

Abstract
Objective
Hearing loss and enlargement of the vestibular aqueduct (EVA) can be inherited as an autosomal recessive trait caused by mutant alleles of the SLC26A4 gene. In some other families, EVA does not segregate in a typical autosomal recessive pattern. The goal of this study was to characterize the SLC26A4 genotypes and phenotypes of extended families with atypical segregation of EVA.
Study Design
Prospective study of cohort of families ascertained between 1998 and 2014 at the National Institutes of Health Clinical Center.
Methods
Study subjects were members of eight families segregating EVA in at least two members who were not related as siblings. Evaluations included pure-tone audiometry, temporal bone imaging, SLC26A4 nucleotide sequence analysis, SLC26A4-linked marker genotype and haplotype analysis, and pedigree analysis.
Results
One family had members with EVA caused by different etiologies, and two families had pseudo-dominant inheritance of recessive mutations of SLC26A4. In five families, the etiology remained unknown and could include inheritance of mutant alleles at another genetic locus, non-genetic influences, or a combination of these factors.
Conclusions
Familial EVA can demonstrate a variety of atypical segregation patterns. Pseudo-dominant inheritance of SLC26A4 mutations or recessive alleles of other hearing loss genes may be more likely to occur in families in which deaf individuals have intermarried. The etiologic basis of atypical segregation of EVA without detectable SLC26A4 mutations remains unknown. Future studies of these families may reveal novel genes for EVA.
Keywords: assortative mating, deafness, genetic, hearing loss, pseudo-dominant, SLC26A4
INTRODUCTION
Enlargement of the vestibular aqueduct (EVA) is the most common malformation observed in imaging studies of temporal bones of children with sensorineural hearing loss1. EVA was originally defined as a diameter >1.5 mm at the midpoint of the vestibular aqueduct2, but recent studies have shown that a midpoint diameter >1.0 mm is specifically associated with hearing loss3. The hearing loss associated with EVA can have a pre-, peri- or even post-lingual onset and can affect one or both ears4. In patients with bilateral EVA, the hearing loss is usually asymmetric. A distinctive clinical feature of EVA is sudden loss or fluctuation in hearing. In some EVA ears, sudden loss or fluctuation can be precipitated by minor head trauma or barotrauma.
Mutations of the SLC26A4 gene are one etiology of EVA. SLC26A4, which is located on chromosome 7, was initially discovered as the gene underlying Pendred syndrome5, an autosomal recessive disorder originally described as the combination of congenital deafness and thyroid goiter6. Numerous studies have further elucidated the auditory and thyroid phenotypic spectrum associated with SLC26A4 mutations. Ultrasonography is a non-invasive and quantitative method to detect the abnormalities of texture (e.g., nodules) and volume in Pendred syndrome thyroid glands7. However, the goiter is not always present and, when it occurs, usually does not manifest until the second decade of life7. Therefore, many EVA patients present with nonsyndromic hearing loss8. Serologic thyroid function tests are not sensitive for the early detection of Pendred syndrome, whereas the perchlorate discharge test is a sensitive method to detect the presymptomatic iodine organification defect in the thyroid glands of patients with Pendred syndrome glands7. However, SLC26A4 mutation testing and temporal bone imaging have essentially replaced the perchlorate discharge test for the diagnosis of Pendred syndrome. EVA is now recognized to be the most penetrant manifestation of SLC26A4 mutations9.
Approximately 25% of North American-European Caucasian patients with EVA have two detectable mutant SLC26A4 alleles (termed M2), as expected for an autosomal recessive trait10–12. Another 25% of patients have only one detectable mutant SLC26A4 allele (termed M1), which is an incompletely diagnostic result. Co-segregation of SLC26A4-linked markers with EVA in M1 families suggests that undetected SLC26A4 mutations underlie the indistinguishable familial recurrence risk observed in M1 versus M2 families12. In contrast, the significantly lower recurrence risk and discordant segregation of SLC26A4-linked markers with EVA in M0 families suggests that other etiologies account for EVA in those patients12.
In order to identify additional genes underlying EVA, we focused our study recruitment on families with multiple affected members. Among those families, we identified 8 with atypical segregation of EVA: i.e. affected members in at least two sibships or generations in an extended family. We use the term segregation instead of inheritance since we could not be certain of a genetic etiology in all of the families. The SLC26A4 genotypes, phenotypes, and segregation patterns in our study families provide insight into possible etiologies of EVA and the foundation for investigations to identify novel genes for EVA.
SUBJECTS AND METHODS
Subjects, methods, and their description are essentially as reported7, 10, 11, 13, 14. Our study was approved by the Combined Neuroscience Institutional Review Board, National Institutes of Health (NIH), Bethesda, Maryland. Written informed consent was obtained for all subjects. Self-reported race and ethnicity were classified according to our institutional review board reporting guidelines.
