–5), where they are believed to be involved in regulation of DNA replication and transcription, genetic recombination, maintaining chromosome stability and other cellular processes (6–9). In particular, the formation of G4 motifs at the single-stranded telomeric 3′-overhang leads to telomerase inhibition (10) and competes with the binding of the ssDNA overhang by POT1, a component of shelterin protein complex protecting chromosomal ends (11–13). Accordingly, G4-stabilizing agents, such as telomestatin, have been shown to impair telomere homeostasis and thereby to induce growth arrest or apoptosis in cultured cells (14–16). As a result, G4 DNA is now considered a promising target for anticancer therapy (17–19). G4-forming sequences are also found to be over-represented in other regulatory regions of the genome, including promoters (20,21), introns (22), mitotic and meiotic double-strand break sites (6), immunoglobulin switch regions (9) and 5′-UTRs (23). To fully understand the role of G4 in cell biology and to allow the efficient use of G4-interactive ligands, it is necessary to understand structural diversity, conformational dynamics and molecular determinants of the stability of these non-canonical DNA structural motifs.
A common structural feature of G4 DNA is the presence of at least two stacked guanine tetrads (G-tetrads) with four co-planar guanine bases, interconnected through cyclic Hoogsteen-type hydrogen bonding and additionally stabilized by coordination of monovalent cations to guanine O6 carbonyl groups (Figure 1A) (24). In intramolecular G4, the stacked G-tetrads are formed by (typically) contiguous guanine runs separated by intervening loops of variable length and sequence. Based on the relative G-strand orientation, G4 motifs are classified into parallel, antiparallel and hybrid conformational classes, in which a variety of different folding topologies are possible (24). In particular, human telomeric G4 has been shown by x-ray crystallography and nuclear magnetic resonance spectroscopy to adopt six distinct conformations containing three (or in one case, two) G-tetrads and the three-nucleotide TTA loops, and differing in strand orientation, glycosidic conformation of guanines, loop arrangement, etc. (25–30).
Figure 1.

(A) Initial conformation of the simulated parallel-stranded G-quadruplex (G4) of human telomeric sequence, taken from the x-ray structure (26): guanine residues forming the 3′-terminal G-tetrad are indicated in blue (I), green (II), purple (III) and red (IV); the two remaining G-tetrads are shown in gray (middle) and orange (5′-terminal), thymine and adenine residues in brown and the sugar–phosphate backbone in yellow; two potassium ions coordinated in the central channel of the G4 are presented as cyan van der Waals spheres. The same color-coding is used throughout the article. (B) Chemical structure of a single G-tetrad in which four guanine nucleobases are linked together through cyclic Hoogsteen-type hydrogen bonding. Black and gray arrows show the orientation of the dipole moment of guanine IV and the guanine located directly below it, respectively. (C) Schematic representation of the parallel-stranded G4 showing the location of the 3′-terminal G-tertrad guanines (I–IV) along the DNA chain.
Calorimetric and thermal denaturation measurements combined with spectroscopic data show, on the one hand, that biologically relevant G4 motifs are characterized by a high thermodynamic stability (24,31,32), similar in magnitude to the stability of the competing Watson–Crick pairing (33). On the other hand, the unfolding free energies do not vary greatly among accessible conformational states (24), and consequently the equilibrium between them depends sensitively on various factors, including the G-tract length (34), the size and base composition of the loops (33,35), type of cations (24,26), molecular crowding (36,37) and extent of hydration (36,37). Since the transitions between different states are slow, ranging from seconds to hours (37–39), G4 folding in cellular processes might be under kinetic rather than thermodynamic control (40), adding another layer of complexity to the study of G4 conformational behavior.
Recent extensive MD simulations allowed to sample conformational dynamics of different G4 topologies and confirmed their stability on μs time scale (41–43). The balance of inter- and intra-molecular forces underlying this stable behavior is, however, much less understood. Quantum chemical and force field calculations on model systems (oligonucleotides or nuclebases assemblies) provided an important insight on how the relative stability of the G-tetrad stack depends on such parameters as the strand polarity, glycosidic conformations and stacking distance, angle and orientation (44–46). These studies predicted energetic preference for certain guanine arrangements, in agreement with the distribution of conformations found in experimentally solved structures. Only recently, it has become possible to directly assess individual contributions to G4 stabilization using single-molecule experiments. Notably, Ghimire et al. used click-chemistry-assisted laser-tweezer stretching to show that loop interactions might be at least as important for the G4 stability as guanine association and stacking (47). Despite the above efforts, a complete and comprehensive picture of G4 stabilization, based on structural information and providing molecular-level understanding of driving forces for G4 folding, is yet to emerge.
To address this problem, here we probe the determinants of G4 DNA stability by computing the first, to our knowledge, free energy profile for the dissociation of a single G-tetrad in the context of a complete G4 structure. To this end, we use explicit-solvent, all-atom molecular dynamics combined with umbrella sampling (US), with a total simulation time of 35 μs, to capture conformational fluctuations of a flexible DNA molecule. As a model system, we chose the 3′-terminal G-tetrad in a parallel-stranded telomeric G4, which was shown to be the most stable conformation under crowded conditions and therefore may be predominant form of G4 in genomic DNA (36,37). Based on the free energy profile, we estimate the thermodynamics and kinetics of G-tetrad dissociation and re-assembly, and predict that both proceed by a sequential and cooperative mechanism dependent on cation binding. The G-tetrad formation is found to be enthalpically driven, with the dominant favorable contribution provided by the interactions involving the sugar-phosphate backbone rather than guanine residues. This overall enthalpic gain is largely compensated by a decrease in entropy due to the association of guanines and optimization of the backbone geometry.
MATERIALS AND METHODS
Molecular systems and simulation procedure
Initial coordinates of the parallel-stranded G4 were taken from the 2.1-Å resolution crystal structure of the 22-bp long single-stranded DNA with the human telomeric sequence d[AGGG(TTAGGG)3], (PDB code: 1KF1) (Figure 1A and C) (26). The DNA structure was solvated with 11 469 TIP3P water molecules (48) in dodecahedron box with a minimal distance between any solute atom and the edge of the box of 1.2 nm, and at physiological ionic strength (150 mM KCl). The CHARMM27 force field (49) was used for DNA and ions.
The MD simulations were run using Gromacs (50) in the isothermal-isobaric (NPT) ensemble with the reference temperature and pressure of 300 K and 1 bar, respectively. Periodic boundary conditions were applied in 3D, and electrostatic interactions were calculated using the particle mesh Ewald (PME) method with a real-space cutoff of 1 nm and a Fourier grid spacing of 0.1 nm. A cut-off of 1 nm was used for Lennard–Jones interactions. Bond lengths were constrained using P-LINCS (51) for DNA and SETTLE (52) for water. The equations of motion were integrated using the leap-frog algorithm with a 2 fs time step. Prior to the actual free energy calculations, the system was subject to 1 μs equilibrium MD simulation. All molecular images were created using VMD (53).
