

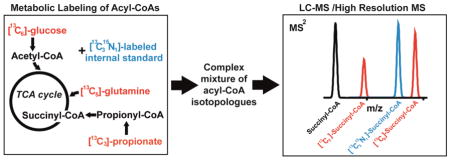
Abstract
Acyl-coenyzme A thioesters (acyl-CoAs) are evolutionarily conserved, compartmentalized, and energetically activated substrates for biochemical reactions. The ubiquitous involvement of acyl-CoAs in metabolism, including the tricarboxylic acid cycle, fatty acid metabolism, amino acid degradation, and cholesterol metabolism highlights the broad applicability of applied measurements of acyl-CoAs. However, quantitation of acyl-CoA levels provides only one dimension of metabolic information and a more complete description of metabolism requires the relative contribution of different precursors to individual substrates and pathways. Using two distinct stable isotope labeling approaches, acyl-CoAs can be labeled with either a fixed [13C315N1] label derived from pantothenate into the CoA moiety, or via variable [13C] labeling into the acyl-chain from metabolic precursors. Liquid chromatography-hybrid quadrupole/Orbitrap high resolution mass spectrometry using parallel reaction monitoring, but not single ion monitoring, allowed the simultaneous quantitation of acyl-CoAs by stable isotope dilution using the [13C315N1] label and measurement of the incorporation of labeled carbon atoms derived from [13C6]-glucose, [13C515N2]-glutamine and [13C3]-propionate. As a proof-of-principle we applied this method to human B-cell lymphoma (WSU-DLCL2) cells in culture, to precisely describe the relative pool size and enrichment of isotopic tracers into acetyl-, succinyl-, and propionyl-CoA. This method will allow highly precise, multiplexed, and stable isotope resolved determination of metabolism to refine metabolic models, characterize novel metabolism, and test modulators of metabolic pathways involving acyl-CoAs.
Keywords: Stable isotope labeling, mass spectrometry, coenzyme A, metabolism, bioanalytical methods
Graphical Abstract
Introduction
Acyl-coenzyme A (acyl-CoA) thioesters are ubiquitous substrates for cellular biochemistry[1]. Since they are conserved across evolution, acyl-CoA mediated metabolism is critical to biomedical, agricultural, and bio-industrial processes. In humans, normal physiology is regulated by acyl-CoAs[2], and extreme perturbations of acyl-CoA metabolism, often due to inborn errors of metabolism, results in diseases of high morbidity and mortality[3, 4]. Evidence for a role of acyl-CoAs in metabolic reprogramming in cancer[5] as well as post-translational modifications of proteins in a variety of pathological states[6, 7] reflects the increasing recognition of the importance of these metabolites across biochemistry. Additionally, the synthesis of lipids important in food and biofuel production is mediated via acyl-CoA intermediates. Finally, structurally complex natural products derived from prokaryotic organisms[8] and insects are ubiquitously derived from, and modified by, acyl-CoA substrates[9, 10]. By determining the intracellular concentration of these critical CoA thioesters, we may be able to better characterize metabolic pathway activity. Although the accurate measurement of acyl-CoAs remains a challenge, the two parameters needed for functional characterization of these metabolites include (1) their absolute concentration and (2) the relative atomic contribution of their precursors[11].
The current gold standard to determine the absolute concentration of intracellular metabolites is by using a stable isotope dilution (SID) liquid chromatography-mass spectrometry (LC-MS) based approach. LC-MS has been shown to provide increasingly sensitive and specific measurements of acyl-CoAs over previous LC-UV based methods. By using SID, in which an isotopically labeled analog of a target analyte (preferably 13C- or 15N-labeled) is spiked into the sample, quantitation is improved by accounting for analytical variability during extraction and analysis[12]. Stable isotope labeled analogs can also increase sensitivity through a carrier effect, competing with the light analyte for losses during processing. In addition to a limited number of commercially available labeled CoA internal standards, these can also be generated biosynthetically by incubating cells in [13C315N1]-pantothenic acid to efficiently generate a biosynthetically tunable library of stable isotope labeled analogs[13, 14].
To determine the relative atomic contribution of CoA precursors, 13C-isotopic labeling offers the most robust approach. In this methodology, cells are incubated in the presence of a 13C-labeled substrate and the metabolites are analyzed after a given amount of time for incorporation of the label, typically represented as a percentage of the metabolite pool. With stable isotope labeling coupled to detection by GC-MS or LC-MS, this approach improves ease of use, sensitivity, specificity, and dimensionality of information over traditional radiolabeling approaches. Advances in bioinformatics have even enabled routine isotope tracing experiments with a targeted[15] or untargeted approach[16].