Subjects were evaluated at the NIH Clinical Center as previously described7, 10, 11, 13, 14. We relied upon records from outside health care providers for subjects who did not come to the NIH Clinical Center. All MRI or CT scans were reviewed by the same neuroradiologist (JAB) and otolaryngologist-head and neck surgeon (AJG). We originally defined a vestibular aqueduct (VA) as enlarged if its diameter exceeded 1.5 mm at the midpoint between the posterior cranial fossa and the vestibule of the inner ear10. We subsequently revised our midpoint diameter criterion to >1.0 mm. Auditory evaluations included pure-tone and speech audiometry and CT and MRI of the temporal bones14. We defined normal hearing as air conduction thresholds of ≤15 db HL for children (≤18 years old), and ≤25 db HL for adults, at the 6 octave test frequencies from 250 Hz to 8 kHz. For study subjects with known hearing loss, we considered their hearing loss to be inconsistent with EVA if they had an imaging study of the temporal bones that was negative for EVA, or if the loss had an onset during adulthood or was otherwise phenotypically distinguishable from EVA. Evaluation of the thyroid gland included: (1) measurement of peripheral venous blood levels of thyrotropin, thyroxine, free thyroxine, triiodothyronine, thyroglobulin, anti-thyroid peroxidase and anti-thyroglobulin antibodies, and thyroid-binding globulin; (2) ultrasonographic assessment of thyroid volume and texture; and, in some cases, (3) perchlorate discharge testing7. Ultrasonographic examinations were all analyzed by the same radiologist (THS), and perchlorate discharge studies were all performed according to our standard protocol7. The results of serologic, ultrasonographic and perchlorate discharge studies were all interpreted according to our objective published criteria7.
We identified eight families with at least two or more non-sibling members with EVA (Figure 1). We thus excluded families in which EVA was present only in a single sibship. We included families in which there was EVA present in a proband and parent (388, 442), grandparent (151), or great aunt (414), as well as families with more complex patterns of segregation. We included family 443 because 3 siblings had EVA and the father had bilateral sensorineural hearing loss with bilateral cochlear modiolar hypoplasia, thought by some authors to be a forme fruste of ears with EVA15. The race of the 8 families was white/Caucasian and ethnicity was non-Hispanic.
Figure 1. Study families segregating EVA.

Pedigrees are shown for families 151, 414, 347, 317, 388, 442, 443, and 372. Enlargement of the vestibular aqueduct(s) is indicated by black symbols. Hearing loss consistent with EVA but not confirmed by radiologic imaging is indicated by gray symbols. Hearing loss that is known or suspected, based upon clinical presentation, to be unassociated with EVA is indicated by striped symbols. SLC26A4 genotypes are shown below the subject identification number. N324Y is shown for family 388 (E) although it is considered to be a benign variant.
We extracted genomic DNA and sequenced the 21 exons and flanking intronic regions of SLC26A410, 11, 13. We performed haplotype analyses of SLC26A4-linked short tandem repeat markers (D7S496, D7S2459 and D7S2456) to track segregation of wild type alleles of SLC26A4. Zygosity of the twins (2107, 2108) in family 388 and parental relationships were determined by analysis of unlinked short tandem repeat markers spanning the rest of the genome12. The trans configuration of all compound heterozygous variants was confirmed by sequence analysis of parental DNA.
We classified the pathogenic potential of SLC26A4 variants according to American College of Medical Genetics guidelines16. To assess the pathogenic potential of p.V186F and p.N324Y, we generated complementary DNA (cDNA) expression constructs encoding wild type or mutant variants of SLC26A4 fused at its C-terminus to green fluorescent protein, transfected COS-7 cells, and evaluated intracellular localization by confocal microscopy11, 13. We evaluated anion exchange activity of the p.V186F and p.N324Y variants, fused at their C-terminus to a poly-histidine tag, 11, 13 encoded by complementary RNA (cRNA) injected into Xenopus oocytes. Chlorine-36 (36Cl−) influx and efflux were measured and analyzed as reported previously11, 13. SLC26A4 mutation c.2320-2A>G (IVS20-2A>G) was considered pathogenic due to its predicted effect on a consensus splice acceptor site.
RESULTS
Family 151 (Figure 1A) included a previously reported female proband (1387) with bilateral EVA and an indeterminate thyroid phenotype10. Phenotypes of all affected subjects are summarized in Table 1. Her paternal grandparents (1437, 1438) were reported to have severe to profound deafness since childhood but were not available for temporal bone imaging. Her parents (1364, 1365) had normal hearing and her father (1364) had a temporal bone CT scan with normal findings. The proband was compound heterozygous for the pathogenic p.G209V and p.Y530S mutations of SLC26A4. Assuming the paternal grandmother had bilateral EVA, the vertical segregation of EVA in family 151 can be explained by transmission of p.G209V through the paternal lineage.
Table 1.
