

Abstract
Purpose
Previous studies indicated hyperlipidemia may be a risk factor for Alzheimer's disease, but the contributions of postprandial triglyceride‐rich lipoprotein (TGRL) are not known. In this study, changes in blood–brain barrier diffusional transport following exposure to human TGRL lipolysis products were studied using MRI in a rat model.
Methods
Male Sprague‐Dawley rats (∼180–250 g) received an i.v. injection of lipoprotein lipase (LpL)‐hydrolyzed TGRL (n = 8, plasma concentration ≈ 150 mg human TGRL/dL). Controls received i.v. injection of either saline (n = 6) or LpL only (n = 6). The 1H longitudinal relaxation rate R1 = 1/T1 was measured over 18 min using a rapid‐acquired refocus‐echo (RARE) sequence after each of three injections of the contrast agent Gd‐DTPA. Patlak plots were generated for each pixel yielding blood‐to‐brain transfer coefficients, Ki, chosen for best fit to impermeable, uni‐directional influx or bi‐directional flux models using the F‐test.
Results
Analysis from a 2‐mm slice, 2‐mm rostral to the bregma showed a 275% increase of mean Ki during the first 20 min after infusion of human TGRL lipolysis product that differed significantly compared with saline and LpL controls. This difference disappeared by 40 min mark.
Conclusion
These results suggest human TGRL lipolysis products can lead to a transient increase in rat BBB permeability. Magn Reson Med 76:1246–1251, 2016. © 2015 The Authors. Magnetic Resonance in Medicine published by Wiley Periodicals, Inc. on behalf of International Society for Magnetic Resonance in Medicine. This is an open access article under the terms of the Creative Commons Attribution NonCommercial License, which permits use, distribution and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Keywords: hyperlipidemia, triglyceride‐rich lipoprotein, permeability, blood–brain barrier, Gd‐DTPA
INTRODUCTION
Alzheimer's disease (AD) is a progressive neurodegenerative disease characterized by irreversible memory loss, deposition of amyloid plaques, neurofibrillary tangles, and brain atrophy 1. In 2012 alone, one in eight Americans over 65 was afflicted with AD, i.e., 5.4 million patients, or approximately 2% of the U.S. population 2. AD and cardiovascular diseases (CVD) share very similar risk factors, including hypertension, apolipoprotein‐E4 genotype and type 2 diabetes 3. Furthermore, lipid profiles of AD patients are also similar to those who are at risk of CVD, and in particular high plasma triglyceride is correlated with vascular dementia and AD 4, 5, 6. Yet clinical trials aimed to lower triglyceride content using fibrate drugs have had mixed results 7, 8. There is also increased recognition that blood–brain barrier (BBB) dysfunction could be a contributing factor 6, 9, 10, 11. Simply put, there is still no consensus on the etiology of AD.
While numerous studies have demonstrated elevated fasting triglyceride concentration is a risk factor for AD, the contribution of postprandial hypertriglyceridemia to AD is unknown. Dietary lipids are absorbed in the intestine and secreted into the circulation as chylomicrons within 3 to 4 h after ingesting a moderate to high fat meal 12. Alternately, endogenously synthesized triglycerides are transported by very‐low density lipoproteins (VLDL) that are produced in the liver. Together chylomicrons and VLDL are classified as triglyceride‐rich lipoproteins (TGRL). Their compositions vary within a range of 50–90% triglyceride content and sizes vary between 25 and 1000 nm. Circulating TGRLs are hydrolyzed by endothelial‐bound lipoprotein lipase (LpL) that have been found on brain microvessels 13. Hydrolyzing TGRLs releases a conglomerate of free fatty acids and phospholipids known as lipolysis products. At high physiological to pathophysiological concentrations, these lipolysis products are harmful to human aortic endothelial cells (HAEC) and pathophysiological effects include rearrangement of junction proteins that lead to increased endothelial permeability, inflammation and apoptosis 14. Although HAEC and cerebrovascular cells are different, it is likely they can both be injured by lipolysis products. However, it is unknown if TGRL lipolysis products cause permeability changes of the BBB, even if transiently.