Steered molecular dynamics
To prepare the initial configurations of the system for the free energy simulations, the entire 3′-terminal G-tetrad of the parallel-stranded G4 structure (guanine residues denoted as I, II, III and IV in Figure 1C) was forced to dissociate over 100 ns by applying an external harmonic potential (with the force constant of 2092 kcal/(mol·nm2)) to the collective variable defined as the total number of native hydrogen bonds (h-bonds), Nhb, formed within this G-tetrad (Figure 1B). This procedure was carried out using the PLUMED 1.3 plugin (54) coupled to Gromacs 4.5.5. Technically, Nhb was approximated as a sum of switching functions (see Supplementary Figure S1 in Supplementary Data) for all eight donor-acceptor pairs (N1–O6 and N2–N7) forming the native h-bonds in the G-tetrad plane and therefore it varied between 8 (for the fully formed G-tetrad) and 0 (for the fully dissociated G-tetrad).
Free energy simulations
To examine the stability of the 3′-terminal G-tetrad, we used the US method to determine the free energy profile along the collective coordinate chosen as the total number of native h-bonds within the G-tetrad. To sample the natural range of this reaction coordinate (0–8 h-bonds) we extracted 33 initial equally-spaced G4 configurations from a trajectory generated in the steered MD run. In each of these US windows, the system was simulated for 1 μs, using the harmonic potential with a force constant of 1255.2 kcal/(mol·nm2) to restrain the system along the collective coordinate. Free energy profiles were determined from the last 900 ns of thus obtained trajectories using the standard weighted histogram analysis method (WHAM)(55). Uncertainties were estimated using bootstrap error analysis taking into account the correlation in the analyzed time series.
RESULTS AND DISCUSSION
Free energy profile for the guanine tetrad dissociation and re-assembly
To examine the conformational stability of a common structural unit of G4 DNA, we computed the free energy profile for the dissociation of the 3′-terminal G-tetrad in the telomeric parallel-stranded G4 structure (Figure 1). As the the collective coordinate describing the studied conformational transition, we used the total number of native h-bonds formed within the G-tetrad (Nhb).
It can be seen from the free energy profile in Figure 2A that the 3′-terminal G-tetrad in its associated state, defined by 8 native h-bonds (state 4 in Figure 2), is ∼12 kcal/mol more stable than the fully dissociated state with 0 native h-bonds (state 1 in Figure 2). Figure 2B and Supplementary Figure S2 show that in the latter state, the G-tetrad-forming guanine residues are typically exposed to water while often engaging into stacking interactions with thymines in the TTA loops (in brown), with the neighboring dissociated guanines, and with the middle G-tetrad (in gray). It should be also noted that even after complete dissociation of the 3′-terminal G-tetrad, the rest of the G4 structure is very stable on the μs-timescale, with no significant perturbations observed within the guanine core, in agreement with the experimental data reporting the formation of stable G4 structures containing only two G-tetrads (35).
Figure 2.

(A) Free energy profile for the dissociation/re-assembly of the 3′-terminal G-tetrad in the parallel-stranded G4 structure along the collective coordinate defined as the total number of the native h-bonds between guanines I–IV, Nhb. Schematic representations (1–4) show typical structures of four conformational states of G4 along Nhb. (B) Representative ensembles of 20 overlaid snapshots corresponding to states 1–4 depicting the changes in the structure and dynamics of the G4 along the dissociation/re-assembly pathway.
Figure 2A also demonstrates that during the transition from the global free energy minimum at 8 h-bonds (state 4) to a fully dissociated state at 0 h-bonds (state 1) the system passes through two local minima around the 4 h-bonds (state 3) and 2 h-bonds (state 2). As might be expected, the cost of detachment of the first guanine (6 kcal/mol), associated with breaking 4 h-bonds, is approximately two times greater than for the two subsequent ones (3 kcal/mol) for which only 2 h-bonds are being broken. Accordingly, we find, by integrating the probability density ρ(Nhb)∝exp [ − βG(Nhb)], where G(Nhb) is the free energy profile, that equilibrium probability of a single guanine dissociating from the fully formed G-tetrad due to thermal fluctuations is about 0.01 % whereas the probability that the next guanine will dissociate is markedly larger and is equal to 0.6 %.
The observed shape of the free energy profile further suggests that the dissociation as well as the re-assembly of the 3′-terminal G-tetrad proceed in a sequential manner where the individual guanine residues detach/attach one by one. The mean first passage time of 250 ns estimated for the detachment of the first guanine (see Supplementary Data for details) shows that this is the slowest step of the sequential dissociation, with the rate constant koff = 0.004 ns−1, and hence it determines the kinetic stability of the tetrameric structure. The largest tendency to dissociate from the fully formed G-tetrad is observed for guanine I (blue in Figure 2), located closest to the 5′-end of the DNA chain, with guanine IV (the 3′-end residue, red in Figure 2) contributing only very slightly to the state with a single detached guanine. These observations are consistent with the results of the additional 4 μs equilibrium simulation in which a spontaneous dissociation of guanine I was observed after 750 ns, as seen from the hydrogen bonding evolution in Supplementary Figure S3. Although the free energy profile suggests that the detachment of the second guanine (guanine II or, to a lesser extent, guanine IV; Figure 2) is one order of magnitude faster, with the mean first passage time of ∼40 ns (koff = 0.025−1), we did not observe such dissociation events in our equilibrium simulation. Closer inspection revealed that this unexpected stability results from the formation of guanine triplet in which each guanine residue participates in three, generally non-native, h-bonds with the two other residues (Supplementary Figure S4). This suggests that the intermediate state with three associated guanines may actually be more stable than indicated by the free energy profile along the Nhb coordinate, which encourages a guanine triplet (state 3 in Figure 2) with two flanking guanines (II and IV) forming only 2 h-bonds with the central one (guanine III). Consequently, dissociation of the second guanine might in fact be significantly slower than that inferred from the free energy profile.
Mechanism of the G-tetrad dissociation
In order to investigate in detail, the sequence of events in the stepwise mechanism of dissociation of the 3′-terminal G-tetrad, we calculated how the probabilities of formation of the native guanine–guanine h-bonds depend on Nhb. To this end, the original (biased) US data were reweighted to recover the unbiased probabilities, using weights of the form exp [(Vi(Nhb) − Fi)/kBT], where Vi and Fi are the biasing potential and the WHAM-derived free energy constant, respectively, both corresponding to the i-th window.