Clinical phenotypes of study subjects
| Family | Subject | Age (y) | Gender | Hearing Loss | EVA | Goiter | Thyroid stimulating hormone | Perchlorate discharge | SLC26A4 allele 1 | SLC26A4 allele 2 |
|---|---|---|---|---|---|---|---|---|---|---|
| 151 | 1387 | 3 | F | Bilateral | Bilateral | ND | Normal | ND | G209V | Y530S |
| 151 | 1437 | 72 | M | Bilateral | ND | ND | ND | ND | G209V | L236P |
| 317 | 1904 | 6 | F | Bilateral | Bilateral | ND | High | ND | V186F | IVS20-2A>G |
| 317 | 2249 | 40 | M | Bilateral | Bilateral | Yes | Normal | ND | IVS8+1G>A | E384G |
| 317 | 1906 | 36 | F | Bilateral | ND | ND | Low | ND | V186F | IVS8+1G>A |
| 317 | 1908 | 61 | F | Bilateral | ND | ND | ND | ND | IVS8+1G>A | T416P |
| 347 | 1991 | 6 | F | Left | Bilateral | No | Normal | Normal | wt | wt |
| 347 | 1992 | 3 | M | Right | Bilateral | No | Normal | ND | wt | wt |
| 347 | 2044 | 17 | M | Bilateral | Bilateral | No | Normal | Normal | wt | wt |
| 372 | 2059 | 29 | F | Bilateral | Bilateral | No | Normal | ND | wt | wt |
| 372 | 2060 | 1-5/12 | F | Bilateral | Bilateral* | No | Normal | ND | wt | wt |
| 372 | 2058 | 30 | M | Left | Left** | No | Normal | ND | wt | wt |
| 372 | 2061 | 29 | M | Bilateral | Bilateral | ND | ND | ND | wt | wt |
| 388 | 2106 | 41 | F | Bilateral | Bilateral | No | Normal | Normal | N324Y | wt |
| 388 | 2107 | 8 | M | Bilateral | Bilateral | No | Normal | Normal | N324Y | wt |
| 388 | 2108 | 8 | M | Bilateral | Bilateral | No | Normal | Normal | N324Y | wt |
| 414 | 2186 | 7 | M | Left | Bilateral | No | Normal | ND | G209V | F335L |
| 414 | 2173 | 46 | F | Bilateral | Bilateral | No | High*** | ND | wt | wt |
| 414 | 2188 | 1-1/12 | M | Suspected**** | ND | ND | ND | ND | G209V | F335L |
| 442 | 2238 | 38 | F | Bilateral | Bilateral | No | Normal | ND | wt | wt |
| 442 | 2240 | 3 | M | Bilateral | Bilateral | No | Normal | ND | wt | wt |
| 443 | 2246 | 9 | M | Bilateral | Bilateral | No | Normal | ND | wt | wt |
| 443 | 2247 | 4 | M | Bilateral | ND | No | Normal | ND | wt | wt |
| 443 | 2248 | 1-0/12 | M | Bilateral | Bilateral | No | Normal | ND | wt | wt |
ND, not done.
Left endolymphatic duct diameter = 1.5 mm; right endolymphatic duct diameter > 1.5 mm.
Endolymphatic duct diameter = 1.3 mm.
Repeat measurement was normal.
Passed newborn hearing screen; history of delayed speech and language development; serial audiologic assessments unable to detect hearing loss.
Family 414 (Figure 1B) included a male proband (2186) with bilateral EVA and a normal thyroid phenotype assessed by ultrasonography and serologic tests of thyroid function. He had normal-hearing parents (2184, 2185) and a 46-year old paternal great aunt (2173) with bilateral EVA and a normal thyroid phenotype assessed by ultrasonography. Her thyroid stimulating hormone (TSH) level, 4.21 mIU/L, was slightly elevated (normal range = 0.40–4.00 mIU/L). A second sample was drawn and the TSH level was within the normal range. The proband (2186) was compound heterozygous for the pathogenic variant p.G209V and the likely pathogenic variant p.F335L of SLC26A4. The paternal great aunt (2173) had no detectable SLC26A4 variants, indicating that EVA had different etiologies in these different family members.
Family 347 (Figure 1C) included two maternally related sibships with three first cousins (1991, 1992, 2044) with bilateral EVA and normal thyroid phenotypes. Three of the parents (1993, 1994, 2043) had normal hearing. One father (2004) had a unilateral mild to moderate sensorineural hearing loss at 4000 to 8000 Hz with a notched audiometric configuration, consistent with his history of exposure to recreational music and fireworks. We were unable to obtain an MRI scan. The grandparents were reported to have age-appropriate normal hearing and this was confirmed by pure-tone audiometry in the grandfather. Subjects 1991, 1992, and 2044 had no detectable SLC26A4 mutations. Haplotype analyses of SLC26A4-linked short tandem repeat markers in 1991, 1992, 1993, and 1994 indicated that 1991 and 1992 inherited different SLC26A4 alleles from their parents, suggesting that their EVA may have been caused by X-linked or autosomal recessive alleles at another locus. The etiology of EVA in their first cousin (2044) remained unclear.