To date, absolute quantification and 13C-isotopic labeling are performed independently due to the inability to resolve the spiked internal standards from the isotopically enriched metabolites on most traditional triple quadrupole instruments. This leads to higher variability and issues of analytical performance[17]. High resolution mass spectrometry (HRMS), on the other hand, is capable of resolving power of over 200,000 at scan speeds compatible with LC, allowing much greater analytical specificity. Sensitivity of analysis by these methods is also excellent, with the capability of quantifying hundreds of metabolites including relatively low abundance analytes[18]. The higher mass resolution allows differentiation, for example of 13C from 15N labels[19], which cannot easily be achieved using traditional approaches. Therefore, by leveraging the biochemistry and fragmentation pattern of acyl-CoAs and the resolution of a hybrid quadrupole-Orbitrap Q Exactive Plus HRMS, we demonstrate here, for the first time, simultaneous SID quantitation using stable isotope labeled analogs combined with 13C-isotope metabolic tracing.
Materials and Methods
Cell culture and metabolic tracer incubation
The human B cell lymphoma line WSU-DLCL2 was maintained in RPMI 1640 media (Invitrogen, Carlsbad, CA) with 10% FBS (Invitrogen). For isotopic tracer experiments, the media was replaced with glutamine and glucose free RPMI 1640 supplemented with the labeled tracers including 5 mM [13C6]-glucose, 200 μM [13C515N2]-glutamine, or 100 μM [13C3]-propionate. An unlabeled control was treated with fully unlabeled substrates at the same concentrations, and was used for isotopic correction as described below. Glutamine and propionate tracers were incubated in the presence of 5 mM unlabeled glucose to assess changes in pool size with additional substrate enrichment, and to provide a carbon source for the extended time points. Cell counts were performed manually with a hemocytometer.
Acyl-CoA extraction
Acyl-CoA extraction was performed as previously described[20] after spiking in 100 μL of acyl-CoA internal standards derived from pan6 deficient Saccharomyces cerevisiae grown in [13C315N1]-pantothenic acid containing media as previously described[21]. Since this acyl-CoA internal standard is biologically derived, some batch to batch variation is expected, but our previous calculations on yield estimate 100 μL of acyl-CoA internal standards produced by this method to contain 200 ng of [13C315N1]-acetyl-CoA, 20 ng of [13C315N1]-succinyl-CoA, and 5 ng of [13C315N1]-propionyl-CoA as well as varying amounts of other [13C315N1]-acyl-CoAs not included in this study.
LC-HRMS and LC-MS/HRMS analysis
Acyl-CoAs were analyzed on an Ultimate 3000 Quaternary UHPLC coupled to a Q Exactive Plus mass spectrometer operating in the positive ion mode with a heated ESI probe in an IonMax Source housing. Samples were kept in a temperature controlled autosampler at 6 °C and LC separation was performed as previously described on a Waters XBridge 3.5 μm particle size C18 2.1 × 150 mm column. LC conditions were as follows modified from previous studies[22, 20]; column oven temperature 25 °C, solvent A water with 5 mM ammonium acetate, solvent B 95:5 acetonitrile: water with 5 mM ammonium acetate, solvent C (wash solvent) 80:20 acetonitrile: water with 0.1% formic acid. The gradient was as follows: 0.2 mL/min flow at 98% A and 2% B for 1.5 min, 80% A 20% B at 5 min, 100% B at 12 min, 0.3 mL/min 100% B at 16 min, 0.2 mL/min 100% C at 17 min, held to 21 min, then re-equilibrated at 0.2 mL/min flow at 98% A and 2% B from 22 to 28 min. Flow from 4–18 minutes was diverted to the instrument. Operating conditions on the mass spectrometer were as follows; auxiliary gas 10 arbitrary units (arb), sheath gas 35 arb, sweep gas 2 arb, spray voltage 4.5 kV, capillary temperature 425 °C, S-lens RF-level 50, aux gas heater temperature 400 °C, in-source CID 5 eV. Scan parameters were optimized during the experiments, but final conditions were alternating full scan from 760–1800 m/z at 140,000 resolution and data independent acquisition (DIA) looped 3 times with all fragment ions multiplexed at a normalized collision energy (NCE) of 20 at a resolution of 280,000. An isolation width of 7 m/z with an offset of 3 m/z was used to capture all relevant isotopologues for targeted acyl-CoAs. Data was processed in Xcalibur, TraceFinder (Thermo), and then isotopic enrichment was calculated by the method of Fernandez, et al., to compensate for the non-linearity of isotopic enrichment[23]. Statistical analysis and graphical plots were generated in Prism v6 (GraphPad, LaJolla, CA). Using these conditions, we re-validated the precision of this LC-MS/HRMS method for acyl-CoAs. We conducted validation for inter- (n=3) and intra-day (n=3) precision for low (LQC), medium (MQC) and high quality control (HQC) levels of 12.5, 50, and 250 ng on column, respectively. Co-efficient of variation (inter-/intra-day) for LQC, MQC, and HQC of acetyl-CoA were 16.9%/16.1%, 2.7%/6.2%, 18.2%/13.5%, respectively. Values for succinyl-, propionyl- and other short chain acyl-CoAs were likewise below 20% for inter- and intra-day variation.