To test this hypothesis, we used MR contrast agent (MRCA)‐assisted MRI as used extensively in assessing BBB injury after stroke both clinically and experimentally 15, 16. MRCA‐MRI has the advantage of achieving relatively high resolution and being relatively noninvasive, allowing a subject to be monitored over time. Here in this study, MRCA‐MRI was used to measure the blood‐to‐brain influx coefficient Ki as a measure of BBB integrity in healthy Sprague‐Dawley rats after injection of saline, LpL, or prelipolyzed human TGRL (final blood concentration of 150 mg/dL TGRL, hereafter designated 150TL).
METHODS
Preparation of Human Triglyceride‐Rich Lipoprotein Lipolysis Products
As approved by the UC Davis Institutional Review Board, plasma samples were collected from healthy volunteers 3.5 h after consuming a high fat meal. TGRL from these plasma samples was isolated by ultracentrifugation at 40,000 rpm at 14°C for 18 h with a density gradient solution (ρ = 1.0063 g/mL), followed by dialysis against a 0.9% NaCl + 0.01% ethylenediaminetetraacetic acid solution overnight. Triglyceride content was assessed using an enzymatic assay kit (Sigma, St. Louis, MO). Before animal infusion, TGRL was concentrated with Amicon 3000 MWCO centrifugal filters (Millipore, Billerica, MA) before incubation for at least 30 min with LpL added at 2 U/mL per 150 mg/dL.
Animal Preparation
Twenty Sprague‐Dawley rats (Charles River Laboratories, Wilmington, MA) weighing 180–250 g were studied as approved by the Animal Use and Care Committee at the University of California, Davis. Animals were anesthetized with sodium pentobarbital (60 mg/kg body weight, i.p) and surgical plane anesthesia maintained with a quarter dose (15 mg/kg body weight, i.p) every 30 to 45 min thereafter. Body temperature was maintained by an electric heating pad during surgery and a water heating pad (Gaymar Inc., Orchard Park, NY) during MRI as described previously 17. The left femoral vein was cannulated with PE‐50 polyethylene tubing for infusion of saline, LpL or 150TL, and gadopentetate dimeglumine (Magnevist; Bayer Healthcare Pharmaceuticals, Wayne, NJ).
Blood Chemistry
Blood samples from a subset of animals were drawn from the venous cannula at the conclusion of MRI data acquisition and analyzed for electrolytes, pH, PCO2, PO2, hemoglobin, and hematocrit using the i‐STAT device (Abbott Laboratories, Abbott Park, IL).
MRI Analysis of Brain Permeability Changes
MRI data were collected using a 7 Tesla (T) Bruker Biospec MRS/MRI system (Bruker Biospin MRI, Inc., Billerica, MA). Anesthetized rats were placed supine on a custom made Plexiglas stage. The stage and rat were then placed into a 72‐mm radio frequency probe inside the 7T magnet centering 2 mm rostral to the bregma [slice 44 in the Scalable Brain Atlas 18]. A previously described rapid‐acquisition refocused‐echo (RARE) sequence was used to create T1 maps 19. In brief, images were captured using a 64‐mm field‐of‐view with a matrix of 128 × 128 pixels with a 2‐mm thickness in 1.8 min. The imaging parameters were echo time (TE) = 10.26 ms; repetition time (TR) array = 168.4, 500.2, 927.1, 1525.9, 2538, and 7500 ms; and RARE factor = 16. After a baseline T1 map was acquired, a 0.5 mL/kg bolus of Magnevist (250 μmol/kg) was injected i.v. over approximately 4 s. The time course of contrast created by each bolus was then followed to acquire a total of 10 T1 maps over a period of 18 min for each of 3 Magnevist injections. The regions of interest (ROIs) were two symmetrical ROIs, one from each hemisphere, chosen to maximize the number of pixels sampled while excluding pixels in the median eminence or outside the brain in the manner described by Nagaraja et al 20. A single pixel sampling the superior sagittal sinus (SSS) was selected to follow the change in R1 = 1/T1 for the arterial input function of each Magnevist injection as previously described 19, 21. Matlab (MathWorks, Natick, MA) was used to convert T1 maps to R1 maps and generate Patlak plots for each pixel to calculate the blood to brain transfer rate coefficient, Ki (min−1), constrained using F‐tests to select the best fit between impermeable, uni‐directional influx and bi‐directional flux models 21, 22.