The female proband (1904) of family 317 (Figure 1D) had bilateral EVA and an indeterminate thyroid phenotype. Her parents (1906, 1907), maternal grandparents (1908, 1909), and two maternal uncles all had severe-to-profound hearing loss. One uncle (2249) had bilateral EVA and a goiter. The other uncle was not available for temporal bone imaging or thyroid evaluations. A brother (1905) with normal hearing at 8 years of age (Figure 2A) was unavailable for temporal bone imaging or follow-up audiologic evaluation. There were six pathogenic SLC26A4 variants segregating in the family: the pathogenic mutations c.1151A>G (p.E384G), c.1246A>C (p.T416P), c.1001+1G>A (IVS8+1G>A) and c.1588T>C (p.Y530H)11, the missense variant c.556G>T (p.V186F), and the splice site variant c.2320-2A>G (IVS20-2A>G). The prevalence of p.E384G, p.T416P and IVS8+1G>A11 among European Americans was 0.0349%, 0.0116%, and 0.0581%, respectively, as compiled in the Exome Variant Server accessed June 19, 2014 (http://evs.gs.washington.edu/EVS/). The p.V186F, p.Y530H and IVS20-2A>G variants were not annotated in the Exome Variant Server, indicating that they are very rare. There were no GJB2 mutations detected in the participating family members. The mutant allele product SLC26A4V186F failed to traffic to the plasma membrane of COS-7 cells (Figure 3). Furthermore, the mutant exhibited minimal or undetectable anion transport activity in Xenopus oocytes as measured by either unidirectional 36Cl− influx (Figure 4), or by 36Cl− efflux under conditions of Cl−/HCO3− exchange or Cl−/I− exchange (Supplemental Figure 1), indicating that this mutation is pathogenic.
Figure 2. Pure-tone audiometry.

Subject 1905 was an eight year-old male with normal pure-tone hearing thresholds. Subject 2244 was a 35 year-old male with bilateral mild-to-moderate sensorineural hearing loss.
Figure 3. Trafficking of SLC26A4 variants in COS-7 cells.

Confocal epifluorescence micrograph of COS-7 cells transiently transfected with cDNA encoding wild type or variant SLC26A4-GFP. Texas Red-conjugated concanavalin A counterstain (channel 2, upper row) binds the external surface of the cell to highlight the plasma membrane. SLC26A4V186F-GFP was retained in intracellular compartments, whereas wild type SLC26A4-GFP and SLC26A4N324Y-GFP trafficked to the plasma membrane (channel 1, lower row).
Figure 4. Anion transport activity of SLC26A4 variants.

36Cl− influx into Xenopus ooyctes previously injected with 1 ng cRNAs encoding either wild-type SLC26A4 or its variants Y186F and N324Y. Uninjected oocytes serve as negative control. Values are means ± s.e.m. for 10 oocytes. *, p < 0.05 by ANOVA with Tukey post-hoc test. Shown is one of two similar experiments, using oocytes from different frogs, showing indistinguishable reductions in relative uptake activities of the variant proteins.
We conclude that IVS20-2A>G is also pathogenic based upon its predicted disruption of the canonical splice acceptor site sequence. The proband 1904 was compound heterozygous for p.V186F and IVS20-2A>G. Her mother (1906), father (1907), maternal grandmother (1908), maternal grandfather (1909), and maternal uncle (2249) were also compound heterozygous for pathogenic mutations of SLC26A4, and were thus likely to have had EVA even though it was not documented. The proband’s normal-hearing brother (1905) shared the same compound heterozygous mutant genotype, but we lacked sufficient clinical data to distinguish conclusively between non-penetrance and presymptomatic EVA. We conclude that the vertical transmission of hearing loss, and documented or presumed EVA, in a pseudo-dominant pattern resulted from segregation of SLC26A4 mutations in family 317. Pseudo-dominant inheritance refers to the appearance of a dominant mode of inheritance when, in reality, the phenotype is associated with inheritance of two autosomal recessive alleles.
Family 388 (Figure 1E) comprised a mother (2106) and her monozygotic twin sons (2107, 2108), all with bilateral EVA. Subject 2106 had a normal-sized thyroid gland with a 3.8 × 5.4 × 5.1 mm solid nodule. Subjects 2107 and 2108 had normal-sized thyroid glands with a small cyst and a probable collapsed cyst, respectively. All three subjects had normal thyroid function and normal perchlorate discharge test results. The maternal grandmother had hearing loss detected in the 5th decade of life and required a cochlear implant in the 8th decade. Computed tomography of her temporal bones was interpreted as negative for EVA. The scan was not available for our review. A maternal uncle was reported to have progressive hearing loss since the age of 10 years. He currently uses bilateral hearing aids but no imaging studies or other results were available. The mother and sons (2106, 2107, 2108) were all heterozygous for c.970A>T (p.N324Y), which we previously reported as nonpathogenic13. The allele frequencies calculated from Exome Variant Server data (http://evs.gs.washington.edu/EVS/; accessed April10, 2015) were 0.05% among European Americans and 2.7% among African Americans. The variant allele product SLC26A4N324Y trafficked to the plasma membrane of COS-7 cells in a pattern similar to that of wild type SLC26A4 (Figure 3). Moreover, SLC26A4N324Y exhibited normal anion transport activity as measured in Xenopus oocytes by 36Cl− influx (Figure 4) and by 36Cl− efflux in conditions of Cl−/HCO3− exchange and Cl−/I− exchange (Supplemental Figure 2). These data strongly suggest that the p.N324Y mutation is benign. The segregation of EVA in family 388 may reflect inheritance of a mutant allele at another locus. Alternatively, EVA in twin offspring (2107, 2108) may reflect our previous observation of a potential causal association of twinning with EVA12. Thus it is possible that the etiology of EVA differs between the mother and sons in family 388.
