

Abstract
Introduction
Metabolites are underappreciated for their effect on coagulation. Taurocholic acid (TUCA), a bile acid, has been shown to regulate cellular activity and promote fibrin sealant degradation. We hypothesize that TUCA impairs whole blood clot formation and promotes fibrinolysis.
Methods
TUCA was exogenously added to whole blood obtained from volunteers. A titration from 250 μM to 750 μM was utilized due to biologic relevance. Whole blood mixtures were assayed using thrombelastography (TEG) for clot strength (MA) and fibrinolysis (LY30) quantification. Tranexamic acid (TXA) was used to block plasmin mediated fibrinolysis. Platelet microfluidics were performed. A proteomic analysis was completed on citrated plasma obtained from a shock and resuscitation rat model.
Results
Fibrinolysis increased, when 750 μM TUCA was added to whole blood (median LY30 0.08 to 5.7, p=0.010), and clot strength decreased (median MA of 53.3 to 43.8, p=0.010). The addition of TXA, to a 750 μM TUCA titration, partially reversed the induced fibrinolysis (LY30: without 7.7 vs. with 2.7) and the decrease in clot strength (MA: without 48.2 vs. with 53.2), but did not reverse the effects to whole blood levels. Platelet function reduced by 50% in the presence of 100 μM TUCA. Rats had a median 52-fold increase in TUCA, following a shock state that stayed elevated following resuscitation.
Conclusion
TUCA reduces clot strength and promotes fibrinolysis. The clot strength reduction is attributable to platelet inhibition. This metabolic effect on coagulation warrants further investigation, as localized areas of the body, with high levels of bile acid, may be at risk for postoperative bleeding.
Keywords: taurocholic acid, bile salt, thrombelastography, fibrinolysis, clot strength, platelet dysfunction
1. Introduction
Bile salts are unique amphipathic molecules that have been found to enhance tissue plasminogen activator (tPA) mediated fibrinolysis [1]. Normal levels of bile acids in systemic circulation are on the micromolar level [2]. However, bile acids such as taurocholic acid (TUCA) have been found to be elevated by 1000 fold during pathologic conditions [3]. In vitro TUCA causes angiogenesis and cell proliferation [4]. Elevated blood TUCA levels have also been implicated with heart arrhythmias [5], suggesting that this is a highly active biologic molecule. In rat models, it has been shown that serum taurocholic acid levels markedly rise following acute liver injury [6,7].
Previous studies evaluating the effect of TUCA on coagulation have not used whole blood assays. Hoffman and Monroe [8] have elucidated the cell based model of hemostasis, emphasizing the key role of platelets. Because of the many receptors on platelets and biologic activity of TUCA, we hypothesize that this bile acid impairs whole blood clot formation via platelet inhibition, that it promotes fibrinolysis, and that this metabolite can be released following hemorrhagic shock due to oxygen sensitivity of the liver.
2. Methods
2.1 Subjects
After obtaining informed consent under an institution review board approved protocol (COMIRB # 14-0366) blood samples were obtained from healthy volunteers with no known abnormalities in the coagulation or fibrinolytic system; none of these individuals were taking salicylic acid or other non-steroidal anti-inflammatory medications within 120 hours of the experiment. All subjects were male with a median age of 28 years, range 20–37.
2.2 Taurocholic Acid (TUCA)
Lyophilized taurocholic acid, purchased from sigma Aldrich (Product # 86993, St. Louis, MO), was reconstituted to 0.125 M in normal saline. Aliquots of 40 μL of this solution were stored at 4 °C until use.
2.3 Thrombelastography TUCA Challenge (TEG TUCA Challenge)
Blood was collected in 3.3ml citrated blood tubes from venous puncture. Citrated samples were kept at room temperature and assayed between 20 minutes and 2 hours after blood draw, in accordance with manufacturer recommendations. Whole blood was mixed with TUCA to reach a final volume of 500 microliters in individual Eppendorf tubes (Hamburg, Germany). The concentration of TUCA ranged from 0–750 micromolar (μM). TUCA was added to whole blood mixtures within 5 minutes of initiating the assay. Citrated native thrombelastography (TEG) assays were re-calcified and run according to the manufacturer’s instructions on a TEG 5000 Thrombelastograph Hemostasis Analyzer (Haemonetics, Niles IL). The following parameters were recorded from the tracings of the TEG: R time (seconds), angle (α, degrees), maximum amplitude (MA, mm), and lysis 30 minutes after MA (LY30, %). To detect the presence of plasmin-mediated fibrinolysis, tranexamic acid (TXA) at a concentration of 20mg/ml was mixed with samples.