Statistical Analysis
Unless otherwise stated, data are reported as mean ± SEM. Analysis of variance for repeated measures was used to test for differences between treatments using the criterion of P < 0.05. When differences between treatments were found, the Student‐Newman‐Keuls (SNK) posthoc test for multiple comparisons was used to determine which and when treatments were different.
RESULTS
Physiological Data
Venous blood samples from a subset of rats (n = 9) were analyzed and summarized in Table 1. Mean PCO2 was 57.51 ± 5.23 mmHg and there were no significant differences between treatment groups. Mean PO2 was 39.29 ± 3.66 mmHg. Because physiological venous PO2 is near 40 mmHg and PCO2 is between 40 and 50 mmHg, the rats were slightly hypoxic and hypercapnic.
Table 1.
Physiologic Data†
| Na (mmol/L) | 140.78 ± 1.33 |
|---|---|
| K (mmol/L) | 4.71 ± 0.29 |
| TCO2 (mmol/L) | 33.00 ± 1.66 |
| Ca (mmol/L) | 1.30 ± 0.05 |
| Hematocrit (%PCV) | 37.44 ± 0.60 |
| Hemoglobin (g/DL) | 12.74 ± 0.21 |
| pH | 7.35 ± 0.02 |
| PCO2 (mmHg) | 57.51 ± 5.23 |
| PO2 (mmHg) | 39.29 ± 3.66 |
| (mmol/L) | 31.14 ± 1.50 |
Venous blood samples from a subset of animals were analyzed using i‐STAT (N = 9).
Ki Was Significantly Increased in Rats Treated with TGRL Lipolysis Products
Ki was calculated on a pixel‐by‐pixel basis and then averaged over two ROIs selected symmetrically about the midline from each hemisphere away from the median eminence and ventricles. Representative images of Ki for an animal injected with 150TL are shown in Figure 1. (Data were collected from rats lying supine but the images were inverted for illustration purposes.) Representative ROIs are highlighted by green border and the SSS used for the arterial input function is indicated by the white arrow. Mean Ki for each treatment group was plotted against time (Fig. 2) demonstrating Ki for 150TL increased 275% from baseline control. Analysis of variance (ANOVA) for repeated measures showed mean Ki for 150TL was significantly greater than LPL and saline when results for all three time points were included. The SNK posthoc analysis showed Ki for 150TL was significantly greater than for LpL alone for the 0‐ to 20‐min interval but SNK showed no significant difference between treatments for the 20‐ to 40‐min interval. Standard t‐tests for the 0‐ to 20‐min interval comparing 150TL with saline and LpL infusions gave P = 0.024 and 0.035, respectively. Finally, in an effort to quantify the number of leaky sites, for each animal we counted the number of pixels with Ki 2 SDs greater than the mean of its own control Ki. Consistent with findings described above, ANOVA for repeated measures showed there was an increased number of high Ki pixels in the 150TL group compared with both LpL alone as well as for saline. Standard ANOVA followed by SNK analysis showed the significant difference between treatments occurred for the 0‐ to 20‐min interval (Fig. 3). Although SNK analysis did not show significant differences between individual treatments at any particular time, standard t‐tests comparing 150TL with saline and LpL infusions had P‐values of 0.052 and 0.026, respectively for the 0‐ to 20‐min interval and 0.081 and 0.043, respectively, for the 20‐ to 40‐min interval.
Figure 1.
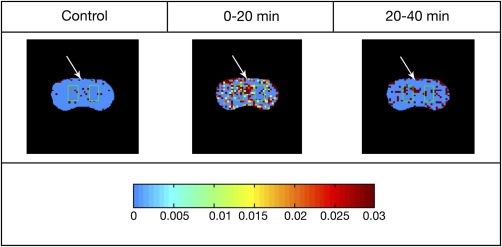
Representative images of rat brain treated with 150TL at different time points. The color bar below the images shows changes in blood–brain barrier transfer coefficient Ki varying from 0 to 0.03 min− 1. Pixels with Ki > 0.03 min− 1 are assigned the same color as Ki = 0.03 min− 1. For each map, representative ROIs in each hemisphere are highlighted by green border and the white arrow points to the SSS. Rats were lying supine during data collection and images were inverted for illustration purposes. Please see text for further detail.
Figure 2.