Results and Discussion
Resolution of 13C and 15N isotopic labels derived from pantothenate or substrates of carbon metabolism
Initial method development focused on acetyl-, succinyl- and propionyl-CoA as both acetyl- and succinyl-CoA are substrates in central carbon metabolism via the Krebs cycle. Furthermore, acetyl-CoA is produced from glucose, acetate, β-oxidation of fatty acids, amino acid metabolism, and is also a substrate for fatty acid synthesis. Succinyl-CoA serves as the entrance point into the Krebs cycle for the various anaplerotic metabolic pathways through propionyl-CoA, including cholesterol, odd chain fatty acids, and branched chain amino acid metabolism. Thus, with a limited set of analytes, a variety of isotopically labeled substrates representing major bioenergetics processes can be studied (Figure 1A). Further, MS analysis of CoAs are well established from multiple methodological studies and applications[20, 24–30], and the positive ion fragmentation of acyl-CoAs produces a high abundance neutral loss of 507, leaving the acyl-chain as well as the pantothenate derived label on the product ion (Figure 1B).
Figure 1.

Metabolism, isotopic tracing, and mass spectral identification of acyl-CoAs. (A) Isotopic tracers incorporated into acyl-CoA species, including acetyl-, succinyl-, and propionyl-CoA. (B) Structure and fragmentation of acyl-CoAs.
However, all previous studies have used lower resolution instruments, usually tandem MS/MS on triple quadrupoles at unit resolution, except Liu et al., that adapted a high resolution MS approach to label free quantitation[27]. Theoretical isotopic patterns of the [M+H]+ of succinyl-CoA using natural isotopic abundance predicted that baseline resolution of the most important M4 isotopologues containing [13C4] and [13C315N1] labels would not be achieved even at maximal resolution of 280,000 (Figure 2A). Using the [M-507+H]+ product ion was predicted to achieve baseline resolution of a number of the possible isotopes (Figure 2B). Simulating a 1:1 abundance of the [13C4] and [13C315N1] labels gave complete resolution at 280,000 resolution, and resolution at less than 10% of peak at 140,000 resolution (Figure 2C).
Figure 2.

Theoretical and experimental resolution of M4 isotopologue of succinyl-CoA demonstrates that both tandem MS and ultra-high resolution are required for stable isotope resolution of acyl-CoAs. Theoretical Gaussian resolution (FWHM) with 15 points per sample are shown for 280000,140000, 70000, 35000, 17500 resolution, predicted centroid masses at each resolution are indicated on the graph for (A) the [M+H]+ of succinyl-CoA and (B) the [M-507+H]+ fragment ion of succinyl-CoA and (C) a 1:1 complex mixture of 13C4 and 13C315N1 labeled succinyl-CoA. (D) Experimentally determined resolutions [R] by LC-HRMS (broken blue line) and LC-MS/HRMS (solid black line) of the M4 isotopologue from cell extract of DLCL2 cells grown with 13C515N2 glutamine for (top) [M+H]+ of succinyl-CoA and (bottom) the [M-507+H]+ fragment ion of succinyl-CoA. (E) LC-HRMS incompletely resolves the 13C4 and 13C315N1 labels, resulting in an erroneous quantification of the 13C4 label. (F) LC-MS/HRMS completely resolves the 13C4 and 13C315N1 labels correctly quantifying the isotopologue distribution. Chromatograms have been offset for clarity.