The h-bond probabilities shown in Figure 3A confirm a sequential nature of the dissociation process with the simultaneous breaking of 4 h-bonds for the first detaching guanine and 2 h-bonds for the two subsequent ones. As it was already mentioned, the highest tendency to dissociate from the fully formed G-tetrad is seen for guanine I (blue in Figure 3C). Detachment of guanine I is followed, with 0.8 probability, by the dissociation of guanine II (green) and, finally, by breaking the contact between guanine III (purple) and guanine IV (red).
Figure 3.

(A) Probabilities of formation of 24 native h-bonds between guanines as a function of Nhb. H-bonds were identified via geometric criteria, i.e. donor–acceptor distance < 3.5 Å and donor–H–acceptor angle < 40.0°. (B) Probabilities of finding K+ ion in four ion-binding sites along the G4 central channel as a function of Nhb. Numbering of the h-bonds within the 3′-terminal G-tetrad (1–8) and the ion-binding sites (1–4) is shown in panels (C) and (D), respectively. Gray and orange colors represent the middle and 5′-terminal G-tetrad, respectively. Green arrows show states 1–4, as defined in Figure 2.
Figure 3 a also shows that in response to dissociation of the first and second guanine from the 3′-terminal G-tetrad the probability of forming the remaining h-bonds within the partially assembled G-tetrad gradually decreases down to ∼0.5. This decrease indicates that the G-tetrad-stabilizing h-bonds have a cooperative nature and, consequently, planar structures formed by less than four guanines are generally characterized by reduced structural stability. The two other G-tetrads in G4 (orange and gray in Figure 3A) generally remain intact throughout the dissociation process, even though a slight reduction of the h-bonds probabilities can be observed, especially around the detachment events.
There are two types of h-bonds stabilizing the G-tetrad: (i) h-bonds formed between the N2 amino group and N7 acceptor (‘outer h-bonds’ in Figure 1B, labeled as 2, 4, 6 and 8 in Figure 3C), and (ii) h-bonds between the N1 imino group and the O6 acceptor (‘inner h-bonds’, labeled 1, 3, 5 and 7). Figure 3A reveals that the two types of hydrogen bonds are not equivalent in terms of their stability during the dissociation process. More specifically, only in the fully formed G-tetrad the outer and inner h-bonds form with equally high probability. For the intermediate states, however, the outer h-bonds are considerably more stable. This stability seems to be related to the geometric constraints within the Hoogsteen-bonded G-tetrad that result in the outer h-bonds being shorter (the average donor–acceptor distance of 2.98 Å) and more collinear (the average acceptor–donor–hydrogen angle of 15°) than it is the case for the inner h-bonds (3.10 Å and 24°, respectively). Involvement of the O6 atoms in coordinating K+ ion may also partially reduce their h-bonding potential.
It has been shown that monovalent cations, such as K+, Na+ or NH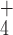 , are essential for the formation of G4, as they tend to coordinate between the stacked G-tetrads to the eight O6 carbonyl groups exposed to the central pore (see Figure 1), and thereby provide additional stabilization (57–59). To investigate changes in the occupancy of the ion-binding sites during the transition from the dissociated state to the fully formed G-tetrad, we calculated the probability of individual sites being occupied by a K+ cation as a function of the conformational coordinate Nhb. The unbiased probabilities were computed from the original US data using the same reweighting procedure as applied above for the h-bonds. A given binding site was considered occupied if any K+ ion was found within 0.15 nm from the geometrical center of the site. Since in our equilibrium simulations of the parallel-stranded G4 K+ showed a certain tendency to bind to the four O6 carbonyl groups of the external G-tetrads (positions 1 and 4 in Figure 3D), these two sites were also included in the analysis.
, are essential for the formation of G4, as they tend to coordinate between the stacked G-tetrads to the eight O6 carbonyl groups exposed to the central pore (see Figure 1), and thereby provide additional stabilization (57–59). To investigate changes in the occupancy of the ion-binding sites during the transition from the dissociated state to the fully formed G-tetrad, we calculated the probability of individual sites being occupied by a K+ cation as a function of the conformational coordinate Nhb. The unbiased probabilities were computed from the original US data using the same reweighting procedure as applied above for the h-bonds. A given binding site was considered occupied if any K+ ion was found within 0.15 nm from the geometrical center of the site. Since in our equilibrium simulations of the parallel-stranded G4 K+ showed a certain tendency to bind to the four O6 carbonyl groups of the external G-tetrads (positions 1 and 4 in Figure 3D), these two sites were also included in the analysis.
It can be seen from the probabilities in Figure 3B that when the 3′-terminal G-tetrad is completely dissociated (state 1), the binding site formed by the two remaining G-tetrads (position 3) is occupied with probability close to 1, as expected. Remarkably, upon association of the first two guanines of the 3′-terminal G-tetrad, i.e. upon reaching state 2 in Figure 2, the occupancy probability of position 2, between the 3′-terminal guanine plane and the middle plane increases markedly up to ca. 0.7, largely at the expense of binding site 3. This result indicates that the presence of six O6 carbonyl groups within binding site 2 is apparently sufficient to effectively attract a potassium ion from position 3. Attachment of the next two guanines results in a further increase of the binding site 2 occupancy, with the probability eventually approaching unity for the fully formed 3′-terminal G-tetrad in state 4. Concomitant with this increase, the occupancy probability of binding site 3 drops to almost zero. A clear preference to occupy position 2 rather than position 3 was also seen in our equilibrium simulations which showed that coordination of a single K+ ion in the central channel is sufficient to keep the parallel-stranded G4 stable on the microsecond time scale (Supplementary Figure S5). To further evaluate this finding, we computed the free energy profile for the transition of a single K+ between site 2 and 3 using an additional metadynamics simulation (see Supplementary Data). The resulting free energy function, shown in Supplementary Figure S6, confirms that both sites are not equivalent in terms of their affinity for K+ cations, with binding at site 2 being by ∼2 kcal/mol more favorable relative to site 3. Interestingly, the prevalence of the native structure (state 4) with a single ion bound in the central pore indicates that coordination of K+ in one site considerably reduces the ion binding affinity of another, due to electrostatic repulsion. It should be kept in mind that this ion exclusion is probably overemphasized in our model, as most additive force fields tend to overestimate cation–cation repulsion, as described by Islam et al.(43).
Nevertheless, the observation that a G-tetrad can be stable on the microsecond time scale even in the absence of a monovalent ion directly coordinated to it raises a question about the determinants of this stability. It is worth mentioning, however, that the presence of at least one K+ inside the channel is found necessary for stabilizing the parallel G4 in our simulations and whenever the overall occupancy probability falls below 1 (e.g. Nhb in the range of 0.7–1.2 or 2.2–2.6), the stability of the entire structure is reduced, as reflected in the inter-guanine h-bond probabilities in Figure 3A.