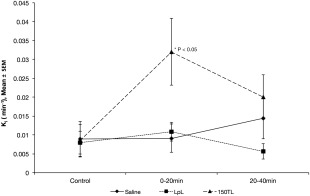
Mean blood to brain transfer rate coefficients Ki are plotted against time. ROIs are selected from brain hemispheres and combined to calculate a mean for each animal. Then mean Ki for all animals in each treatment is plotted at three different time points representing the time for data acquisition (control, 0–20 min and 20–40 min). ANOVA for repeated measures showed a significant difference (P < 0.05) in Ki between animals infused with 150TL (n = 8) and those infused with either saline (n = 6) or with LpL alone (n = 6). Posthoc analysis showed in particular that a difference between 150TL and LpL alone occurred at the 0–20 min time point (*P < 0.05).
Figure 3.
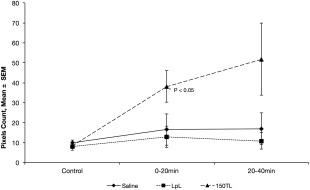
Mean number of pixels having a blood to brain transfer coefficient Ki more than 2 SDs greater than the same animal's control plotted against each time point. Using the criteria outlined by Nagaraja et al 20, pixels are considered to be very leaky if the Ki is more than 2SDs greater than control. ANOVA for repeated measures shows animals infused with 150TL have more leaky pixels by this criterion compared with those infused with saline or LpL alone (P < 0.05). Standard ANOVA for multiple comparisons showed the effect of treatment was significant for the 0–20 min interval (*P < 0.05).
DISCUSSION
The neurovascular unit remains a central focus of AD research. It is composed of such components as endothelial cells, basement membrane, astrocytes, pericytes, and neurons. The endothelial cells normally maintain a barrier to segregate blood‐borne substances from the brain extracellular fluid, i.e., the BBB, while regulating the entry of endocrine factors, immune cells and cytokines, and the secretion of cytokines 23, 24. BBB permeability can be increased by perturbations such as hypoxia, ischemia/reperfusion, inflammation, and brain tumors and assessing these changes is important for understanding the etiology of cerebrovascular diseases 25, 26.
AD is one such disease in which BBB integrity seems to play an important role. In the AD mouse model Tg2576 BBB integrity was compromised as early as 4 months in some areas of the cerebral cortex 27. In humans, disturbances in the cerebral‐spinal fluid albumin index (a measure of BBB integrity) are positively correlated with AD progression 28. Damage to the BBB in the hippocampus is also correlated with AD in the early stage 29. In an analogous system, loss of endothelial integrity in the systemic circulation is also correlated with atherosclerosis 30. In addition, risk factors for CVD also coincide with AD. These shared risk factors include hypertension, high LDL cholesterol, low HDL cholesterol, diabetes, and high triglycerides 3, 5, 31. However, evidence also exists for no overlap of these risk factors 4 and clinical trials lowering high plasma triglyceride with fibrate drugs have yielded mixed results 7, 8.
Some of these mixed results could be due to the difference between triglyceride and triglyceride lipolysis products. Vascular lipoprotein lipase hydrolyzes TGRLs and releases neutral and oxidized free fatty acids. We previously demonstrated that these lipolysis products of TGRLs caused extensive rearrangement of junctional proteins such as ZO‐1 and occludins in HAEC 14 and increased reactive oxygen species production 32. In contrast, un‐hydrolyzed TGRL did not induce similar effects, suggesting that it is the released contents from TGRL hydrolysis and not the plasma concentration of triglyceride that is deleterious to the vasculature. Therefore, prelipolyzed triglyceride was used to study the acute effect on the BBB in this study.
Our study here shows that after normal rats were fed regular chow, infusing human lipolysis products to obtain a blood level of 150 mg/dL TGRL transiently increased the blood‐to‐brain transfer coefficient (Ki) for Gd‐DTPA (Fig. 2). That is, the time course of our experiment suggests Ki increased within 20 min after injecting 150TL and then fell toward baseline within 40 min afterward. This rise‐and‐fall trend, however, does not appear to be true for the mean number of pixels having Ki more than 2 SDs above control (Fig. 3). While the change is not statistically significant using the Student‐Newman‐Keuls test, the difference in the trends could be due to variability of the pixels sampled with the MRI. While the majority of pixels made low to moderately leaky by the 150TL infusion were no longer significantly different from those treated with saline or LpL after 40 min, t‐test analysis suggests a trend showing the mean number of very leaky pixels (i.e., > 2 SD greater than baseline) is greater 40 min after 150TL treatment than after LpL infusion. One interpretation of this result is that lower permeability sites recover while more leaky sites do not recover within the same time interval. This, however, remains speculative and requires further study.