Due to these theoretical predictions, a MS/HRMS based approach was adopted using the higher collision energy dissociation (HCD) cell of the Q Exactive Plus to perform fragmentation of the acyl-CoAs before isotopologue analysis. A comparison of cell extract from cultures in unlabeled media and spiked with [13C315N1]-acyl-CoAs versus cell extract from cultures with [13C515N2]-glutamine and spiked with [13C315N1]-acyl-CoAs was conducted. As predicted, the [M+H]+ ion of the two resulting M+4 isotopologues at m/z 872.145 was incompletely resolved at maximum resolution of the instrument (set 280,000, observed 138,672 max) (Figure 2D, upper). However, the product ions of the two different labels were completely resolved at maximum resolution (set 280,000, observed 219,466 max) (Figure 2D, lower). The lower experimental resolution in both cases is likely due to the decay of Orbitrap resolution at higher masses, where the theoretical resolution is that expected at m/z 200[31]. Additionally, consistent with the retention of both isotopic labels and the high resolution accurate mass of the product ions our data provides support to the fragmentation proposed by Gao, et al., from lower resolution triple quadrupole MS/MS spectra[25].
This study was limited only to the relatively hydrophilic short-chain acyl-CoAs, essentially below a carbon chain length of 6. Thus, for MS/HRMS, an isolation window of 7 m/z with an offset of 3 m/z was used to include all isotopes detected under experimental conditions. Therefore, for acetyl-, succinyl-, and propionyl-CoA the isolation windows were 813.1, 871.1, and 827.1 +/- 3.5 m/z, respectively. To avoid systematic bias in quantitation driven by non-linear relationship between isotopologues with increased labeling, transitions including up to at least 2 additional 13C isotopes past the maximum acyl-chain length were monitored (e.g. up to [13C4]- for acetyl-CoA). The sum of isotopologues past that range are expected to contribute well below 0.2% of the total signal based on the isotopic distribution of unlabeled or [13C4]-succinyl-CoA and thus are negligible even with a fully labeled acyl moiety. Although we are able to resolve the [13C4] and [13C315N1] labels, we still refer to each isotopologue by the M0, M1, M2, etc. convention, indicating the sequential incorporation of an increasing number of labeled atoms, as we do not know the exact position of the isotopes and this naming is widely used.
The effects of increased resolution on quantitation of labels was examined using the same system. The theoretical m/z of [13C1], [13C2], [13C3], [13C4] and [13C315N1] labeled succinyl-CoA and the [M-507+H]+ fragment were extracted from chromatograms with a 3 ppm window. This window would be sufficient to exclude overlapping isotopologues derived from [13C4] and [13C315N1] labels if complete resolution of the spectral peaks were obtained (Figure 2E). Co-elution between all isotopologues was observed, with no apparent interfering peaks in the time window since previous method development had excluded other acyl-CoAs from interference. Integration of the [M+H]+ ions resulted in an underestimate of the abundance of the [13C4] label, likely due to interference of the [13C315N1] label in quantitation (Figure 2E). No such interference was observed for the [M-507+H]+ fragment (Figure 2F).
Since all isobaric acyl-CoAs examined in this study were separated by chromatography, and no overlapping fragments were observed, all fragment ions were multiplexed into the Orbitrap for scanning. This was facilitated by the routine fragmentation of acyl-CoAs, such that even with less specific isolation, no overlap in fragment ions was observed for any of the acyl-CoAs in this study. The duty cycle of the instrument under experimental conditions with a full scan and multiplexed MS/HRMS scan was under 2 seconds, with approximately 15 scans across a 30 second peak. Performance of the Orbitrap for the purposes of this study were adequate but could be improved. First, increased scan speed of the Orbitrap utilizing the higher field design more recently deployed and coupled to a similar hybrid platform[32, 33] allowing higher resolution with additional scans across a chromatographic peak, would likely improve precision and may enable faster chromatographic runs at higher backpressures. Performance of time-of-flight (ToF) instruments using this method was not examined, but hybrid triple quadrupole-ToF instruments may provide similar capabilities depending on selection of product ion and resolution. Second, increased sensitivity of the instrument is a requisite in isotopic tracer experiments where increasing contribution of noise to the lower abundance isotopes can be a source of measurement error and variation. Thus, adoption of nanospray ESI or the increasing sensitivity of MS instruments may also improve the capabilities of this approach.