The affected members of family 442 (Figure 1F) were a mother (2238) and son (2240) with bilateral EVA. Their thyroid function testing and ultrasonography results were normal. They had no detectable SLC26A4 mutations and the results of outside OtoChip™ testing for Hearing Loss and Usher Syndrome were negative for diagnostic variants in known hearing loss genes. The segregation of EVA in family 442 may reflect dominant, pseudo-dominant, or X-linked inheritance at another locus, different etiologies in the mother and son, or a combination of these possibilities.
The three affected male offspring (2246, 2247, 2248) of family 443 (Figure 1G) all had bilateral sensorineural hearing loss. The siblings 2246 and 2248 had bilateral EVA. Sibling 2247 had not had temporal bone imaging. Their mother (2245) had normal hearing but their father (2244) had bilateral mild to moderate sensorineural hearing loss (Figure 2B). Magnetic resonance imaging of his temporal bones revealed bilateral hypoplasia of the cochlear modiolus but no EVA (Figure 5). Since modiolar hypoplasia has been suggested to be a forme fruste of EVA15, the father (2244) could be considered to have the same phenotype as his sons, albeit with reduced severity. Hearing was reported to be normal for members of the extended family. There were no SLC26A4 mutations detected in 2244, 2246, 2247, or 2248. The segregation of EVA in family 443 may reflect semi-dominant or pseudo-dominant inheritance at another locus or different etiologies in the father and sons.
Figure 5. Magnetic resonance imaging.
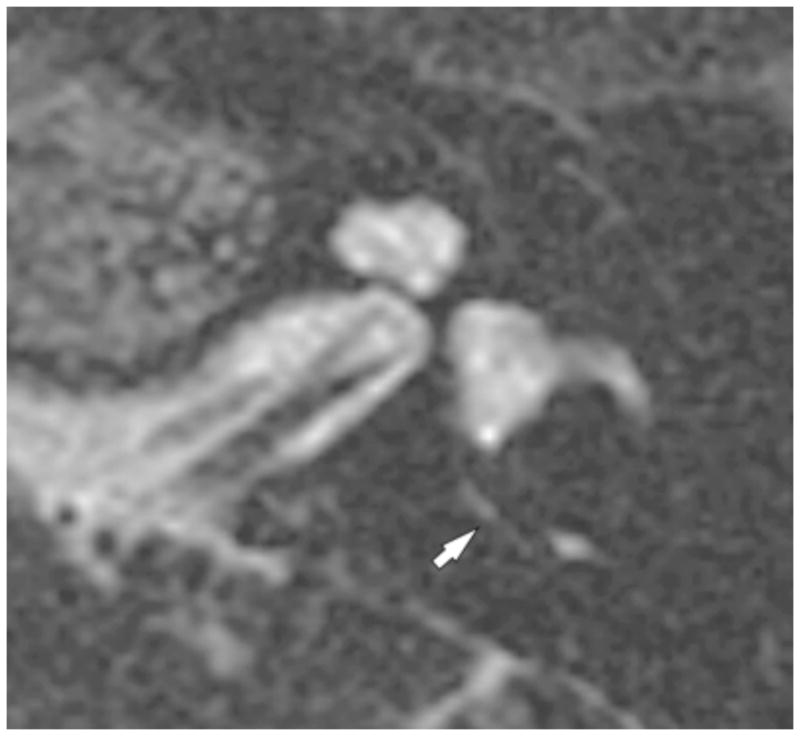
The left temporal bone of subject 2244 demonstrated on 3D T2 VISTA MRI reformatted along the axis of the modiolus. The normal-caliber endolymphatic duct is indicated (white arrow). Subject 2244 had bilateral hypoplasia of the cochlear modiolus but no enlargement of the endolymphatic sacs or ducts.
Family 372 (Figure 1H) segregated EVA in a complex pattern. The proband (2060) had bilateral EVA. Her father (2058) had unilateral EVA whereas her mother (2059) and a maternal uncle (2061) had bilateral EVA. There were no reports of hearing loss or EVA among other members of the extended family. A two year-old male sibling (2225) without documented hearing loss was reported to have had a single episode of unexplained, sudden-onset ataxia requiring hospitalization, with a reportedly normal magnetic resonance imaging study of the temporal bones at another facility. He was not available for evaluation at the NIH Clinical Center. We detected no SLC26A4 mutations in subjects 2058, 2059, 2060, 2061 or 2225. The segregation of EVA in the mother (2059) and maternal uncle (2061) are consistent with autosomal recessive or X-linked inheritance at another locus. If EVA in the father (2058) and mother (2059) was caused by mutant alleles at the same locus, the segregation of EVA in the proband (2060) would reflect pseudo-dominant or dominant inheritance at that locus. A shared etiology remained possible, since the parents shared remote ethno-religious ancestry. However, we cannot rule out different etiologies in different members of the family17.
DISCUSSION
We have ascertained and described eight families, each with EVA in at least two members who were not related as siblings. In our experience and in the literature, occurrence of EVA in at least two sibships of an extended family is rare. In some cases, this represented multiple etiologies within the same family, as we showed for family 414. Intra-familial etiologic heterogeneity was also a possibility in five of the other families in our study. In two families, 151 and 317, the vertical segregation of EVA was the result of pseudo-dominant inheritance of recessive mutations of SLC26A4. This pattern of inheritance of SLC26A4 mutations and Pendred syndrome has been previously reported18,19,20.
