

Abstract
Vascular endothelial growth factor (VEGF) plays an important role in Kaposi’s sarcoma (KS). We administered PTC299, a post-transcriptional inhibitor of pathogenic VEGF, to persons with HIV-related KS. Seventeen participants received three different doses of PTC299. Adverse events typically observed with VEGF-inhibition were absent. Three participants had partial tumor responses and 11 had stable disease. There were no differences in exposure to PTC299 by antiretroviral regimen. Serum VEGF, but not KSHV DNA, decreased on treatment. Given redundancies in the VEGF feedback loop, future trials should consider combining PTC299 with agents that inhibit different pathways implicated in KS and KSHV proliferation.
Keywords: Kaposi’s sarcoma (KS), Vascular Endothelial Growth Factor (VEGF), Vascular Endothelial Growth Factor Inhibitor (VEGF inhibitor), Kaposi’s sarcoma-associated herpes virus (KSHV/HHV-8), HIV/AIDS-related malignancy, pharmacokinetics (PK)
INTRODUCTION
Although Kaposi’s sarcoma (KS), the most common AIDS-defining malignancy worldwide [1, 2], can be treated with antiretroviral therapy (ART) alone or with chemotherapy added [3-5], responses are often incomplete. Cytotoxic chemotherapy may induce acute and chronic adverse events and pharmacokinetic interactions with ART, and access to chemotherapy is limited in high-incidence, low-resource regions, especially sub-Saharan Africa [6, 7]. Thus, alternative treatments are needed.
Vascular endothelial growth factor (VEGF) is highly over-expressed in KS tumors and enhances KS-associated herpesvirus (KSHV) entry and KS gene expression within target cells [8, 9]. In laboratory studies, inhibiting VEGF expression or its signaling pathways results in tumor growth inhibition [10-14]. KSHV-encoded proteins promote vascular endothelial reprogramming and immortalization through up-regulation of VEGF and VEGF receptors (VEGFr) [15, 16]. Additionally, tumor hypoxia induces p53-mediated apoptosis and HIF-1α, further promoting VEGF production [17]. HIF-1α and hypoxemic responses are additionally modulated directly by KSHV, and hypoxia further promotes lytic KSHV replication [18-21].
PTC299 (PTC Therapeutics Inc, South Plainfield, NJ) an orally-bioavailable VEGF inhibitor, acts via post-transcriptional regulation of VEGF mRNA under conditions of cellular stress. Transcriptional regulation of VEGF in normal endothelial cells occurs mainly through cap-dependent translation of its mRNA. In contrast, cellular stress and hypoxemia augment transcription of VEGF in a cap-independent fashion [22-24]. Because PTC299 targets cap-independent pathways of mRNA translation that predominate during stress such as hypoxia and oncogenic transformation, VEGF production in normal endothelium is spared, potentially avoiding undesirable off-target effects of generalized VEGF blockade observed with other VEGF and VEGFr tyrosine kinase inhibitors (TKIs). PTC299 activity was demonstrated in a variety of tumor cell lines and mouse xenograft-tumor models and was safe and well tolerated in several phase I clinical trials [25-28].
These considerations led the AIDS Malignancy Consortium (AMC) to evaluate the safety, dosing, antitumor activity, and pharmacokinetics of PTC299 in patients with HIV-associated KS, and to describe its effects on VEGF expression and KSHV replication.
METHODS
Study Population
Eligible participants were HIV-infected adults with biopsy-proven KS not requiring urgent systemic chemotherapy for symptomatic visceral disease. Participants receiving ART were required to be on a stable regimen ≥ 12 weeks without improvement in KS during that time, not pregnant or breastfeeding, and to have received no KS treatment within one month. Exclusions included Karnofsky score <60, life expectancy <3 months, history of bleeding or clotting diathesis, severe hepatic or renal dysfunction, or other active serious medical conditions.
After providing informed consent, three participants were sequentially enrolled into each of three oral dosage levels, 40mg twice daily (BID), 80mg BID, and 100mg BID, once the preceding dose had been administered without dose limiting toxicities (DLT) for one cycle. Eight additional participants then received 100mg BID. PTC299 was supplied as 20mg capsules. A plan to estimate the maximum tolerated dosage (MTD) was not performed because the sponsor suspended enrollment due to change in drug formulation. Each drug cycle lasted 28 days. KS response was assessed every 28 days as previously described [29, 30], and categorized as complete (CR), partial (PR), stable (SD) or progression (PD). Participants were considered evaluable for response if they completed 1 cycle of PTC299. Participants without CR or PR were removed from study after 6 cycles; treatment was likewise discontinued if KS progressed on therapy or DLT occurred. Participants were continued on therapy for an additional 2 cycles following CR, or up to 12 cycles for PR.