Quantification and isotoplogue analysis from mammalian cell culture
WSU-DCLC2 cells growing in suspension culture were incubated with either unlabeled and/or labeled metabolic substrates over 10 hours with collections of 1 mL aliquots of culture for acyl-CoA analysis at 60, 240, and 600 minutes. Unlabeled/labeled substrate pairs of 5 mM glucose/[13C6]-glucose, 200 μM glutamine/[13C515N2]-glutamine, and 100 μM propionate/[13C3]-propionate were used due to physiological relevance, commercial availability of tracers, and differential incorporation of each of the labels into metabolic pathways of interest. An unlabeled control was also incubated with fully unlabeled tracers at the same concentrations. To assess changes in relative metabolite pool size with additional substrate addition, for the [13C515N2]-glutamine and [13C3]-propionate tracers, 5 mM glucose was added. Quenching and extraction were performed in the presence of [13C315N1]-acyl-CoA internal standards in ice-cold 10% TCA, followed by LC-MS/HRMS and isotopologue analysis (Table 1).
Table 1.
Simultaneous quantification and isotopologue analysis of acetyl-, succinyl- and propionyl-CoA. The relative pool size (relative abundance of each acyl-CoA) and % enrichment of each major isotopologue of each acyl-CoA is shown as the mean +/− the standard error of the mean (n=3).
| Acetyl-CoA | Succinyl-CoA | Propionyl-CoA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Relative Pool Size | M0 (%) | M2 (%) | Relative Pool Size | M0 (%) | M2 (%) | M4 (%) | Relative Pool Size | M0 (%) | M3 (%) | |
| 60 Minutes | ||||||||||
| Glucose | 1.07 ± 0.04 | 68.94 ± 1.77 | 33.10 ± 0.69 | 0.88 ± 0.03 | 98.26 ± 1.60 | 2.72 ± 1.09 | −0.03 ± 0.09 | 0.30 ± 0.05 | 103.08 ± 1.47 | −0.01 ± 0.01 |
| Glutamine | 1.02 ± 0.03 | 83.59 ± 0.79 | 17.16 ± 0.56 | 1.17 ± 0.04 | 47.16 ± 1.52 | 1.94 ± 0.33 | 54.64 ± 1.57 | 1.73 ± 0.37 | 91.89 ± 1.36 | 0.03 ± 0.01 |
| Propionate | 0.90 ± 0.06 | 99.34 ± 0.70 | 0.10 ± 0.16 | 1.00 ± 0.07 | 100.04 ± 1.64 | 0.28 ± 0.41 | 0.00 ± 0.03 | 1.71 ± 0.22 | 71.35 ± 1.89 | 22.22 ± 3.57 |
| 240 Minutes | ||||||||||
| Glucose | 1.23 ± 0.05 | 73.82 ± 0.26 | 27.31 ± 0.45 | 1.04 ± 0.03 | 93.35 ± 0.82 | 7.12 ± 1.39 | 0.04 ± 0.09 | 1.03 ± 0.04 | 91.38 ± 0.87 | −0.01 ± 0.02 |
| Glutamine | 0.93 ± 0.04 | 63.06 ± 0.53 | 38.37 ± 1.07 | 1.09 ± 0.02 | 35.63 ± 0.37 | 5.79 ± 0.52 | 61.72 ± 0.21 | 1.12 ± 0.03 | 91.11 ± 0.77 | 0.07 ± 0.05 |
| Propionate | 0.84 ± 0.04 | 99.76 ± 0.79 | 0.17 ± 0.18 | 0.99 ± 0.08 | 100.96 ± 0.76 | 0.30 ± 0.23 | 0.02 ± 0.02 | 1.06 ± 0.10 | 67.23 ± 0.23 | 24.60 ± 0.64 |
| 600 Minutes | ||||||||||
| Glucose | 1.01 ± 0.03 | 66.64 ± 0.33 | 35.28 ± 0.44 | 0.79 ± 0.06 | 84.05 ± 1.54 | 13.76 ± 1.59 | −0.16 ± 0.07 | 0.82 ± 0.03 | 90.97 ± 0.33 | 0.01 ± 0.01 |
| Glutamine | 1.15 ± 0.05 | 68.10 ± 0.37 | 33.29 ± 0.53 | 1.38 ± 0.02 | 33.31 ± 0.44 | 13.75 ± 0.48 | 52.56 ± 0.49 | 1.26 ± 0.15 | 89.88 ± 0.39 | 0.29 ± 0.04 |
| Propionate | 0.97 ± 0.06 | 99.88 ± 0.88 | 0.21 ± 0.12 | 1.21 ± 0.10 | 99.40 ± 0.45 | 0.45 ± 0.48 | −0.01 ± 0.02 | 1.25 ± 0.05 | 64.58 ± 1.26 | 26.34 ± 0.75 |