Furthermore, the infusion of human TGRL lipolysis products is physiologically relevant despite the acute nature of the infusion. Even though blood TGRL in humans rises steadily from initial fat assimilation in the intestine to peak at 3 h after ingesting a particular meal 12, allometric considerations suggest this is accelerated in rats. In addition, it is well documented that LpL can be found within brain microvessels as well as brain and nervous system 13, 33. Therefore incubating TGRL with lipoprotein lipase (LPL) before injection would likely accelerate the observed effects of TGRL lipolysis products. As a corollary, injecting TGRL without LpL incubation would likely just delay the onset of the permeability changes. Further studies will be required to test this assertion as well.
While our current study does not tell us the mechanism of the observed blood‐to‐brain transfer coefficient changes, it would likely involve the transforming growth factor‐β/SMAD pathway as suggested by previous observations in HAEC with similar treatments (unpublished results). Nonetheless certain artifacts could be ruled out. First, the Patlak model for the blood‐to‐brain transfer rate coefficient Ki (as the product of capillary permeability and surface area per tissue volume) could depend on the flow rate, as Ewing et al estimated a perfusion rate less than 50 times Ki could create a flow‐limited situation 21. The reason being that changes in blood flow could be associated with changes in surface area and/or measured permeability if contrast agent delivery is flow limited. Using an MRI sequence to generate perfusion weighted images from our samples, more than 90% of our dataset had a cerebral blood flow above this limit (data not shown).
Furthermore, to the extent that Ki measurements are flow‐limited, this will underestimate Ki. Therefore the increases we measured in the 150TL group are a conservative estimate and any possible flow‐limitation will not invalidate the conclusion that 150TL infusion increases the Ki. Second, even though the experimental animals were slightly hypoxic and hypercapnic, likely due to the use of Nembutal that is known to suppress ventilation without procedural intervention, studies have shown that only after 3 h of acute and severe hypercapnia (PACO2 > 65 mmHg), but not after continuous hypercapnia, was BBB function disturbed in rabbits 34. Mean PCO2 in the experimental animals is well below this level of hypercapnia (Table 1) and no correlation between venous PCO2 and Ki could be found (data not shown). Therefore, the observed changes in blood‐to‐brain transfer coefficient are not likely due to blood flow changes in the brain or hypoxia or hypercapnia.
While pinpointing the exact lipolysis product that caused the observed changes is beyond the scope of this study, there can be possible candidates. Apart from transporting lipid, TGRL can also transport amyloid‐beta (Aβ). Chylomicrons, a class of TGRL produced in the intestine to facilitate triglyceride transport from dietary lipid, are carriers for Aβ 35, 36. Elevated triglyceride also preceded Aβ deposition in two AD mouse models 37. Takechi et al hypothesized the increased Aβ‐chylomicron load that occurs during chronic high fat feeding might lead to more intestinally produced Aβ deposition in the brain 38. It is possible that TGRL lipolysis released this Aβ fraction that injured the BBB and led to the study observations. Further studies will be needed to distinguish the effects, if any, between TGRL lipolysis products and Aβ in our model.
In conclusion, we have demonstrated in this study that RARE‐MRI measurement of changes in T1 following contrast agent infusion is a useful technique to monitor the changes of blood–brain barrier function in vivo following TGRL lipolysis product infusion and that under the conditions of this study, TGRL lipolysis products transiently increase the blood‐to‐brain transfer coefficient for Gd‐DTPA. To our knowledge this is the first use of this technique to examine this phenomenon in vivo. This result provides important insight into the effects of lipids on BBB permeability and this technique could be a valuable tool for testing the efficacy of lipid‐modulating drugs in maintaining BBB function.
Correction added after online publication 26 April 2016. The author corrected their name from Hnin Aung to Hnin Hnin Aung.