Figure 3B also shows that position 4, outside the 5′-terminal G-tetrad, is also quite frequently occupied by K+, especially in the native state (Nhb in the range 7–8), in which position 3 is generally not occupied and therefore does not contribute unfavorably to the ion binding energy at position 4. Coordination of K+ in position 4 seems to be facilitated by the adenine residues that tend to stack onto the 5′-terminal G-tetrad (see Figure 2B and Supplementary Figure S2), thereby forming the binding cavity and providing additional ion-coordinating groups (see Supplementary Figure S7 for a typical structure). Indeed, when such stacking does not occur, as it is the case at the 3′-terminal G-tetrad due to steric reasons, no significant ion binding can be observed (Supplementary Figure S5).
Energetic determinants of the G-tetrad stability
To identify the major energetic determinants of the stability of G4, we decomposed the enthalpy of formation of the 3′-terminal G-tetrad, i.e. the enthalpy difference between states 4 and 1 in Figure 2A, into contributions due to interactions between individual structural elements of G4. The contributions were calculated as the differences of the electrostatic and van der Waals interaction energies averaged over the unbiased ensembles in the respective states and are shown in Supplementary Figure S8 and 9.
It can be seen from Supplementary Figure S8 that the 4-fold symmetry of the intramolecular parallel-stranded G4 (with the symmetry axis passing through a central channel) is clearly reflected in the individual enthalpic contributions. Indeed, most symmetry-equivalent interaction energies, e.g. between the guanine moiety and the adjacent guanines in the G-tetrad or between the phosphate group and the rest of the DNA backbone, have similar values for all four guanine nucleotides (I–IV). Therefore, to characterize the average energetic stabilizing force felt by a single guanine nucleotide, we averaged the symmetry-equivalent energy contributions shown in Supplementary Figure S8 and thus decomposed the average enthalpy change accompanying the attachment of a single guanine nucleotide to the G-tetrad (Figure 4).
Figure 4.

Contributions to the enthalpy change upon G-tetrad formation due to interactions between guanine nucleotides I–IV, divided into guanine (G), phosphate (P) and deoxyribose (dR), and the rest of the system. Symmetry-equivalent interaction energies were averaged to get the enthalpic contributions corresponding to the attachment of a single guanine nucleotide to the G-tetrad. Subscripts ‘wc’ and ‘h’ denote the interaction with the guanine nucleotide that contacts with a given one with its Watson–Crick and Hoogsteen edge, respectively. Subscript ‘d’ denotes the interaction with diagonally opposite guanine nucleotide, and no subscript denotes the intra-nucleotide term. The rest of the G4 was divided into: guanine residues of the middle (PLANE2) and 5′-terminal (PLANE3) G-tetrads, adenine (ADE) and thymine (THY) residues, and the sugar–phosphate backbone (DNA BB).
Both the local contributions due to the guanine nucleotides I–IV (Figure 4) and the global contributions involving the rest of the system (Supplementary Figure S9) reveal that the enthalpy of formation of the G-tetrad is a result of a fine balance between competing interactions. As expected, the guanine–guanine h-bonds stabilize the G-tetrad plane by 11 kcal/mol per residue, with the the Hoogsteen and Watson–Crick edges of a given guanine providing the same stabilization, due to the symmetry of the structure. This energetic gain is, however, more than offset by the loss of energetically favorable interactions between guanines and water (15 kcal/mol per residue). Unexpectedly, also the interactions between the guanines I–IV and the middle and 5′-terminal G-tetrads is unfavorable, with the energy changes of 2.0 and 1.0 kcal/mol per residue, respectively. This increase stems from a parallel arrangement of the large dipole moments of stacked guanines (6.71 D; see Figure 1B) resulting in electrostatic repulsion, which is only partially counterbalanced by attractive van der Waals interaction. Therefore, an additional stabilization of 7.5 kcal/mol per residue provided by coordination of K+ ion between the planes (position 2 in Figure 3D) can in fact be necessary for promoting the G-tetrad formation, in accordance with the well-known experimental fact (38,57,59). This ion-induced stabilization might in fact be even more pronounced, given the possible underestimation of the guanine-cation attraction (43). Importantly, a delicate balance in the interactions between guanine residues and their immediate environment resulting in the overall energetic stabilization of the G-tetrad (∼2 kcal/mol) should be largely independent of the G4 topology and details of a dissociation pathway and hence can be expected to be crucial for the stability of other G-tetrads as well.
Notably, the above stabilization due to local guanine interactions is only marginal—even more so given that the conformational entropy change upon the G-tetrad formation is necessarily unfavorable (see also the next section). Therefore, longer-range interactions, possibly involving the intervening loops, might be at least equally important for the G4 stability, as has been also reported by others (47). Accordingly, we find in particular that the formation of the 3′-terminal G-tetrad in the the parallel-stranded G4 is additionally promoted by the favorable orientation of the guanine dipole moment with respect to the phosphate groups of the Hoogsteen-bound and, to a lesser extent, Watson–Crick-bound adjacent nucleotides Figure 4.
The significance of the long-range energy terms for the adoption of a native G4 topology is further reflected in the backbone interactions. Because of electrostatic repulsion, the interactions between phosphate groups I–IV could be expected to destabilize the G-tetrad relative to its dissociated state. Unexpectedly, however, our data demonstrate that these interactions contribute favorably to the stability of the G-tetrad with the average energy change of −1.5 kcal/mol, −1.5 kcal/mol and −2.5 kcal/mol due to the interaction with the phosphate of the Hoogsteen-bound, Watson–Crick-bound and diagonally opposite nucleotides, respectively. This result clearly indicates that the phosphate groups I–IV move away from each other upon G-tetrad formation. To further investigate this structural change, we computed how the pairwise distances between all phosphate groups and their radius of gyration depend on the position along the Nhb coordinate. The results presented in Figure 5A show that not only do phosphates I–IV move away from each other and from the other phosphates (on average by 5.6%) but also the separations between most of the other phosphate groups increase markedly (by 9.6%) reaching the maximum value for the fully formed G-tetrad. This repulsion-driven relative displacement of phosphate groups results in the large changes of interaction energy between phosphates I–IV and the rest of DNA backbone (Figure 4) and within the backbone itself (Supplementary Figure S9). Consequently, it can be proposed that, by assuming the energetically optimal conformation, the DNA backbone increases the kinetic stability of the parallel-stranded G4, as it opposes local perturbations of the structure such as (partial) dissociation of the G-tetrad.
Figure 5.