The design of the tracer experiments reflects incorporation of labeled carbon into the acyl chains of each acyl-CoA by time through % enrichement of the major isotopologues in each acyl-CoA. Thus, at 60 minutes, carbons from glucose and glutamine were incorporated into acetyl-CoA, with acetyl-CoA M2 enrichment of 33.10% and 17.16%, respectively. Succinyl-CoA however, was highly enriched in carbon derived from glutamine, with 54.64% enrichment in M4 and a 1.17-fold increase in relative pool size. Perhaps most intriguingly, the relative pool size of propionyl-CoA increased massively in both propionate and glutamine labeled experiments. However, in contrast to the enrichment of M3 in propionate labeled cells, no enrichment was observed in the glutamine labeled cells despite the increase in propionyl-CoA pool size. This finding may reflect the diversion of anaplerosis from propionyl-CoA to succinyl-CoA when anaplerosis from glutamine is more significant. Also worth future study, labeled propionate did not increase succinyl-CoA or acetyl-CoA labeling in our experimental conditions. Considering physiologic concentrations of propionate range from 6 μM in peripheral blood, to 0.1 mM- 0.3 mM in the portal vein, and to 4–6 mM in patients with propionic academia in crisis, the biochemical ability to shunt excess propionate is metabolically relevant[34, 35].
Labeling patterns at 240 minutes largely reflected those observed at 60 minutes, except an increase in the incorporation of M2 label derived from glutamine in acetyl-CoA to 38.37% from 17.16%. This relatively long period for accumulation of label likely reflects the differential spread of a tracer through metabolic pathways, in this case, the route of carbon derived from glutamine around the tricarboxylic acid cycle.
Conclusion
Stable isotope dilution allows precise quantitation in complex bioanalytical workflows. However, quantitation describes only one dimension of metabolic information from a sample. Tracer experiments with isotopic labeling provides orthogonal information on routes and mechanisms of metabolic control. We combined these two labeling methods to quantify and metabolically trace the ubiquitous metabolic substrates, acetyl-, succinyl-, and propionyl-CoA, using a hybrid quadrupole/Orbitrap high resolution mass spectrometer. The proof-of-principle experiments include tracing of the carbon sources glucose, glutamine, and propionic acid into acyl-CoA intermediates. Combining metabolic substrate labeled acyl-CoAs, pantothenate labeled acyl-CoAs, and universally labeled [13C]-acyl-CoAs as recently reported[36], may enable future multiplexed experimental design, including neutron encoded labeling to elegantly and efficiently study new CoA synthesis, turnover, or compartmentalization. Thus, this method provides an approach to interrogate normal and pathologic metabolism as well as optimize bio-industrial chemistry.
Acknowledgments
This work was supported by a Pennsylvania Department of Health Commonwealth Universal Research Enhancement grant.
Footnotes
Conflict of Interest
The authors declare no competing financial or non-financial interests.