REFERENCES
- 1. Altman R, Rutledge JC. The vascular contribution to Alzheimer's disease. Clin Sci 2010;119:407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alzheimer's disease facts and figures. Alzheimers Dement 2012;8:131–168. [DOI] [PubMed] [Google Scholar]
- 3. Stampfer MJ. Cardiovascular disease and Alzheimer's disease: common links. J Intern Med 2006;260:211–223. [DOI] [PubMed] [Google Scholar]
- 4. Sabbagh M, Zahiri HR, Ceimo J, Cooper K, Gaul W, Connor D, Sparks DL. Is there a characteristic lipid profile in Alzheimer's disease? J Alzheimers Dis 2004;6:585–589; discussion 673–581. [DOI] [PubMed] [Google Scholar]
- 5. Raffaitin C, Gin H, Empana JP, Helmer C, Berr C, Tzourio C, Portet F, Dartigues JF, Alperovitch A, Barberger‐Gateau P. Metabolic syndrome and risk for incident Alzheimer's disease or vascular dementia: the Three‐City Study. Diabetes Care 2009;32:169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bowman GL, Kaye JA, Quinn JF. Dyslipidemia and blood‐brain barrier integrity in Alzheimer's disease. Curr Gerontol Geriatr Res 2012;2012:184042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rizos E, Mikhailidis DP. Are high density lipoprotein (HDL) and triglyceride levels relevant in stroke prevention? Cardiovasc Res 2001;52:199–207. [DOI] [PubMed] [Google Scholar]
- 8. Ancelin ML, Carriere I, Barberger‐Gateau P, Auriacombe S, Rouaud O, Fourlanos S, Berr C, Dupuy AM, Ritchie K. Lipid lowering agents, cognitive decline, and dementia: the three‐city study. J Alzheimers Dis 2012;30:629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pallebage‐Gamarallage MM, Takechi R, Lam V, Galloway S, Dhaliwal S, Mamo JC. Post‐prandial lipid metabolism, lipid‐modulating agents and cerebrovascular integrity: implications for dementia risk. Atheroscler Suppl 2010;11:49–54. [DOI] [PubMed] [Google Scholar]
- 10. Bell RD, Winkler EA, Singh I, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012;485:512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Erickson MA, Banks WA. Blood‐brain barrier dysfunction as a cause and consequence of Alzheimer's disease. J Cereb Blood Flow Metab 2013;33:1500–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hyson DA, Paglieroni TG, Wun T, Rutledge JC. Postprandial lipemia is associated with platelet and monocyte activation and increased monocyte cytokine expression in normolipemic men. Clin Appl Thromb Hemost 2002;8:147–155. [DOI] [PubMed] [Google Scholar]
- 13. Wang H, Eckel RH. Lipoprotein lipase in the brain and nervous system. Annu Rev Nutr 2012;32:147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eiselein L, Wilson DW, Lame MW, Rutledge JC. Lipolysis products from triglyceride‐rich lipoproteins increase endothelial permeability, perturb zonula occludens‐1 and F‐actin, and induce apoptosis. Am J Physiol Heart Circ Physiol 2007;292:H2745–H2753. [DOI] [PubMed] [Google Scholar]
- 15. Nagaraja TN, Knight RA, Ewing JR, Karki K, Nagesh V, Fenstermacher JD. Multiparametric magnetic resonance imaging and repeated measurements of blood‐brain barrier permeability to contrast agents. Methods Mol Biol 2011;686:193–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Taheri S, Gasparovic C, Shah NJ, Rosenberg GA. Quantitative measurement of blood‐brain barrier permeability in human using dynamic contrast‐enhanced MRI with fast T1 mapping. Magn Reson Med 2011;65:1036–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lam TI, Anderson SE, Glaser N, O'Donnell ME. Bumetanide reduces cerebral edema formation in rats with diabetic ketoacidosis. Diabetes 2005;54:510–516. [DOI] [PubMed] [Google Scholar]