In general, pseudo-dominant inheritance of recessive hearing loss alleles is rare in outbred populations due to non-assortative (random) mating and the low prevalence of mutant alleles. Pseudo-dominant inheritance of hearing loss in the deaf population can result from assortative mating and a higher prevalence of hearing loss alleles21. It is not surprising to observe pseudo-dominant inheritance of recessive mutations of SLC26A4, since they appear to be the second most common known cause of genetic deafness across the globe18.
Five study families (347, 372, 388, 442, and 443) did not segregate detectable SLC26A4 mutations. Furthermore, EVA did not co-segregate with SLC26A4-linked markers, suggesting that EVA was not caused by undetected mutations of SLC26A4. These M0 families may segregate mutations of another gene(s) at another locus or loci. The non-complementary mating in family 372 raises the possibility of a major gene or mutation accounting for EVA in affected members of these families and others. The low recurrence risk of EVA in M0 families indicates that mutations of another gene or other genes are a rare cause of EVA, have low penetrance, or both12.
Variants of FOXI1, KCNJ10, and the 5′-untranslated region of SLC26A4 have been proposed by Yang et al. to cause EVA22,23, but those hypotheses have not been supported in other studies to date24,25,26,27. It remains possible that the low recurrence risk of EVA in M0 families reflects a non-genetic etiology. Congenital cytomegalovirus infection is a common cause of childhood hearing loss, but it is not a major cause of EVA28. There is also an association of twinning with EVA12, but twinning cannot account for EVA in most of the families in this study.
The primary weakness of our study was incomplete clinical data for some of the members of the study families. For families 151, 317 and 414, the lack of phenotypic data did not affect the interpretation of segregation. Additional clinical data could potentially clarify the phenotype in some members of the five M0 families for whom the underlying etiology remains unknown. However, it seems unlikely the additional data would affect our interpretation of the segregation pattern.
Although the etiology of M0 EVA remains unknown, it deserves further investigation since approximately 50% of all EVA probands have no detectable SLC26A4 mutations. Identifying other etiologies of EVA will facilitate counseling of patients and families, our understanding of the etiology of hearing loss in EVA, as well as stratification of patients for potential interventions to prevent, reduce, or reverse the hearing loss associated with EVA.
CONCLUSIONS
Segregation of EVA with hearing loss can demonstrate a variety of atypical patterns in families. SLC26A4 mutation testing is a critical diagnostic test in families with atypical patterns of segregation of EVA. Pseudo-dominant inheritance of SLC26A4 mutations can account for atypical segregation of EVA in families with intermarriage among deaf individuals. The etiologic basis of atypical segregation of EVA in families without SLC26A4 mutations remains unknown. These families may allow discovery of novel genes and mechanisms for EVA.
Supplementary Material
Supplemental Figure 1. 36Cl− efflux measurements of pendrin V186F-mediated Cl−/HCO3− exchange and Cl−/I− exchange.
A. Traces of 36Cl− efflux from a representative uninjected oocyte and from individual oocytes previously injected with cRNA encoding either wild-type pendrin or pendrin mutant V186F. Oocytes were exposed sequentially to baths containing 72 mM NaCl plus 24 mM Na cyclamate, 72 mM NaCl plus 24 mM NaHCO3, 72 mM Na cyclamate plus 24 mM NaHCO3, and 96 mM Na cyclamate. B. 36Cl− efflux rate constants of oocytes subjected to the experimental protocol of panel A. C. 36Cl− efflux rate constants from oocytes previously injected with water or with cRNAs encoding wild type (WT) or mutant (V186F) pendrin. Oocytes were exposed sequentially to baths containing 96 mM NaCl (ND96), 91 mM Na cyclamate plus 5 mM Na iodide (I−), and 96 mM Na cyclamate in an experimental format similar to that shown in panel A. Values are means ± s.e.m. for (n) oocytes. Statistical significance was determined by ANOVA with Bonferroni post-hoc test. (*, p <0.05 vs. WT pendrin; NS, not significant, for (n) oocytes as indicated).
Supplemental Figure 2. 36Cl− efflux measurements of pendrin N324Y-mediated Cl−/HCO3− exchange and Cl−/I− exchange.
A. Traces of 36Cl− efflux from a representative uninjected oocyte and from individual oocytes previously injected with cRNA encoding either wild-type pendrin or pendrin mutant N324Y. Oocytes were exposed sequentially to baths containing 24 mM NaCl plus 72 mM Na cyclamate (24 Cl−), 24 mM NaHCO3 plus 72 mM Na cyclamate (24 HCO3−), and 96 mM Na cyclamate. B. 36Cl− efflux rate constants of oocytes subjected to the experimental protocol of panel A. C. 36Cl− efflux rate constants from uninjected oocyte and from oocytes previously injected with cRNAs encoding wild type (WT) or mutant (N324Y) pendrin. Oocytes were exposed sequentially to baths containing 1) 96 mM NaCl (ND96), 91 mM Na cyclamate plus 5 mM Na iodide (I−), and 96 mM Na cyclamate. Values are means ± s.e.m. for (n) oocytes. Statistical significance was determined by ANOVA with Bonferroni post-hoc test. (*, p <0.05 vs. WT pendrin; NS, not significant, for (n) oocytes as indicated
Acknowledgments
Funding: NIH intramural research funds Z01-DC-000060-13, Z01-DC-000064-13 and Z01-DC-000082-02.