(A) Dependence of the separation distances between all 21 phosphate groups in the DNA backbone on the conformational state along the dissociation/re-assembly pathway. Values of the radius of gyration for all phosphate groups (Rgyr), averaged separately for each of the four states, confirm that the phosphates move away from each other upon G-tetrad re-assembly. (B) Solvent-accessible surface area for the entire G4 DNA (red) and its individual components, labeled consistently with Figure 4. (C) Number of K+ ions in contact (r < 0.3 nm) with G4 DNA as a function of Nhb.
The increase of the phosphate–phosphate distances upon the G-tetrad formation is also reflected in the expansion of the solvent accessible surface area (SASA) of the DNA backbone, which dominates the SASA of the entire G4 causing it to increase by ∼2 nm2 even though at the same time the SASA of the associating guanines is naturally decreasing (Figure 5B). As can be seen from Figure 4 and Supplementary Figure S9, the observed increase in the backbone surface facilitates the hydration of the phosphate groups and leads to additional energetic stabilization of the G-tetrad, which, in the case of phosphates I–IV, amounts to ∼7 kcal/mol per group and is clearly smaller for the rest of the backbone whose conformation is less distorted by the enforced dissociation (∼4 kcal/mol per group).
Figure 4 and Supplementary Figure S9 further show that the energetic contribution of the phosphate–phosphate and phosphate–water interaction to the G-tetrad stability is compensated by the loss of electrostatic interactions between the phosphate groups and K+ ions present in the solvent. This finding is quite surprising, given the increased exposure of the phosphates to the solvent upon the G-tetrad formation. To understand the mechanism behind this unexpected result, we first calculated how the number of K+ ions in direct contact with G4 changes with Nhb coordinate. Figure 5C shows that the formation of the G-tetrad is indeed associated with a loss of ∼1 K+ ion bound to the G4. The probabilities of finding K+ around the G4, shown in Figure 6A, demonstrate that upon transition from the fully dissociated (Nhb =0) to the native (Nhb =8) state the spatial distribution of ions changes markedly. In the native state, the ions bind mainly in the central channel and in the cavities formed by the phosphates groups of the TTA-loops (Figure 6C), as also observed in the previous molecular dynamics simulations (41,43,60). Instead, in the dissociated state, there are four well-defined ion-binding sites around the 5′-terminal G-tetrad formed mainly by the N7 atoms of guanine and (stacked) adenine residues and the O4’ atom of deoxyribose (Figure 6B). These newly-formed binding sites are largely responsible for binding, on average, one additional K+ ion to the G4.
Figure 6.

(A) Spatial distribution of potassium ions around the G4 with the dissociated (top) and fully formed (bottom) 3′-terminal G-tetrad, represented as probability density isosurface at a value 20 times greater than in bulk solution. Two additional well-defined K+-binding sites, indicated by red and blue boxes, were identified outside the central channel and are blown up in the panels below. (B) Potassium-binding site around the 5′-terminal G-tetrad. (C) Potassium-binding site formed by the phosphates groups of the TTA-loops.
Entropic cost of the G-tetrad formation
The above decomposition of the interaction free energy into pairwise contributions suggested that the formation of the G-tetrad is enthalpically favorable. However, this enthalpic gain is expected to be at least partially offset by a decrease in conformational entropy due to the loss of conformational freedom upon the association of guanines. Unfortunately, direct determination of the entropy change involved in the formation of the entire G-tetrad is quite challenging, as it would require well-converged values of the relevant association free energy computed at several temperatures. Therefore, we first focused our analysis on the association of a single guanine nucleobase. This approach reduced the computational cost by an order of magnitude while still allowing meaningful conclusions to be drawn for the entire G-tetrad, due to its symmetry. Specifically, to estimate the entropy/enthalpy compensation in the G-tetrad formation, we used the adaptive biasing force method (61) to calculate the free energy profile for the dissociation of a single guanine residue (IV in Figure 1C) from the 3′-terminal G-tetrad of the parallel-stranded G4 structure, at three different temperatures (295, 300 and 305 K; see Supplementary Data for details). The obtained dependence of the free energy profile on the temperature (Supplementary Figure S10) allowed us to decompose the association free energy into enthalpic (ΔH) and entropic (−ΔS) components, shown in Figure 7A along with the free energy profile at 300 K.
Figure 7.

(A) Enthalpic (ΔH) and entropic (−TΔS) contributions to the free energy profile (ΔG) for the dissociation of a single guanine (IV in Figure 1) from the 3′-terminal G-tetrad of the parallel-stranded G4. (B) Conformational entropy changes upon G-tetrad dissociation/re-assembly calculated within quasi-harmonic approximation for the entire G4 DNA and its individual components, labeled consistently with Figure 4 (solid lines). It should be emphasized that due to correlations individual contributions are not additive. Additionally, per-atom entropy profiles are shown as dashed lines. For the convergence of entropy values see Supplementary Figure S11.
It is worth noting that the free energy gain accompanying the association of guanine IV to G-triplex (−7 kcal/mol) agrees well with the free energy change upon the transition from state 4 to state 3 in Figure 2A (−6 kcal/mol), even though previously the transition involved a different guanine (mostly I) and that the reaction coordinate employed was also different (the number of native h-bonds versus the center-of-mass separation distance used here). This consistency confirms that the profiles are well converged and that the energetics of association reflects the symmetric structure of the G-tetrad. Most notably, Figure 7A shows that the overall association free energy is indeed a result of the compensation between favorable enthalpic term (ΔH = −35 kcal/mol) and unfavorable entropic term (−TΔS = +29 kcal/mol), which also strongly suggest that the guanine–guanine binding in G4 and, hence, the G-tetrad formation are enthalpically driven. Similar energy/entropy compensation in G4 folding was previously shown based on thermodynamic analysis of calorimetric data (32,62).
As can be seen from Figure 7A, the enthalpy and entropy changes for the guanine detachment are relatively large compared to the energetic stabilization of the G-tetrad by interactions between the guanine residues and their immediate environment (estimated above to be ∼2 kcal/mol). This disparity in energy scales indicates that the detachment of a guanine residue from the G-tetrad requires more extensive rearrangements of the G4 conformation involving the structure of the sugar–phosphate backbone. This conclusion is consistent with the above-described energetic optimization of the backbone geometry through maximizing the inter-phosphate distances. Apparently, this local optimization of the backbone geometry in the native state (Nhb = 8) contributes significantly to a decrease in conformational entropy of the G4 upon guanine association.