2.4 Platelet Function Assay
A previous validated PDMS flow device [9] was used to create eight evenly spaced flow channels (250 μm in width, 60 μm in height) that are perpendicularly deposited over pre-patterned collagen matrix (250 μm in width). PPACK (irreversible thrombin inhibitor, 100 μM) treated (+/− taurocholic acid) whole blood was perfused over collagen fibrillar at venous shear rate (100 s−1). Dynamic change of platelet (labeled with anti-CD 61 Ab) deposition on collagen was monitored with fluorescence microscopy (Olympus IX81).
2.5 Animal Shock Resuscitation Model
Sprague Dawley rats were subjected to hemorrhagic shock (IACUC # 90814), and citrated plasma samples were collected for proteomic and metabolic analysis. In brief, rats were anesthetized with pentobarbital and underwent tracheostomy and femoral artery cannulation. Blood was drawn through the femoral artery. After baseline blood was obtained, animals were hemorrhaged to a mean arterial pressure (MAP) of 25 mm hg for 30 minutes, followed by an additional blood draw. Animals were then resuscitated with normal saline, and blood samples were obtained 15 minutes after animals obtained a MAP greater than 30 mm hg. The volume of resuscitation was limited to less than 20%EBV. A post hoc analysis to assess TUCA was preformed on previously run samples corresponding to baseline, shock, and reperfusion plasma.
2.6 TUCA Quantification Via Mass Spectroscopy
Plasma samples (10 μl) were immediately extracted in ice-cold lysis/extraction buffer (methanol:acetonitrile:water 5:3:2) at 1:25 dilutions. Samples were then agitated at 4°C for 30 min and centrifuged at 10,000g for 15min at 4°C. Protein and lipid pellets were discarded, while supernatants were stored at −80°C prior to metabolomics analyses. Ten μl of sample extracts were injected into an UPLC system (Ultimate 3000, Thermo, San Jose, CA, USA) and run on a Kinetex XB-C18 column (150×2.1 mm i.d., 1.7 μm particle size – Phenomenex, Torrance, CA, USA) using a 9 min gradient at 250 μl/min (mobile phase: 5% acetonitrile, 95% 18 mΩ H2O, 0.1% formic acid). The UPLC system was coupled online with a QExactive system (Thermo, San Jose, CA, USA), scanning in Full MS mode (2 μscans) at 70,000 resolution in the 60–900 m/z range, 4kV spray voltage, 15 sheath gas and 5 auxiliary gas, operated in negative and then positive ion mode (separate runs). Calibration was performed before each analysis against positive or negative ion mode calibration mixes (Piercenet – Thermo Fisher, Rockford, IL, USA) to ensure sub ppm error of the intact mass. Taurocholate assignments was performed using the software Maven [10] (Princeton, NJ, USA), upon conversion of .raw files into .mzXML format through MassMatrix (Cleveland, OH, USA). The software allows for peak picking, feature detection, and metabolite assignment against the KEGG pathway database. TUCA assignment was further confirmed against chemical formula determination (as gleaned from isotopic patterns and sub 1 ppm error on accurate intact mass), and retention times were confirmed against commercially available hydrophobic standard libraries (SIGMA Aldrich, St. Louis, MO, USA; IROATech, Bolton, MA, USA).
2.7 Statistical Analysis
SPSS software version 22 (IBM, Armonk, NY) was used for statistical analysis. The TEG value LY30 and MA in addition to TUCA concentrations had a skewed, non-normal distribution. Spearman’s Rho was used to assess the correlation of TEG values and concentration of TUCA in whole blood (0, 250, 500, 750 μM). Comparisons between groups LY30 with and with out TUCA and TXA were performed with the non-parametric paired Friedman’s test with the stepwise adjustment for multiple comparisons. TUCA plasma levels in rat from baseline, shock, and reperfusion were contrasted with the same statistical methodology. Platelet fluorescent intensity in microfluidic assay was averaged across three clotting events (± SD, shaded area). Significance was set at two-tailed alpha of 0.05 for all statistical tests.