- 18. Papp EA, Leergaard TB, Calabrese E, Johnson GA, Bjaalie JG. Waxholm Space atlas of the Sprague Dawley rat brain. Neuroimage 2014;97:374–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walton JH, Ng KF, Anderson SE, Rutledge JC. MRI measurement of blood‐brain barrier transport with a rapid acquisition refocused echo (RARE) method. Biochem Biophys Res Commun 2015;463:479–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nagaraja TN, Karki K, Ewing JR, Croxen RL, Knight RA. Identification of variations in blood‐brain barrier opening after cerebral ischemia by dual contrast‐enhanced magnetic resonance imaging and T 1sat measurements. Stroke 2008;39:427–432. [DOI] [PubMed] [Google Scholar]
- 21. Ewing JR, Knight RA, Nagaraja TN, Yee JS, Nagesh V, Whitton PA, Li L, Fenstermacher JD. Patlak plots of Gd‐DTPA MRI data yield blood‐brain transfer constants concordant with those of 14C‐sucrose in areas of blood‐brain opening. Magn Reson Med 2003;50:283–292. [DOI] [PubMed] [Google Scholar]
- 22. Ewing JR, Brown SL, Lu M, et al. Model selection in magnetic resonance imaging measurements of vascular permeability: Gadomer in a 9L model of rat cerebral tumor. J Cereb Blood Flow Metab 2006;26:310–320. [DOI] [PubMed] [Google Scholar]
- 23. Erickson MA, Dohi K, Banks WA. Neuroinflammation: a common pathway in CNS diseases as mediated at the blood‐brain barrier. Neuroimmunomodulation 2012;19:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Banks WA. Brain meets body: the blood‐brain barrier as an endocrine interface. Endocrinology 2012;153:4111–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abbott NJ. Inflammatory mediators and modulation of blood‐brain barrier permeability. Cell Mol Neurobiol 2000;20:131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood‐brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol 2006;1:223–236. [DOI] [PubMed] [Google Scholar]
- 27. Ujiie M, Dickstein DL, Carlow DA, Jefferies WA. Blood‐brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation 2003;10:463–470. [DOI] [PubMed] [Google Scholar]
- 28. Bowman GL, Kaye JA, Moore M, Waichunas D, Carlson NE, Quinn JF. Blood‐brain barrier impairment in Alzheimer disease: stability and functional significance. Neurology 2007;68:1809–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Montagne A, Barnes SR, Sweeney MD, et al. Blood‐brain barrier breakdown in the aging human hippocampus. Neuron 2015;85:296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation 2007;115:1285–1295. [DOI] [PubMed] [Google Scholar]
- 31. Luchsinger JA, Mayeux R. Cardiovascular risk factors and Alzheimer's disease. Curr Atheroscler Rep 2004;6:261–266. [DOI] [PubMed] [Google Scholar]
- 32. Wang L, Gill R, Pedersen TL, Higgins LJ, Newman JW, Rutledge JC. Triglyceride‐rich lipoprotein lipolysis releases neutral and oxidized FFAs that induce endothelial cell inflammation. J Lipid Res 2009;50:204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brecher P, Kuan HT. Lipoprotein lipase and acid lipase activity in rabbit brain microvessels. J Lipid Res 1979;20:464–471. [PubMed] [Google Scholar]
- 34. Pakulski C. [The influence of acute hypercapnia on the permeability of the blood‐brain barrier for gentamycin under conditions of general anesthesia in rabbits]. Ann Acad Med Stetin 1998;44:285–296. [PubMed] [Google Scholar]
- 35. Mamo JC, Jian L, James AP, Flicker L, Esselmann H, Wiltfang J. Plasma lipoprotein beta‐amyloid in subjects with Alzheimer's disease or mild cognitive impairment. Ann Clin Biochem 2008;45(Pt 4):395–403. [DOI] [PubMed] [Google Scholar]
- 36. Galloway S, Jian L, Johnsen R, Chew S, Mamo JC. beta‐amyloid or its precursor protein is found in epithelial cells of the small intestine and is stimulated by high‐fat feeding. J Nutr Biochem 2007;18:279–284. [DOI] [PubMed] [Google Scholar]
- 37. Burgess BL, McIsaac SA, Naus KE, et al. Elevated plasma triglyceride levels precede amyloid deposition in Alzheimer's disease mouse models with abundant A beta in plasma. Neurobiol Dis 2006;24:114–127. [DOI] [PubMed] [Google Scholar]
- 38. Takechi R, Galloway S, Pallebage‐Gamarallage MM, Mamo JC. Chylomicron amyloid‐beta in the aetiology of Alzheimer's disease. Atheroscler Suppl 2008;9:19–25. [DOI] [PubMed] [Google Scholar]