Laboratory Methods
Biopsies of non-indicator KS lesions were performed at baseline and during week 4, cycle 1, and evaluated for changes in expression of viral, angiogenesis and proliferation markers (see Supplemental Material).
KSHV DNA was quantified in plasma at baseline, cycles 2 and 5, and treatment discontinuation, using competitive DNA PCR [31]. Whole blood CD4+ T cell counts and plasma HIV RNA levels were determined at these same time points.
VEGF-A and IL-6 levels were quantified by ELISA (Quest Laboratories, San Juan Capistrano, CA) in serum and plasma on Day 1 of cycles 1-6, on Cycle 1, Day 15 and at treatment discontinuation.
Serial blood samples for pharmacokinetic analysis were collected immediately preceding and at 1, 2, 3, 4, 5, 6, and 8 hours following the morning administration of PTC299 on Cycle 1, Days 1 and 28. Additional trough samples were obtained on Cycle 1, Day 15 and Cycle 2, Day 28. Samples were analyzed and pharmacokinetic parameters estimated as described (see Supplemental Material).
Statistical Considerations
Descriptive statistics were used to summarize adverse events and response rates.
Effects of PTC299 on serum and plasma VEGF, VEGFR and cytokine profiles were evaluated comparing pre-treatment values with Day 28 and with the lowest value subsequent to Day 28, and differences tested with Wilcoxon signed rank tests. Methods for analyzing other correlative laboratory endpoints are described in Supplemental Materials.
RESULTS
Seventeen volunteers were enrolled: 3 at 40 mg BID, 3 at 80 mg BID and 11 at 100 mg BID. Baseline characteristics, including extent of KS, ART regimen, HIV parameters, and prior KS therapy are described in Table 1, as are details of study treatment.
Table 1.
Baseline Participant Characteristics and Study Therapy Received
| Characteristic | N=17 | |||
|---|---|---|---|---|
| Age median (range) | 45 (31,57) | |||
| Male | 17 (100%) | |||
| Race/Ethnicity | ||||
| White, non-Hispanic | 8 (47) | |||
| Black | 4 (24) | |||
| Hispanic | 3 (18) | |||
| Asian/Pacific Islander/Other | 2 (12) | |||
| KS Disease Characteristics | ||||
| Oral lesions | 1 (6) | |||
| ≥50 lesions | 16 (88) | |||
| Tumor-associated edema | 16 (88) | |||
| Prior treatment for KS* | 16 (94) | |||
| Cytotoxic Chemotherapy | 14 (82) | |||
| Local therapies (topical, radiation, or cryotherapy) | 4 (24) | |||
| Imatinib | 4 (24) | |||
| Other/experimental therapies | 6 (35) | |||
| Baseline KSHV Plasma Log-copies/ml, median (IQR) | 4.8 (2.5, 5.8) | |||
| CD4 T-cell Count, median (range) | 489 (133, 1132) | |||
| HIV Viral load undetectable | 13 (76) | |||
| Current Antiretroviral regimens | N =17 (%) | |||
| Nucleoside Analog Backbone | ||||
| ABC/d4T | 1 (6) | |||
| ABC/3TC/ddI | 1 (6) | |||
| TDF/FTC or TDF/3TC | 12 (71) | |||
| 3TC | 1 (6) | |||
| AZT/3TC | 1 (6) | |||
| Nucleoside-sparing regimen | 1 (6) | |||
| Non-Nucleoside Agents | 6 (35) | |||
| EFV | 6 (35) | |||
| Protease Inhibitors** | 10 (59) | |||
| Other Classes (raltegravir, maraviroc) | 5 (29) | |||
| PTC299 Cycles Received | 40mg BID | 80mg BID | 100mg BID | Total |
| N=3 | N=3 | N=11 | N=17 | |
| median (range) | 7 (6,13) | 3 (2,5) | 4 (1,10) | 5 (1,13) |
Abbreviations: KS, Kaposi sarcoma; HIV, human immunodeficiency virus; KSHV, Kaposi Sarcoma-associated herpes virus; IQR, interquartile range; ABC, abacavir; d4T, stavudine; 3TC, lamivudine; ddI, didanosine; TDF, tenofovir; FTC, emtricitabine; AZT, zidovodine; EFV, efavirenz.