To further evaluate individual contributions to the entropic cost of the G-tetrad formation, the quasi-harmonic analysis (63) was applied to estimate conformational entropy changes along the Nhb coordinate. To this end, we used the reweighted US data (see above) to compute and diagonalize the covariance matrix of atomic fluctuations as a function of Nhb, for the entire G4 and, separately, for its major structural elements. The resulting quasi-harmonic entropy profiles shown in Figure 7B confirm that the formation of the G-tetrad is accompanied by a large decrease in entropy (−TΔS = +130 kcal/mol), which, due to the symmetry, is approximately four times as large as that estimated above for a single guanine from the temperature dependence of the association free energy. Even though individual contributions to the quasi-harmonic entropy are not strictly additive due to correlations, the similar changes in the entropy of the DNA backbone and the guanine residues (−TΔS = +40 kcal/mol) indicate that reduced conformational fluctuations of these elements contribute about equally to the overall entropy decrease. In contrast, the conformational entropy of the loop thymine and adenine residues increases upon the G-tetrad formation (−TΔS = −25 kcal/mol), as in the native state they are maximally exposed to the aqueous environment and sample multiple conformations.
To provide structural insight into the overall entropy loss, in Supplementary Figure S12 we compared the equilibrium distributions of the dihedral angles defining the geometry of the G-tetrad-forming nucleotides in the native and dissociated states. As expected, all these rotational degrees of freedom are more hindered for the native state, which is reflected in the observed decrease in the conformational entropy, however, no clear conformational transitions are seen upon G-tetrad formation. The largest change occurs for the glycosidic χn angle indicating that formation of the G-tetrad requires adopting a well defined orientation of the guanine with respect to the 2-deoxyribose ring, in the anti conformation (∼120°, as expected for the parallel-stranded G4 topology). The other angles for which a marked narrowing of the distribution is observed are βn, αn and δn governing the local geometry of the DNA backbone, in particular the arrangement of the guanosine moiety with respect to the 5′-phosphate group. Similarly, the deoxyribose ring puckering is better defined in the native state where it is restricted to the C2′-endo (76 %) and C1′-exo (18%) conformations, with the distribution of populated states being significantly broader in the dissociate state (48% C2′-endo, 38% C1′-exo, 6% C3′-exo and 5% O4′-endo).
CONCLUSIONS
In this work, we examined the molecular origins of the formation and stability of a guanine tetrad (G-tetrad) which is a core component of G4, four-stranded DNA secondary structures possibly involved in regulating multiple biological processes and thus considered as potential drug targets. To this end, we characterized the thermodynamics and kinetics of the dissociation of the 3′-terminal G-tetrad of the parallel-stranded G4 in a quantitative manner using molecular dynamics-based free energy calculations. The computed free energy profiles revealed that the dissociation and re-assembly of the G-tetrad occur by sequential and cooperative mechanism that depends on monovalent ion binding in the central channel of the G4. From the decomposition of the dissociation free energy into individual enthalpic contributions, we further predicted that the interactions between guanine residues and their environment provide only marginal stability to the G-tetrad. This is because the stabilizing intra-plane Hoogsteen h-bonds and ion binding interactions are almost entirely offset by the desolvation penalty and by the unfavorable parallel orientation of the dipole moments of the stacked nucleobases. Therefore, according to our simulations, the G-tetrad is further stabilized by the interactions involving the sugar–phosphate backbone and TTA loops, in agreement with the experimental data (47,56,64). Our data indicate that this additional driving force results in particular from the optimization of the backbone geometry through maximizing the inter-phosphate distances. Because any perturbation seems to disrupt the locally optimal distribution of phosphates in the native state, this contribution may also increase the kinetic stability of G4 DNA, consistently with the higher rupture forces measured in single-molecule experiments for unfolding through the loop residues (47). Enthalpy–entropy decomposition of the dissociation free energy further showed that the energetic gain upon G-tetrad formation is largely compensated by a decrease in entropy accompanying the association of guanines and optimization of the backbone structure.
Supplementary Material
Acknowledgments
The authors would like to thank Miłosz Wieczór for critically reading the manuscript and offering helpful suggestions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Foundation for Polish Science (FNP) Homing Plus Programme, co-financed from the European Union's Regional Development Fund within the Operational Programme Innovative Economy (HOMING PLUS/2011-4/3); Funding for open access charge: Gdansk University of Technology; PL-Grid Infrastructure; Academic Computer Centre TASK.
Conflict of interest statement. None declared.
REFERENCES
- 1.Sen D., Gilbert W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature. 1988;334:364–366. doi: 10.1038/334364a0. [DOI] [PubMed] [Google Scholar]
- 2.Gilbert D.E., Feigon J. Multistranded DNA structures. Curr. Opin. Struct. Biol. 1999;9:305–314. doi: 10.1016/S0959-440X(99)80041-4. [DOI] [PubMed] [Google Scholar]
- 3.Schaffitzel C., Berger I., Postberg J., Hanes J., Lipps H.J., Plückthun A. In vitro generated antibodies specific for telomeric guanine-quadruplex DNA react with Stylonychia lemnae macronuclei. Proc. Natl. Acad. Sci. U.S.A. 2001;98:8572–8577. doi: 10.1073/pnas.141229498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henderson A., Wu Y., Huang Y.C., Chavez E.A., Platt J., Johnson F.B., Brosh R.M., Sen D., Lansdorp P.M. Detection of G-quadruplex DNA in mammalian cells. Nucleic Acids Res. 2013;42:860–869. doi: 10.1093/nar/gkt957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biffi G., Tannahill D., McCafferty J., Balasubramanian S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013;5:182–186. doi: 10.1038/nchem.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bochman M.L., Paeschke K., Zakian V.A. DNA secondary structures: stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012;13:770–780. doi: 10.1038/nrg3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murat P., Balasubramanian S. Existence and consequences of G-quadruplex structures in DNA. Curr. Opin. Genet. Dev. 2014;25:22–29. doi: 10.1016/j.gde.2013.10.012. [DOI] [PubMed] [Google Scholar]