We thank Dennis Drayna and Tom Friedman for critical review of the manuscript. We thank the staff members of the NIDCD clinic and NIH Clinical Center for their support of the study and subjects.
Footnotes
Conflicts of Interest: the authors have no relevant financial interests to disclose.
Level of Evidence: NA
References
- 1.Antonelli PJVA, Mancuso AA. Diagnostic yield of high-resolution computed tomography for pediatric sensorineural hearing loss. Laryngoscope. 1999;109:1642–1647. doi: 10.1097/00005537-199910000-00018. [DOI] [PubMed] [Google Scholar]
- 2.Valvassori GE, Clemis JD. The large vestibular aqueduct syndrome. Laryngoscope. 1978;88:723–728. doi: 10.1002/lary.1978.88.5.723. [DOI] [PubMed] [Google Scholar]
- 3.Boston M, Halsted M, Meinzen-Derr J, et al. The large vestibular aqueduct: a new definition based on audiologic and computed tomography correlation. Otolaryngol Head Neck Surg. 2007;136:972–977. doi: 10.1016/j.otohns.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 4.Griffith AJ, Wangemann P. Hearing loss associated with enlargement of the vestibular aqueduct: mechanistic insights from clinical phenotypes, genotypes, and mouse models. Hear Res. 2011;281:11–17. doi: 10.1016/j.heares.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Everett LA, Glaser B, Beck JC, et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS) Nat Genet. 1997;17:411–422. doi: 10.1038/ng1297-411. [DOI] [PubMed] [Google Scholar]
- 6.Pendred V. Deaf-Mutism and Goitre. Lancet. 1896;ii:532. [Google Scholar]
- 7.Madeo AC, Manichaikul A, Reynolds JC, et al. Evaluation of the thyroid in patients with hearing loss and enlarged vestibular aqueducts. Arch Otolaryngol Head Neck Surg. 2009;135:670–676. doi: 10.1001/archoto.2009.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li XC, Everett LA, Lalwani AK, et al. A mutation in PDS causes non-syndromic recessive deafness. Nat Genet. 1998;18:215–217. doi: 10.1038/ng0398-215. [DOI] [PubMed] [Google Scholar]
- 9.Phelps PD, Coffey RA, Trembath RC, et al. Radiological malformations of the ear in Pendred syndrome. Clin Radiol. 1998;53:268–273. doi: 10.1016/s0009-9260(98)80125-6. [DOI] [PubMed] [Google Scholar]
- 10.Pryor SP, Madeo AC, Reynolds JC, et al. SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J Med Genet. 2005;42:159–165. doi: 10.1136/jmg.2004.024208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi BY, Stewart AK, Madeo AC, et al. Hypo-functional SLC26A4 variants associated with nonsyndromic hearing loss and enlargement of the vestibular aqueduct: genotype-phenotype correlation or coincidental polymorphisms? Hum Mutat. 2009;30:599–608. doi: 10.1002/humu.20884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi BY, Madeo AC, King KA, et al. Segregation of enlarged vestibular aqueducts in families with non-diagnostic SLC26A4 genotypes. J Med Genet. 2009;46:856–861. doi: 10.1136/jmg.2009.067892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chattaraj P, Reimold FR, Muskett JA, et al. Use of SLC26A4 mutation testing for unilateral enlargement of the vestibular aqueduct. JAMA Otolaryngol Head Neck Surg. 2013;139:907–913. doi: 10.1001/jamaoto.2013.4185. [DOI] [PubMed] [Google Scholar]
- 14.King KA, Choi BY, Zalewski C, et al. SLC26A4 genotype, but not cochlear radiologic structure, is correlated with hearing loss in ears with an enlarged vestibular aqueduct. Laryngoscope. 2010;120:384–389. doi: 10.1002/lary.20722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lemmerling MM, Mancuso AA, Antonelli PJ, Kubilis PS. Normal modiolus: CT appearance in patients with a large vestibular aqueduct. Radiology. 1997;204:213–219. doi: 10.1148/radiology.204.1.9205250. [DOI] [PubMed] [Google Scholar]
- 16.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rehman AU, Santos-Cortez RL, Drummond MC, et al. Challenges and solutions for gene identification in the presence of familial locus heterogeneity. Eur J Hum Genet. 2014 Dec 10; doi: 10.1038/ejhg.2014.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park HJ, Shaukat S, Liu XZ, et al. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet. 2003;40:242–248. doi: 10.1136/jmg.40.4.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shears DCH, Murakami T, Fukai K, Alles R, Trembath R, Bitner-Glindzicz M. Molecular heterogeneity in two families with auditory pigmentary syndromes: the role of neuroimaging and genetic analysis in deafness. Clin Genet. 2004;65:384–389. doi: 10.1111/j.0009-9163.2004.00235.x. [DOI] [PubMed] [Google Scholar]