Prior treatments included (# of participants): cytotoxic therapies: liposomal doxorubicin (14), paclitaxel (7), vinblastine (1), etoposide (1), vincristine (1), daunomycin (1); Other/experimental therapies included: valproic acid (2), IM-862 (2), rapamycin (1), flavopiridol (1), Col-3 [incyclinide] (3), interferon-alpha (1), VEGF-antisense (1).
Protease inhibitors included were lopinivir/ritonavir, darunavir/ritonavir, and atazanavir with and without ritonavir.
Five participants completed therapy per protocol, 6 terminated study treatment for KS progression, and 4 voluntarily withdrew after a median of 3 cycles. One patient died and one withdrew from study before the first response evaluation. PR was documented in 3 participants (18%), lasting for 2, 3, and 4 months respectively at doses of 40mg BID (n=1) and 100mg BID (n=2). Eleven participants showed SD lasting a median of 3 months (IQR 2-6.5). PD ultimately occurred in 6 participants, of whom 5 initially had either PR or SD.
Safety Assessment
Common adverse events included nausea (41%), vomiting (18%), diarrhea (24%), limb pain (47%), and fatigue (29%); >90% of events were Grade 1 or 2. Limb pain, fatigue, and peripheral edema were prevalent at baseline and consistent with the primary disease. Hyperglycemia (29%), dyslipidemia (53%), elevated creatinine (18%), and proteinuria (18%), were the most frequent laboratory abnormalities. Three participants (18%), all receiving atazanavir, had elevated bilirubin. All participants with elevated creatinine and/or proteinuria received concurrent tenofovir. Serious adverse events were reported in 3 participants: one grade 3 nephrolithiasis, considered unlikely related to study drug; one grade 3 myalgia at the 100 mg/dose level, considered probably PTC299-related; and one death. The death was officially ascribed to hypertensive cardiovascular disease and reported as possibly study drug-related, although autopsy revealed detectable serum levels of several illicit and prescription opiates and sedatives; precise cause of death is therefore uncertain.
Pharmacokinetics
Pharmacokinetics
Pharmacokinetic analysis was available for 15 patients. A post-hoc analysis categorizing participants on whether the ART regimen was known to induce CYP2C19 (ritonavir) or inhibit CYP2C19 (efavirenz) [32] showed no statistically significant alterations in pharmacokinetics of the parent compound, but significant alterations in exposure to the less-active metabolite, des-methyl PTC299 (see Supplemental Material)[33]. There was no correlation between treatment response and PTC-299 or metabolite exposure (p > 0.05)
Effects of PTC299 on Biologic markers
Day 1, cycle 1 levels of serum VEGF were significantly higher than levels at all later time-points (Figure 1); plasma VEGF showed a less sustained decrease that was significant only at intermediate time-points. No changes were observed in serum or plasma IL-6.
Figure 1. Change in mean serum and plasma VEGF levels during treatment with PTC299.
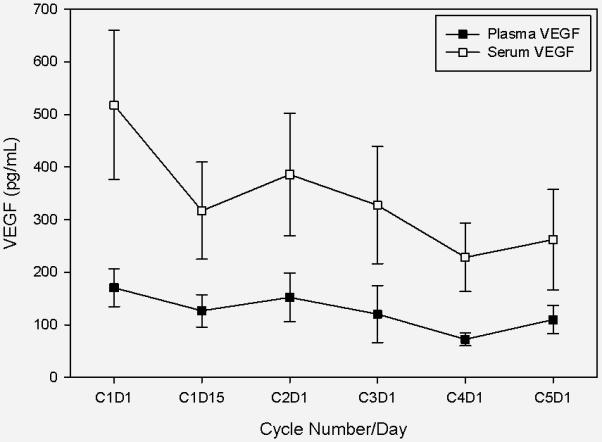
Figure legend: This graph demonstrates mean VEGF levels in serum (open boxes) and plasma (closed boxes) with standard deviations denoted by brackets. Compared with the baseline (C1D1) mean level of 518.1 pg/ml, serum VEGF decreased significantly at all subsequent time points ( to 316.9 pg/ml, p=0.001 at C1D15; and to 261.81 pg/ml, p=0.026 at the final time point: C5D1). Baseline (C1D1) mean plasma VEGF was 170.4 pg/ml and was 126.7 pg/ml at C1D15 (p=0.061). Plasma VEGF decreased significantly between baseline and C3D1 (120.3 pg/ml, p=0.009) and C4D1 only (72.3 pg/ml,p=0.001) and but not at the final time point on C5D1 (109.9 pg/ml, p=0.432).