- 8.Ray S., Bandaria J.N., Qureshi M.H., Yildiz A., Balci H. G-quadruplex formation in telomeres enhances POT1/TPP1 protection against RPA binding. Proc. Natl. Acad. Sci. U.S.A. 2014;111:2990–2995. doi: 10.1073/pnas.1321436111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rhodes D., Lipps H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015;43:8627–8637. doi: 10.1093/nar/gkv862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zahler A.M., Williamson J.R., Cech T.R., Prescott D.M. Inhibition of telomerase by G-quartet DNA structures. Nature. 1991;350:718–720. doi: 10.1038/350718a0. [DOI] [PubMed] [Google Scholar]
- 11.De Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 12.Zaug A.J., Podell E.R., Cech T.R. Human POT1 disrupts telomeric G-quadruplexes allowing telomerase extension in vitro. Proc. Natl. Acad. Sci. U.S.A. 2005;102:10864–10869. doi: 10.1073/pnas.0504744102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H., Nora G.J., Ghodke H., Opresko P.L. Single molecule studies of physiologically relevant telomeric tails reveal POT1 mechanism for promoting G-quadruplex unfolding. J. Biol. Chem. 2011;286:7479–7489. doi: 10.1074/jbc.M110.205641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shammas M.A., Reis R. J.S., Li C., Koley H., Hurley L.H., Anderson K.C., Munshi N.C. Telomerase inhibition and cell growth arrest after telomestatin treatment in multiple myeloma. Clin. Cancer Res. 2004;10:770–776. doi: 10.1158/1078-0432.ccr-0793-03. [DOI] [PubMed] [Google Scholar]
- 15.Tauchi T., Shin-Ya K., Sashida G., Sumi M., Okabe S., Ohyashiki J., Ohyashiki K. Telomerase inhibition with a novel G-quadruplex-interactive agent, telomestatin: in vitro and in vivo studies in acute leukemia. Oncogene. 2006;25:5719–5725. doi: 10.1038/sj.onc.1209577. [DOI] [PubMed] [Google Scholar]
- 16.Gomez D., O'Donohue M.-F., Wenner T., Douarre C., Macadré J., Koebel P., Giraud-Panis M.-J., Kaplan H., Kolkes A., Shin-ya K., et al. The G-quadruplex ligand telomestatin inhibits POT1 binding to telomeric sequences in vitro and induces GFP-POT1 dissociation from telomeres in human cells. J. Cancer Res. 2006;66:6908–6912. doi: 10.1158/0008-5472.CAN-06-1581. [DOI] [PubMed] [Google Scholar]
- 17.Campbell N.H., Patel M., Tofa A.B., Ghosh R., Parkinson G.N., Neidle S. Selectivity in ligand recognition of G-quadruplex loops. Biochemistry. 2009;48:1675–1680. doi: 10.1021/bi802233v. [DOI] [PubMed] [Google Scholar]
- 18.Neidle S. Human telomeric G-quadruplex: the current status of telomeric G-quadruplexes as therapeutic targets in human cancer. FEBS J. 2010;277:1118–1125. doi: 10.1111/j.1742-4658.2009.07463.x. [DOI] [PubMed] [Google Scholar]
- 19.Casagrande V., Salvati E., Alvino A., Bianco A., Ciammaichella A., D'Angelo C., Ginnari-Satriani L., Serrilli A.M., Iachettini S., Leonetti C., et al. N-cyclic bay-substituted perylene G-quadruplex ligands have selective antiproliferative effects on cancer cells and induce telomere damage. J. Med. Chem. 2011;54:1140–1156. doi: 10.1021/jm1013665. [DOI] [PubMed] [Google Scholar]
- 20.Huppert J.L., Balasubramanian S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007;35:406–413. doi: 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu S.-T.D., Varnai P., Bugaut A., Reszka A.P., Neidle S., Balasubramanian S. A G-rich sequence within the c-kit oncogene promoter forms a parallel G-quadruplex having asymmetric G-tetrad dynamics. J. Am. Chem. Soc. 2009;131:13399–13409. doi: 10.1021/ja904007p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eddy J., Maizels N. Conserved elements with potential to form polymorphic G-quadruplex structures in the first intron of human genes. Nucleic Acids Res. 2008;36:1321–1333. doi: 10.1093/nar/gkm1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumari S., Bugaut A., Huppert J.L., Balasubramanian S. An RNA G-quadruplex in the 5′ UTR of the NRAS proto-oncogene modulates translation. Nat. Chem. Biol. 2007;3:218–221. doi: 10.1038/nchembio864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lane A.N., Chaires J.B., Gray R.D., Trent J.O. Stability and kinetics of G-quadruplex structures. Nucleic Acids Res. 2008;36:5482–5515. doi: 10.1093/nar/gkn517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y., Patel D.J. Solution structure of the human telomeric repeat d[AG3(T2AG3)3] G-tetraplex. Structure. 1993;1:263–282. doi: 10.1016/0969-2126(93)90015-9. [DOI] [PubMed] [Google Scholar]
- 26.Parkinson G.N., Lee M. P.H., Neidle S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature. 2002;417:876–880. doi: 10.1038/nature755. [DOI] [PubMed] [Google Scholar]
- 27.Dai J.X., Carver M., Punchihewa C., Jones R.A., Yang D.Z. Structure of the Hybrid-2 type intramolecular human telomeric G- quadruplex in K+ solution: insights into structure polymorphism of the human telomeric sequence. Nucleic Acids Res. 2007;35:4927–4940. doi: 10.1093/nar/gkm522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dai J.X., Punchihewa C., Ambrus A., Chen D., Jones R.A., Yang D.Z. Structure of the intramolecular human telomeric G-quadruplex in potassium solution: a novel adenine triple formation. Nucleic Acids Res. 2007;35:2440–2450. doi: 10.1093/nar/gkm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim K.W., Amrane S., Bouaziz S., Xu W., Mu Y., Patel D.J., Luu K.N., Phan A.T. Structure of the human telomere in K+ solution: a stable basket-type G-quadruplex with only two g-tetrad layers. J. Am. Chem. Soc. 2009;131:4301–4309. doi: 10.1021/ja807503g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lim K.W., Ng V. C.M., Martín-Pintado N., Heddi B., Phan A.T. Structure of the human telomere in Na+ solution: an antiparallel (2+2) G-quadruplex scaffold reveals additional diversity. Nucleic Acids Res. 2013;41:10556–10562. doi: 10.1093/nar/gkt771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chaires J.B. Human telomeric G-quadruplex: thermodynamic and kinetic studies of telomeric quadruplex stability. FEBS J. 2010;277:1098–1106. doi: 10.1111/j.1742-4658.2009.07462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bončina M., Lah J., Prislan I., Vesnaver G. Energetic basis of human telomeric DNA folding into G-quadruplex structures. J. Am. Chem. Soc. 2012;134:9657–9663. doi: 10.1021/ja300605n. [DOI] [PubMed] [Google Scholar]