References
- 1.Grevengoed TJ, Klett EL, Coleman RA. Acyl-CoA metabolism and partitioning. Annu Rev Nutr. 2014;34:1–30. doi: 10.1146/annurev-nutr-071813-105541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao S, Moran TH, Lopaschuk GD, Butler AA. Hypothalamic malonyl-CoA and the control of food intake. Physiol Behav. 2013;122:17–24. doi: 10.1016/j.physbeh.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schulze A, Lindner M, Kohlmuller D, Olgemoller K, Mayatepek E, Hoffmann GF. Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics. 2003;111(6 Pt 1):1399–406. doi: 10.1542/peds.111.6.1399. [DOI] [PubMed] [Google Scholar]
- 4.Bennett MJ, Rinaldo P, Strauss AW. Inborn errors of mitochondrial fatty acid oxidation. Crit Rev Clin Lab Sci. 2000;37(1):1–44. doi: 10.1080/10408360091174169. [DOI] [PubMed] [Google Scholar]
- 5.Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014;20(2):306–19. doi: 10.1016/j.cmet.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pougovkina O, Te Brinke H, Wanders RJ, Houten SM, de Boer VC. Aberrant protein acylation is a common observation in inborn errors of acyl-CoA metabolism. J Inherit Metab Dis. 2014;37(5):709–14. doi: 10.1007/s10545-014-9684-9. [DOI] [PubMed] [Google Scholar]
- 7.Lee JV, Shah SA, Wellen KE. Obesity, cancer, and acetyl-CoA metabolism. Drug Discov Today Dis Mech. 2013;10(1–2):e55–e61. doi: 10.1016/j.ddmec.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schuetz R, Zamboni N, Zampieri M, Heinemann M, Sauer U. Multidimensional optimality of microbial metabolism. Science. 2012;336(6081):601–4. doi: 10.1126/science.1216882. [DOI] [PubMed] [Google Scholar]
- 9.Seitz LM, Ram MS. Metabolites of lesser grain borer in grains. J Agric Food Chem. 2004;52(4):898–908. doi: 10.1021/jf035190m. [DOI] [PubMed] [Google Scholar]
- 10.Brindle PA, Baker FC, Tsai LW, Reuter CC, Schooley DA. Sources of propionate for the biogenesis of ethyl-branched insect juvenile hormones: Role of isoleucine and valine. Proc Natl Acad Sci U S A. 1987;84(22):7906–10. doi: 10.1073/pnas.84.22.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zamboni N, Saghatelian A, Patti GJ. Defining the Metabolome: Size, Flux, and Regulation. Mol Cell. 2015;58(4):699–706. doi: 10.1016/j.molcel.2015.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciccimaro E, Blair IA. Stable-isotope dilution LC-MS for quantitative biomarker analysis. Bioanalysis. 2010;2(2):311–41. doi: 10.4155/bio.09.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Basu SS, Mesaros C, Gelhaus SL, Blair IA. Stable isotope labeling by essential nutrients in cell culture for preparation of labeled coenzyme A and its thioesters. Anal Chem. 2011;83(4):1363–9. doi: 10.1021/ac1027353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snyder NW, Basu SS, Zhou Z, Worth AJ, Blair IA. Stable isotope dilution liquid chromatography/mass spectrometry analysis of cellular and tissue medium- and long-chain acyl-coenzyme A thioesters. Rapid Commun Mass Spectrom. 2014;28(16):1840–8. doi: 10.1002/rcm.6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clasquin MF, Melamud E, Rabinowitz JD. LC-MS data processing with MAVEN: a metabolomic analysis and visualization engine. Curr Protoc Bioinformatics. 2012;Chapter 14(Unit 14):1. doi: 10.1002/0471250953.bi1411s37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang X, Chen YJ, Cho K, Nikolskiy I, Crawford PA, Patti GJ. X13CMS: global tracking of isotopic labels in untargeted metabolomics. Anal Chem. 2014;86(3):1632–9. doi: 10.1021/ac403384n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hellerstein MK, Neese RA. Mass isotopomer distribution analysis at eight years: theoretical, analytic, and experimental considerations. Am J Physiol. 1999;276(6 Pt 1):E1146–70. doi: 10.1152/ajpendo.1999.276.6.E1146. [DOI] [PubMed] [Google Scholar]