Abbreviations: VEGF, vascular endothelial growth factor; CxDx, cycle and day of PTC299 therapy
There were no significant treatment-associated changes in KSHV viral loads, absolute and percent CD4, or immunohistochemical expression of VEGF, VEGFr, phospho-Akt, p53, HIF-1α, or Ki-67, or viral gene expression or cellular gene transcription in tumor biopsies.
DISCUSSION
Recognition of the essential role of VEGF in KS development has prompted evaluation of several VEGF inhibitors in AIDS-associated KS. This study evaluated PTC299, a novel, orally-bioavailable small molecule that inhibits VEGF protein production by preventing translation of pathological VEGF. Because the study was halted early, we were unable to define the MTD in this population, but doses administered were similar to those tested in other Phase I and II studies. Those trials and ours demonstrated a pharmacokinetic profile for PTC299 consistent with maintenance of drug levels well above those required for efficacy in preclinical models.
Participants were allocated to PTC299 dosage levels without respect to the concurrent antiretroviral regimen. We observed no significant variation between participants receiving CYP2C19 inducers and inhibitors in the pharmacokinetics of the parent drug, nor correlation of treatment response with drug and metabolite exposure, although metabolite exposure and metabolite:parent drug ratio differed significantly between these two groups. Without stratified dosage assignments, such as those used in a subsequent AMC trial of sunitinib [34], we were unable to correlate adverse events by ART regimen effects on CYP2C19.
We did not observe many of the medically significant side effects of VEGF inhibitors and TKIs, such as bleeding, hypertension and renal vascular injury [35, 36]. Mild proteinuria was limited to persons receiving tenofovir and/or atazanavir, both known to cause renal tubulopathies [37] and all persons with elevated bilirubin levels were receiving atazanivir [38]. Fewer adverse events were observed than for other classes of VEGF inhibitors/TKIs, suggesting that drugs inhibiting VEGF by targeting tumor-specific mRNA rather than general kinase activity may be less likely to induce off-target effects. The short duration of PTC299 administration in some participants may, however, have precluded observation of potential adverse events.
Although we could not evaluate the planned dosage range of PTC299, moderate serum VEGF inhibition was achieved, though only modest effects on KS growth, consistent with results from other VEGF inhibitors. Many participants had previously received multiple KS treatments, but we did not note a pattern of response with respect to prior receipt of agents with anti-VEGF activity. Other studies of VEGF inhibitors in KS reported a 30% response rate for bevacizumab, 20% for sorafenib, and no better than SD for sunitunib [34, 39-41]. As such, this study highlights a persistent question in the search for improved therapies for KS and many soft tissue sarcomas (STS) which similarly over-express VEGF and its receptors [42]. It is unclear why, despite the apparent reliance of many such tumors on VEGF overproduction, VEGF inhibitors have not proven highly efficacious in these tumors. Five VEGF inhibitors have been studied in other STS, of which four received approval for this indication. However, single-agent PR rates were only 14% for sorafenib [43], 17% for bevacizumab [44], 6% for pazopanib [45] and metabolic PR in 47% of patients receiving sunitinib for varied STS [46]. Despite PTC299 activity in multiple pre-clinical in vivo sarcoma models [33], and a statistically significant decrease in serum VEGF levels in this trial, the response rate of 20% was similar to rates for other TKIs, but inferior to standard-of-care chemotherapy.
One reason that single-point blockade of the VEGF pathway may be insufficient to cause tumor regression, particularly in AIDS-related KS, is redundancy in the human VEGF pathway, which KSHV reinforces at several points [40]. Additionally, while VEGF is necessary for proliferation, it may not be necessary for tumor survival. This is similar to the mTOR pathway, an upstream regulator of VEGF and IL-6 expression [47-50]. Viral IL-6 and KSHV-mediated up-regulation of HIF-1α and VEGFrs may reinforce this autocrine-paracrine loop, such that single-point blockade of the VEGF pathway is easily circumvented. Virus-tumor interactions, allowing up-regulation of alternate pathways, may explain why objective responses to single agent VEGF inhibitors have been modest. Lastly, since pharmacologic growth factor depletion is seldom complete, the typical outcome of single-agent therapy in pre-clinical studies is growth arrest, translating to no better than SD in most patients.