3. Results
3.1 Increasing Whole Blood Concentration of TUCA correlates with LY30 and MA
Whole blood median LY30 was 0.08% (IQR 0–1.35). Increasing the molar concentration of TUCA correlated with an increase in LY30 (Spearman’s Rho = 0.743 p<0.001). LY30 was statistically increased when TUCA concentration was 750 μM (5.7 IQR 5.0–7.7) compared to baseline blood (p=0.010 figure 1). Clot strength also showed a relationship to TUCA concentration. Whole blood median MA was 53.3 (IQR 49.5–58.5). Increasing the concentration of TUCA correlated with a decrease in MA (Spearman’s Rho =−0.479 p=0.018 figure 2). Clot strength was significantly reduced at TUCA concentration of 750μM (MA = 43.8 IQR 41.5–48.3) compared to baseline (p=0.010). Other TEG parameters (R time Spearman’s Rho = −0.098 p=0.613 and Angle Spearman’s Rho =−0.135 p=0.485) did not have a correlation with TUCA concentration.
Figure 1. Fibrinolysis Increases With Increasing Concentration of TUCA.

* = P<0.05, LY30 = Percent fibrinolysis 30 minutes after maximum amplitude, TUCA = Taurocholic Acid. Y-axis represents the percent of lysis 30 minutes after blood clot reaches maximum amplitude. The X-axis represents a progressive concentration of TUCA in whole blood ranging from 0 (left) to 750 micromolar (right).
Figure 2. Clot Strength Decreases With Increasing Concentration of TUCA.

* = P<0.05, MA = Maximum amplitude of clot formation, TUCA = Taurocholic Acid. Y-axis represents the maximum amplitude of formed blood clot. X-axis represents a progressive concentration of TUCA in whole blood ranging from 0 (left) to 750 micromolar (right).
3.2 Tranexamic Acid Partially Reverses LY30 and MA Changes Associated With TUCA
The addition of TXA to whole blood did not change LY30 (without 1.6 IQR0-2 vs. with 1.35 IQR0.1–1.7 p=0.343) or MA (without 56.5 IQR 53.4–58.6 vs. with 56.3 IQR 52.9–57.4 p=0.408 figure 3). Reversal of the effect of TUCA at 750μM was significant with TXA for LY30 (without 7.7 IQR 5.7–14.2 vs. with 2.7 IQR 1.85–3.3 p = 0.028) and MA (without 48.2 IQR 43.8–52.4 vs. with 53.2 IQR 48–56.1 p=0.048). However, LY30 remained significantly higher in the blood with 750 μM concentration TUCA with TXA compared to whole blood (p=0.028) in addition to having a lower MA (p=0.048).
Figure 3. Tranexamic Acid Only Partially Reverses the Affects of Taurocholic Acid on Whole Blood Clot Formation.
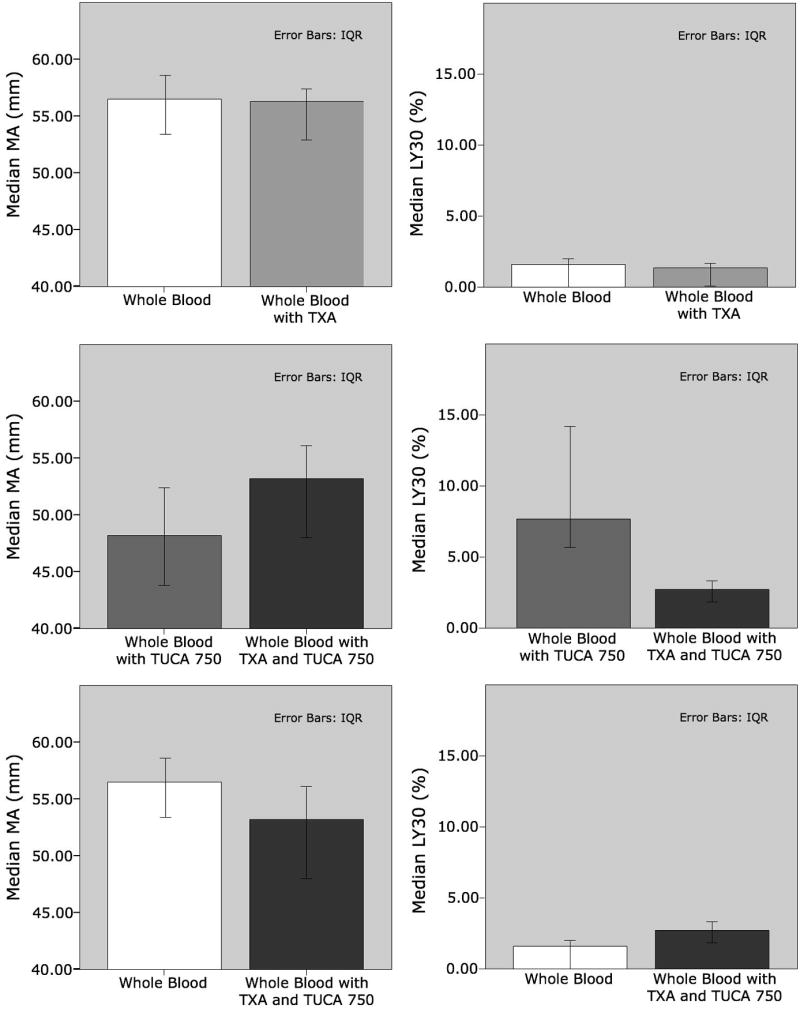
LY30 = Percent fibrinolysis 30 minutes after maximum amplitude, MA = Maximum amplitude of clot formation, TUCA = Taurocholic Acid, TXA = Tranexamic Acid. Row 1 demonstrates MA and LY30, for whole blood without TUCA, without and with TXA. Row 2 demonstrates MA and LY30 for whole blood with TUCA, without and with TXA. Row 3 correlates MA and LY30 for whole blood without TUCA and TXA verses whole blood with TUCA and TXA.
3.3 TUCA Inhibits Platelet Function