- 33.Kumar N., Sahoo B., Varun K., Maiti S., Maiti S. Effect of loop length variation on quadruplex-Watson Crick duplex competition. Nucleic Acids Res. 2008;36:4433–4442. doi: 10.1093/nar/gkn402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rachwal P.A., Brown T., Fox K.R. Effect of G-tract length on the topology and stability of intramolecular DNA quadruplexes. Biochemistry. 2007;46:3036–3044. doi: 10.1021/bi062118j. [DOI] [PubMed] [Google Scholar]
- 35.Fujimoto T., ichi Nakano S., Sugimoto N., Miyoshi D. Thermodynamics-hydration relationships within loops that affect G-quadruplexes under molecular crowding conditions. J. Phys. Chem. B. 2013;117:963–972. doi: 10.1021/jp308402v. [DOI] [PubMed] [Google Scholar]
- 36.Miyoshi D., Nakao A., Sugimoto N. Molecular crowding regulates the structural switch of the DNA G-quadruplex. Biochemistry. 2002;41:15017–15024. doi: 10.1021/bi020412f. [DOI] [PubMed] [Google Scholar]
- 37.Heddi B., Phan A.T. Structure of human telomeric DNA in crowded solution. J. Am. Chem. Soc. 2011;133:9824–9833. doi: 10.1021/ja200786q. [DOI] [PubMed] [Google Scholar]
- 38.Gray R.D., Trent J.O., Chaires J.B. Folding and unfolding pathways of the human telomeric G-quadruplex. J. Mol. Biol. 2014;426:1629–1650. doi: 10.1016/j.jmb.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.You H., Zeng X., Xu Y., Lim C.J., Efremov A.K., Phan A.T., Yan J. Dynamics and stability of polymorphic human telomeric G-quadruplex under tension. Nucleic Acids Res. 2014;42:8789–8795. doi: 10.1093/nar/gku581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xue Y., Liu J.-q., Zheng K.-w., Kan Z.-y., Hao Y.-h., Tan Z. Kinetic and thermodynamic control of G-quadruplex folding. Angew. Chem. Int. Ed. Engl. 2011;50:8046–8050. doi: 10.1002/anie.201101759. [DOI] [PubMed] [Google Scholar]
- 41.Islam B., Sgobba M., Laughton C., Orozco M., Šponer J., Neidle S., Haider S. Conformational dynamics of the human propeller telomeric DNA quadruplex on a microsecond time scale. Nucleic Acids Res. 2013;41:2723–2735. doi: 10.1093/nar/gks1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei D., Husby J., Neidle S. Flexibility and structural conservation in a c-KIT G-quadruplex. Nucleic Acids Res. 2014;43:629–644. doi: 10.1093/nar/gku1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Islam B., Stadlbauer P., Krepl M., Koca J., Neidle S., Haider S., Šponer J. Extended molecular dynamics of a c-kit promoter quadruplex. Nucleic Acids Res. 2015;43:8673–8693. doi: 10.1093/nar/gkv785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cang X., Šponer J., Cheatham T.E. Explaining the varied glycosidic conformational, G-tract length and sequence preferences for anti-parallel G-quadruplexes. Nucleic Acids Res. 2011;39:4499–4512. doi: 10.1093/nar/gkr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Šponer J., Mládek A., Špačková N., Cang X., Thomas E., Cheatham I., Grimme S. Relative stability of different DNA guanine quadruplex stem topologies derived using large-scale quantum-chemical computations. J. Am. Chem. Soc. 2013;135:9785–9796. doi: 10.1021/ja402525c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lech C.J., Heddi B., Phan A.T. Guanine base stacking in G-quadruplex nucleic acids. Nucleic Acids Res. 2013;41:2034–2046. doi: 10.1093/nar/gks1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghimire C., Park S., Iida K., Yangyuoru P., Otomo H., Yu Z., Nagasawa K., Sugiyama H., Mao H. Direct quantification of loop interaction and π−π stacking for G-quadruplex stability at the submolecular level. J. Am. Chem. Soc. 2014;136:15537–15544. doi: 10.1021/ja503585h. [DOI] [PubMed] [Google Scholar]
- 48.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 49.Foloppe N., MacKerell A.D., Jr All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000;21:86–104. [Google Scholar]
- 50.Hess B., Kutzner C., van der Spoel D., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory. Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 51.Hess B. P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory. Comput. 2008;4:116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 52.Miyamoto S., Kollman P.A. Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992;13:952–962. [Google Scholar]
- 53.Humphrey W., Dalke A., Schulten K. VMD—visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 54.Bonomi M., Branduardi D., Bussi G., Camilloni C., Provasi D., Raiteri P., Donadio D., Marinelli F., Pietrucci F., Broglia R.A., et al. PLUMED: a portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009;180:1961–1972. [Google Scholar]
- 55.Kumar S., Rosenberg J.M., Bouzida D., Swendsen R.H., Kollman P.A. Multidimensional free-energy calculations using the weighted histogram analysis method. J. Comput. Chem. 1995;16:1339–1350. [Google Scholar]
- 56.Yu Z., Koirala D., Cui Y., Easterling L.F., Zhao Y., Mao H. Click chemistry assisted single-molecule fingerprinting reveals a 3D biomolecular folding funnel. J. Am. Chem. Soc. 2012;134:12338–12341. doi: 10.1021/ja303218s. [DOI] [PubMed] [Google Scholar]
- 57.Simonsson T. G-quadruplex DNA structures—variations on a theme. Biol. Chem. 2001;382:621–628. doi: 10.1515/BC.2001.073. [DOI] [PubMed] [Google Scholar]
- 58.Miyoshi D., Nakao A., Sugimoto N. Structural transition from antiparallel to parallel G-quadruplex of d (G4T4G4) induced by Ca2 + Nucleic Acids Res. 2003;31:1156–1163. doi: 10.1093/nar/gkg211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Burge S., Parkinson G.N., Hazel P., Todd A.K., Neidle S. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006;34:5402–5415. doi: 10.1093/nar/gkl655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gray R.D., Petraccone L., Trent J.O., Chaires J.B. Characterization of a K+-induced conformational switch in a human telomeric DNA oligonucleotide using 2-aminopurine fluorescence. Biochemistry. 2010;49:179–194. doi: 10.1021/bi901357r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Darve E., Pohorille A. Calculating free energies using average force. J. Chem. Phys. 2001;115:9169–9183. [Google Scholar]
- 62.Sengar A., Heddi B., Phan A.T. Formation of G-quadruplexes in poly-G sequences: structure of a propeller-type parallel-stranded G-quadruplex formed by a G15 stretch. Biochemistry. 2014;53:7718–7723. doi: 10.1021/bi500990v. [DOI] [PubMed] [Google Scholar]
- 63.Andricioaei I., Karplus M. On the calculation of entropy from covariance matrices of the atomic fluctuations. J. Chem. Phys. 2001;115:6289–6292. [Google Scholar]
- 64.Li W., Hou X.-M., Wang P.-Y., Xi X.-G., Li M. Direct measurement of sequential folding pathway and energy landscape of human telomeric G-quadruplex structures. J. Am. Chem. Soc. 2013;135:6423–6426. doi: 10.1021/ja4019176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.