Given the limited therapeutic results of VEGF monotherapy, a logical next step may be to combine mechanistically-distinct VEGF inhibitors [51] or VEGF inhibitors with agents that target either tumor or virus. A current example is an ongoing trial of bevacizumab with liposomal doxorubicin (NCT00923936). Additionally, nucleoside analog inhibitors of viral DNA polymerase with direct gamma herpesvirus activity, despite little anti-KS efficacy as single agents [52, 53], may provide adjuvant effects against virally-mediated paracrine stimulation to remove redundancy in the VEGF pathway. Furthermore, several HIV protease inhibitors have off-target effects on pathways regulating tumor growth, including Akt, NF-κB, and the 20S proteasome [54, 55]. Of particular interest is nelfinavir, which down-regulates HIF-1α and VEGF, and has direct anti-herpesvirus activity [56, 57].
Supplementary Material
ACKNOWLEDGEMENTS
We thank the members of the AMC-059 Study Team [David Aboulafia, Virginia Mason Medical Center, Seattle, WA; Robert Baiocchi, Ohio State Medical Center, Columbus, OH; Elizabeth Y. Chiao, Baylor College of Medicine, Houston, TX; Bruce J. Dezube, Beth Israel Deaconess Hospital, Boston, MA; Ronald T. Mitsuyasu, University of California Los Angeles (UCLA) and UCLA Clinical AIDS Research and Education (CARE) Center, Los Angeles, CA; Erin Gourley Reid, University of California San Diego, Moores Cancer Center, La Jolla, CA; Bruce Shiramizu, University of Hawaii John A. Burns School of Medicine, Honolulu, HI; Joseph A. Sparano, Albert Einstein College of Medicine, Montefiore Medical Center, Bronx, NY; Anil Tulpule, University of Southern California Keck School of Medicine, Los Angeles, CA] for their recruitment and management of the study participants; Anthony Eason and Veenadhari Chavakula (Department of Microbiology and Immunology and Lineberger Comprehensive Cancer Center, University of North Carolina School of Medicine, Chapel Hill, North Carolina, USA) for performing immunohistochemistry and gene expression analyses; PTC Therapeutics for their provision of PTC299 and support for measurements of plasma drug level and serum and plasma cytokines; Julia Lynne, EMMES Corporation, Rockville, MD for her assistance in protocol development; and the study participants for taking part in this trial. Author Contributions: RBI, SEK, and MAR, evaluated the results and wrote the manuscript; SEK principally conceived of and executed the clinical trial; MAR performed the pharmacokinetic analysis; JYL performed the main statistical analysis; RFA performed the KSHV PBMC analyses; DPD performed the tumor IHC and gene expression analyses. All authors have reviewed the final manuscript and agree with its publication. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
FUNDING SOURCES
The project was supported by an NIH/NCI cooperative agreement (UO1CA121947) to the AIDS Malignancy Consortium and by PTC Therapeutics, Inc. Additional support was provided by the Analytical Pharmacology Core of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins (NIH grants P30 CA006973 and UL1TR001079) and CA163217 to DPD. RBI received an NIH interdisciplinary training grant in cancer research (T32-CA080416) to support time given to this project.
Footnotes
Registered on Clinical Trials.gov: NCT00686842
CONFLICT OF INTEREST
RBI, JYL, AS, VK, DPD, RFA, MAR, and SEK declare no conflict of interest.