- 20.Kopp P, Arseven OK, Sabacan L, et al. Phenocopies for deafness and goiter development in a large inbred Brazilian kindred with Pendred’s syndrome associated with a novel mutation in the PDS gene. J Clin Endocrinol Metab. 1999;84:336–341. doi: 10.1210/jcem.84.1.5398. [DOI] [PubMed] [Google Scholar]
- 21.Arnos KSWK, Tekin M, Norris VW, Blanton SH, Pandya A, Nance WE. A comparative analysis of the genetic epidemiology of deafness in the United States in two sets of pedigrees collected more than a century apart. Am J Hum Genet. 2008;83:200–207. doi: 10.1016/j.ajhg.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang T, Gurrola JG, 2nd, Wu H, et al. Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am J Hum Genet. 2009;84:651–657. doi: 10.1016/j.ajhg.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang T, Vidarsson H, Rodrigo-Blomqvist S, Rosengren SS, Enerback S, Smith RJ. Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4) Am J Hum Genet. 2007;80:1055–1063. doi: 10.1086/518314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jonard L, Niasme-Grare M, Bonnet C, et al. Screening of SLC26A4, FOXI1 and KCNJ10 genes in unilateral hearing impairment with ipsilateral enlarged vestibular aqueduct. Int J Pediatr Otorhinolaryngol. 2010;74:1049–53. doi: 10.1016/j.ijporl.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Chen K, Wang X, Sun L, Jiang H. Screening of SLC26A4, FOXI1, KCNJ10, and GJB2 in bilateral deafness patients with inner ear malformation. Otolaryngol Head Neck Surg. 2012;146:972–8. doi: 10.1177/0194599812439670. [DOI] [PubMed] [Google Scholar]
- 26.Landa P, Differ AM, Rajput K, Jenkins L, Bitner-Glindzicz M. Lack of significant association between mutations of KCNJ10 or FOXI1 and SLC26A4 mutations in Pendred syndrome/enlarged vestibular aqueducts. BMC Med Genet. 2013;14:85. doi: 10.1186/1471-2350-14-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pique LM, Brennan ML, Davidson CJ, Schaefer F, Greinwald J, Jr, Schrijver I. Mutation analysis of the SLC26A4, FOXI1 and KCNJ10 genes in individuals with congenital hearing loss. PeerJ. 2014;2:e384. doi: 10.7717/peerj.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pryor SP, Demmler GJ, Madeo AC, et al. Investigation of the role of congenital cytomegalovirus infection in the etiology of enlarged vestibular aqueducts. Arch Otolaryngol Head Neck Surg. 2005;131:388–392. doi: 10.1001/archotol.131.5.388. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. 36Cl− efflux measurements of pendrin V186F-mediated Cl−/HCO3− exchange and Cl−/I− exchange.
A. Traces of 36Cl− efflux from a representative uninjected oocyte and from individual oocytes previously injected with cRNA encoding either wild-type pendrin or pendrin mutant V186F. Oocytes were exposed sequentially to baths containing 72 mM NaCl plus 24 mM Na cyclamate, 72 mM NaCl plus 24 mM NaHCO3, 72 mM Na cyclamate plus 24 mM NaHCO3, and 96 mM Na cyclamate. B. 36Cl− efflux rate constants of oocytes subjected to the experimental protocol of panel A. C. 36Cl− efflux rate constants from oocytes previously injected with water or with cRNAs encoding wild type (WT) or mutant (V186F) pendrin. Oocytes were exposed sequentially to baths containing 96 mM NaCl (ND96), 91 mM Na cyclamate plus 5 mM Na iodide (I−), and 96 mM Na cyclamate in an experimental format similar to that shown in panel A. Values are means ± s.e.m. for (n) oocytes. Statistical significance was determined by ANOVA with Bonferroni post-hoc test. (*, p <0.05 vs. WT pendrin; NS, not significant, for (n) oocytes as indicated).
Supplemental Figure 2. 36Cl− efflux measurements of pendrin N324Y-mediated Cl−/HCO3− exchange and Cl−/I− exchange.
A. Traces of 36Cl− efflux from a representative uninjected oocyte and from individual oocytes previously injected with cRNA encoding either wild-type pendrin or pendrin mutant N324Y. Oocytes were exposed sequentially to baths containing 24 mM NaCl plus 72 mM Na cyclamate (24 Cl−), 24 mM NaHCO3 plus 72 mM Na cyclamate (24 HCO3−), and 96 mM Na cyclamate. B. 36Cl− efflux rate constants of oocytes subjected to the experimental protocol of panel A. C. 36Cl− efflux rate constants from uninjected oocyte and from oocytes previously injected with cRNAs encoding wild type (WT) or mutant (N324Y) pendrin. Oocytes were exposed sequentially to baths containing 1) 96 mM NaCl (ND96), 91 mM Na cyclamate plus 5 mM Na iodide (I−), and 96 mM Na cyclamate. Values are means ± s.e.m. for (n) oocytes. Statistical significance was determined by ANOVA with Bonferroni post-hoc test. (*, p <0.05 vs. WT pendrin; NS, not significant, for (n) oocytes as indicated