- 18.Liu X, Ser Z, Locasale JW. Development and quantitative evaluation of a high-resolution metabolomics technology. Anal Chem. 2014;86(4):2175–84. doi: 10.1021/ac403845u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rhoads TW, Rose CM, Bailey DJ, Riley NM, Molden RC, Nestler AJ, et al. Neutron-encoded mass signatures for quantitative top-down proteomics. Anal Chem. 2014;86(5):2314–9. doi: 10.1021/ac403579s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Basu SS, Blair IA. SILEC: a protocol for generating and using isotopically labeled coenzyme A mass spectrometry standards. Nat Protoc. 2012;7(1):1–12. doi: 10.1038/nprot.2011.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Snyder NW, Tombline G, Worth AJ, Parry RC, Silvers JA, Gillespie KP, et al. Production of stable isotope-labeled acyl-coenzyme A thioesters by yeast stable isotope labeling by essential nutrients in cell culture. Anal Biochem. 2015;474:59–65. doi: 10.1016/j.ab.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Snyder NW, Basu SS, Worth AJ, Mesaros C, Blair IA. Metabolism of propionic acid to a novel acyl-coenzyme A thioester by mammalian cell lines and platelets. J Lipid Res. 2015;56(1):142–50. doi: 10.1194/jlr.M055384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernandez CA, Des Rosiers C, Previs SF, David F, Brunengraber H. Correction of 13C mass isotopomer distributions for natural stable isotope abundance. J Mass Spectrom. 1996;31(3):255–62. doi: 10.1002/(SICI)1096-9888(199603)31:3<255::AID-JMS290>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 24.Dalluge JJ, Gort S, Hobson R, Selifonova O, Amore F, Gokarn R. Separation and identification of organic acid-coenzyme A thioesters using liquid chromatography/electrospray ionization-mass spectrometry. Anal Bioanal Chem. 2002;374(5):835–40. doi: 10.1007/s00216-002-1554-x. [DOI] [PubMed] [Google Scholar]
- 25.Gao L, Chiou W, Tang H, Cheng X, Camp HS, Burns DJ. Simultaneous quantification of malonyl-CoA and several other short-chain acyl-CoAs in animal tissues by ion-pairing reversed-phase HPLC/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;853(1–2):303–13. doi: 10.1016/j.jchromb.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 26.Haynes CA, Allegood JC, Sims K, Wang EW, Sullards MC, Merrill AH., Jr Quantitation of fatty acyl-coenzyme As in mammalian cells by liquid chromatography-electrospray ionization tandem mass spectrometry. J Lipid Res. 2008;49(5):1113–25. doi: 10.1194/jlr.D800001-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, Sadhukhan S, Sun S, Wagner GR, Hirschey MD, Qi L, et al. High-Resolution Metabolomics with Acyl-CoA Profiling Reveals Widespread Remodeling in Response to Diet. Mol Cell Proteomics. 2015;14(6):1489–500. doi: 10.1074/mcp.M114.044859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magnes C, Suppan M, Pieber TR, Moustafa T, Trauner M, Haemmerle G, et al. Validated comprehensive analytical method for quantification of coenzyme A activated compounds in biological tissues by online solid-phase extraction LC/MS/MS. Anal Chem. 2008;80(15):5736–42. doi: 10.1021/ac800031u. [DOI] [PubMed] [Google Scholar]
- 29.Minkler PE, Kerner J, Kasumov T, Parland W, Hoppel CL. Quantification of malonyl-coenzyme A in tissue specimens by high-performance liquid chromatography/mass spectrometry. Anal Biochem. 2006;352(1):24–32. doi: 10.1016/j.ab.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 30.Worth AJ, Basu SS, Deutsch EC, Hwang WT, Snyder NW, Lynch DR, et al. Stable isotopes and LC-MS for monitoring metabolic disturbances in Friedreich’s ataxia platelets. Bioanalysis. 2015;7(15):1843–55. doi: 10.4155/bio.15.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zubarev RA, Makarov A. Orbitrap mass spectrometry. Anal Chem. 2013;85(11):5288–96. doi: 10.1021/ac4001223. [DOI] [PubMed] [Google Scholar]
- 32.Makarov A, Denisov E, Lange O. Performance evaluation of a high-field Orbitrap mass analyzer. J Am Soc Mass Spectrom. 2009;20(8):1391–6. doi: 10.1016/j.jasms.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 33.Michalski A, Damoc E, Hauschild JP, Lange O, Wieghaus A, Makarov A, et al. Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol Cell Proteomics. 2011;10(9):M111 011015. doi: 10.1074/mcp.M111.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut. 1987;28(10):1221–7. doi: 10.1136/gut.28.10.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Al-Lahham SH, Peppelenbosch MP, Roelofsen H, Vonk RJ, Venema K. Biological effects of propionic acid in humans; metabolism, potential applications and underlying mechanisms. Biochim Biophys Acta. 2010;1801(11):1175–83. doi: 10.1016/j.bbalip.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 36.Neubauer S, Chu DB, Marx H, Sauer M, Hann S, Koellensperger G. LC-MS/MS-based analysis of coenzyme A and short-chain acyl-coenzyme A thioesters. Anal Bioanal Chem. 2015;407(22):6681–8. doi: 10.1007/s00216-015-8825-9. [DOI] [PubMed] [Google Scholar]