References
- 1.Whitby D, et al. Detection of Kaposi sarcoma associated herpesvirus in peripheral blood of HIV-infected individuals and progression to Kaposi's sarcoma. Lancet. 1995;346(8978):799–802. doi: 10.1016/s0140-6736(95)91619-9. [DOI] [PubMed] [Google Scholar]
- 2.Schalling M, et al. A role for a new herpes virus (KSHV) in different forms of Kaposi's sarcoma. Nat Med. 1995;1(7):707–8. doi: 10.1038/nm0795-707. [DOI] [PubMed] [Google Scholar]
- 3.Cianfrocca M, et al. Randomized trial of paclitaxel versus pegylated liposomal doxorubicin for advanced human immunodeficiency virus-associated Kaposi sarcoma: evidence of symptom palliation from chemotherapy. Cancer. 2010;116(16):3969–77. doi: 10.1002/cncr.25362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooley T, et al. A randomized, double-blind study of pegylated liposomal doxorubicin for the treatment of AIDS-related Kaposi's sarcoma. Oncologist. 2007;12(1):114–23. doi: 10.1634/theoncologist.12-1-114. [DOI] [PubMed] [Google Scholar]
- 5.Mosam A, et al. A randomized controlled trial of highly active antiretroviral therapy versus highly active antiretroviral therapy and chemotherapy in therapy-naive patients with HIV-associated Kaposi sarcoma in South Africa. J Acquir Immune Defic Syndr. 2012;60(2):150–7. doi: 10.1097/QAI.0b013e318251aedd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adebamowo CA, et al. Challenges in the detection, prevention, and treatment of HIV-associated malignancies in low- and middle-income countries in Africa. J Acquir Immune Defic Syndr. 2014;67(Suppl 1):S17–26. doi: 10.1097/QAI.0000000000000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krown SE, et al. Stage-stratified approach to AIDS-related Kaposi's sarcoma: implications for resource-limited environments. J Clin Oncol. 2014;32(23):2512–3. doi: 10.1200/JCO.2014.55.8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sivakumar R, et al. Kaposi's sarcoma-associated herpesvirus induces sustained levels of vascular endothelial growth factors A and C early during in vitro infection of human microvascular dermal endothelial cells: biological implications. J Virol. 2008;82(4):1759–76. doi: 10.1128/JVI.00873-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamden KE, et al. Raf and VEGF: emerging therapeutic targets in Kaposi's sarcoma-associated herpesvirus infection and angiogenesis in hematopoietic and nonhematopoietic tumors. Leukemia. 2005;19(1):18–26. doi: 10.1038/sj.leu.2403532. [DOI] [PubMed] [Google Scholar]
- 10.Bais C, et al. Kaposi's sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/ KDR. Cancer Cell. 2003;3(2):131–43. doi: 10.1016/s1535-6108(03)00024-2. [DOI] [PubMed] [Google Scholar]
- 11.Mutlu AD, et al. In vivo-restricted and reversible malignancy induced by human herpesvirus-8 KSHV: a cell and animal model of virally induced Kaposi's sarcoma. Cancer Cell. 2007;11(3):245–58. doi: 10.1016/j.ccr.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samaniego F, et al. Vascular endothelial growth factor and basic fibroblast growth factor present in Kaposi's sarcoma (KS) are induced by inflammatory cytokines and synergize to promote vascular permeability and KS lesion development. Am J Pathol. 1998;152(6):1433–43. [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L, et al. Immortalization of primary endothelial cells by the K1 protein of Kaposi's sarcoma-associated herpesvirus. Cancer Res. 2006;66(7):3658–66. doi: 10.1158/0008-5472.CAN-05-3680. [DOI] [PubMed] [Google Scholar]
- 14.Carroll PA, Brazeau E, Lagunoff M. Kaposi's sarcoma-associated herpesvirus infection of blood endothelial cells induces lymphatic differentiation. Virology. 2004;328(1):7–18. doi: 10.1016/j.virol.2004.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang HW, et al. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat Genet. 2004;36(7):687–93. doi: 10.1038/ng1384. [DOI] [PubMed] [Google Scholar]
- 16.Hong YK, et al. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat Genet. 2004;36(7):683–5. doi: 10.1038/ng1383. [DOI] [PubMed] [Google Scholar]
- 17.Catrina S-B, et al. Hypoxia-inducible factor-1α and hypoxia-inducible factor-2α are expressed in Kaposi sarcoma and modulated by insulin-like growth factor-I. Clinical cancer research. 2006;12(15):4506–4514. doi: 10.1158/1078-0432.CCR-05-2473. [DOI] [PubMed] [Google Scholar]
- 18.Carroll PA, et al. Latent Kaposi's sarcoma-associated herpesvirus infection of endothelial cells activates hypoxia-induced factors. Journal of virology. 2006;80(21):10802–10812. doi: 10.1128/JVI.00673-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis DA, et al. Hypoxia induces lytic replication of Kaposi sarcoma–associated herpesvirus. Blood. 2001;97(10):3244–3250. doi: 10.1182/blood.v97.10.3244. [DOI] [PubMed] [Google Scholar]
- 20.Sodhi A, et al. The Kaposi’s sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1α. Cancer research. 2000;60(17):4873–4880. [PubMed] [Google Scholar]
- 21.Haque M, et al. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) contains hypoxia response elements: relevance to lytic induction by hypoxia. Journal of virology. 2003;77(12):6761–6768. doi: 10.1128/JVI.77.12.6761-6768.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huez I, et al. Two independent internal ribosome entry sites are involved in translation initiation of vascular endothelial growth factor mRNA. Molecular and cellular biology. 1998;18(11):6178–6190. doi: 10.1128/mcb.18.11.6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stein I, et al. Translation of vascular endothelial growth factor mRNA by internal ribosome entry: implications for translation under hypoxia. Molecular and cellular biology. 1998;18(6):3112–3119. doi: 10.1128/mcb.18.6.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levy AP, Levy NS, Goldberg MA. Post-transcriptional regulation of vascular endothelial growth factor by hypoxia. Journal of Biological Chemistry. 1996;271(5):2746–2753. doi: 10.1074/jbc.271.5.2746. [DOI] [PubMed] [Google Scholar]
- 25.Packer RJ, et al. Phase I and pharmacokinetic trial of PTC299 in pediatric patients with refractory or recurrent central nervous system tumors: a PBTC study. J Neurooncol. 2015;121(1):217–24. doi: 10.1007/s11060-014-1665-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fong B, et al. The molecular biology and novel treatments of vestibular schwannomas. J Neurosurg. 2011;115(5):906–14. doi: 10.3171/2011.6.JNS11131. [DOI] [PubMed] [Google Scholar]
- 27.Knight D, et al. PTC299, a Novel Regulator of Tumor VEGF Expression, Is Well Tolerated and Achieves Target Plasma Concentrations: Dose-Ranging Results of a Phase 1b Study in Women with Metastatic Breast Cancer. Cancer Research. 2009;69(24 Supplement):6092–6092. [Google Scholar]
- 28.Hirawat S, et al. Phase 1 studies assessing the safety, PK, and VEGF-modulating effects of PTC299, a novel VEGF expression inhibitor. ASCO Annual Meeting Proceedings.2007. [Google Scholar]
- 29.Krown SE, Metroka C, Wernz J. Kaposi's sarcoma in the acquired immune deficiency syndrome: a proposal for uniform evaluation, response, and staging criteria. AIDS Clinical Trials Group Oncology Committee. Journal of Clinical Oncology. 1989;7(9):1201–1207. doi: 10.1200/JCO.1989.7.9.1201. [DOI] [PubMed] [Google Scholar]
- 30.Cianfrocca M, et al. Matrix metalloproteinase inhibitor COL-3 in the treatment of AIDS-related Kaposi's sarcoma: a phase I AIDS malignancy consortium study. J Clin Oncol. 2002;20(1):153–9. doi: 10.1200/JCO.2002.20.1.153. [DOI] [PubMed] [Google Scholar]
- 31.Lin L, et al. Effects of chemotherapy in AIDS-associated non-Hodgkin's lymphoma on Kaposi's sarcoma herpesvirus DNA in blood. J Clin Oncol. 2009;27(15):2496–502. doi: 10.1200/JCO.2008.20.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rudek MA, Flexner C, Ambinder RF. Use of antineoplastic agents in patients with cancer who have HIV/AIDS. Lancet Oncol. 2011;12(9):905–12. doi: 10.1016/S1470-2045(11)70056-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.PTC Therapeutics, I. PTC 299 Investigator Brochure. Ver 6, 12. [Google Scholar]
- 34.Rudek MA, et al. A phase 1/pharmacokinetic study of sunitinib in combination with highly active antiretroviral therapy in human immunodeficiency virus-positive patients with cancer: AIDS Malignancy Consortium trial AMC 061. Cancer. 2014;120(8):1194–202. doi: 10.1002/cncr.28554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayman SR, et al. VEGF inhibition, hypertension, and renal toxicity. Curr Oncol Rep. 2012;14(4):285–94. doi: 10.1007/s11912-012-0242-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kamba T, McDonald DM. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br J Cancer. 2007;96(12):1788–95. doi: 10.1038/sj.bjc.6603813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Post F. Adverse events: ART and the kidney: alterations in renal function and renal toxicity. J Int AIDS Soc. 2014;17(4 Suppl 3):19513. doi: 10.7448/IAS.17.4.19513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanne I, et al. Results of a phase 2 clinical trial at 48 weeks (AI424-007): a dose-ranging, safety, and efficacy comparative trial of atazanavir at three doses in combination with didanosine and stavudine in antiretroviral-naive subjects. J Acquir Immune Defic Syndr. 2003;32(1):18–29. doi: 10.1097/00126334-200301010-00004. [DOI] [PubMed] [Google Scholar]
- 39.Uldrick TS, et al. JOURNAL OF CLINICAL ONCOLOGY. AMER SOC CLINICAL ONCOLOGY; 2318 MILL ROAD, STE 800, ALEXANDRIA, VA 22314 USA: 2013. Phase I and pharmacokinetic study of sorafenib in Kaposi sarcoma. [Google Scholar]
- 40.Uldrick TS, et al. Phase II study of bevacizumab in patients with HIV-associated Kaposi's sarcoma receiving antiretroviral therapy. J Clin Oncol. 2012;30(13):1476–83. doi: 10.1200/JCO.2011.39.6853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noy A, et al. Fatal hepatorenal failure and thrombocytopenia with SU5416, a vascular endothelial growth factor Flk-1 receptor inhibitor, in AIDS-Kaposi's sarcoma. Aids. 2007;21(1):113–5. doi: 10.1097/01.aids.0000254688.06831.28. [DOI] [PubMed] [Google Scholar]
- 42.Park MS, Ravi V, Araujo DM. Inhibiting the VEGF-VEGFR pathway in angiosarcoma, epithelioid hemangioendothelioma, and hemangiopericytoma/solitary fibrous tumor. Curr Opin Oncol. 2010;22(4):351–5. doi: 10.1097/CCO.0b013e32833aaad4. [DOI] [PubMed] [Google Scholar]
- 43.Maki RG, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol. 2009;27(19):3133–40. doi: 10.1200/JCO.2008.20.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Agulnik M, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24(1):257–63. doi: 10.1093/annonc/mds237. [DOI] [PubMed] [Google Scholar]
- 45.van der Graaf WT, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879–86. doi: 10.1016/S0140-6736(12)60651-5. [DOI] [PubMed] [Google Scholar]
- 46.George S, et al. Multicenter phase II trial of sunitinib in the treatment of nongastrointestinal stromal tumor sarcomas. J Clin Oncol. 2009;27(19):3154–60. doi: 10.1200/JCO.2008.20.9890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roy D, et al. mTOR inhibitors block Kaposi sarcoma growth by inhibiting essential autocrine growth factors and tumor angiogenesis. Cancer Res. 2013;73(7):2235–46. doi: 10.1158/0008-5472.CAN-12-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sodhi A, et al. The TSC2/mTOR pathway drives endothelial cell transformation induced by the Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor. Cancer Cell. 2006;10(2):133–43. doi: 10.1016/j.ccr.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 49.Nichols LA, Adang LA, Kedes DH. Rapamycin blocks production of KSHV/HHV8: insights into the anti-tumor activity of an immunosuppressant drug. PLoS One. 2011;6(1):e14535. doi: 10.1371/journal.pone.0014535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang HH, Ganem D. A unique herpesviral transcriptional program in KSHV-infected lymphatic endothelial cells leads to mTORC1 activation and rapamycin sensitivity. Cell Host Microbe. 2013;13(4):429–40. doi: 10.1016/j.chom.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Azad NS, et al. Combination targeted therapy with sorafenib and bevacizumab results in enhanced toxicity and antitumor activity. Journal of Clinical Oncology. 2008;26(22):3709–3714. doi: 10.1200/JCO.2007.10.8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krown SE, Dittmer DP, Cesarman E. Pilot study of oral valganciclovir therapy in patients with classic Kaposi sarcoma. J Infect Dis. 2011;203(8):1082–6. doi: 10.1093/infdis/jiq177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Little RF, et al. A pilot study of cidofovir in patients with kaposi sarcoma. J Infect Dis. 2003;187(1):149–53. doi: 10.1086/346159. [DOI] [PubMed] [Google Scholar]
- 54.Gantt S, Casper C, Ambinder RF. Insights into the broad cellular effects of nelfinavir and the HIV protease inhibitors supporting their role in cancer treatment and prevention. Curr Opin Oncol. 2013;25(5):495–502. doi: 10.1097/CCO.0b013e328363dfee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bernstein WB, Dennis PA. Repositioning HIV Protease Inhibitors as Cancer Therapeutics. Curr Opin HIV AIDS. 2008;3(6):666–75. doi: 10.1097/COH.0b013e328313915d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gantt S, et al. The HIV protease inhibitor nelfinavir inhibits Kaposi's sarcoma-associated herpesvirus replication in vitro. Antimicrob Agents Chemother. 2011;55(6):2696–703. doi: 10.1128/AAC.01295-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gantt S, et al. Nelfinavir impairs glycosylation of herpes simplex virus 1 envelope proteins and blocks virus maturation. Adv Virol. 2015;2015:687162. doi: 10.1155/2015/687162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.