Platelet signal in the initiation phase was identical in control and treated group. Signal of treated group started to deviate from control after the first 2 minutes indicating possible effect of taurocholic acid on platelet secondary aggregation. This effect should be independent of thrombin since thrombin was depleted by PPACK. The reduction in platelet signal with 100 μM TUCA reached 50% by the end of the assay suggesting taurocholic acid is a potent antiplatelet agent (figure 4).
Figure 4. Taurocholic Acid Inhibits Platelet Function.

Y-axis shows the signal production relating to platelet activity. X-axis shows the elapsed time.
3.4 TUCA Concentrations Increase Following Hemorrhagic Shock and Early Resuscitation
Rats’ baseline TUCA was 10,500 AU (IQR 0–31,500). After shock all rats had an increase in median TUCA plasma level to 550,000 AU (IQR 67,000–1,237,500). After resuscitation TUCA levels remained elevated (median 525,500 AU IQR 142,500 – 1,725,000). Pairwise adjustment of groups based on TUCA levels from baseline trended towards significance for shock (p=0.063) and was significant after resuscitation (p=0.012 figure 5).
Figure 5. Taurocholic Acid Increases in Rats Following Shock and Stays Elevated During Resuscitation.

TUCA = Taurocholic Acid. Y-axis represents arbitrary quantitative units of taurocholic acid. X-axis represents the 3 stage points for blood analysis during the model.
4. Discussion
Taurocholic acid is a potent liver metabolite that impairs platelet function and promotes fibrinolysis. In vitro assays have identified a dose like effect in which increasing the concentration of TUCA increases LY30 and reduces MA. Tranexamic acid can partially reverse these effects, but is not capable of completely blocking fibrinolysis and reduction of clot strength. In the absence of fibrin generation, TUCA impairs platelet aggregation by up to 50%, at a much lower concentration than required to impair clotting parameters detected by TEG in whole blood. An animal model of shock and resuscitation supports that TUCA levels can increase within 30 minutes of low blood pressure and remain elevated after resuscitation.
Bile acids have previously been associated with platelet dysfunction [11]. The proposed mechanism was through membrane deformity. Phospholipid membrane on platelets has been implicated as critical for these cells to aggregate and degranulate [12]. However bile acids also activate a number of cellular calcium related channel receptors [5,13,14]. Calcium regulation is critical for multiple platelet functions [15], and may be another mechanism for TUCA inhibition of platelets. We have shown that platelet aggregation is inhibited by TUCA using microfluidics (figure 4). We have also shown that clot strength can be reduced as measured by thrombelastography (figure 2), which is perceived to reflect platelet function [16].
Tranexamic acid partially reverses the TUCA reduction in clot strength, and suggests a potential receptor-mediated mechanism. TXA is perceived to be an antifibrinolytic agent, but is known to interact with multiple receptors [17]. TXA has recently been suggested to improve platelet function in patients on dual antiplatelet therapy [18], but the mechanism remains unknown. The reduction of LY30 with TXA in the presence of TUCA does support some degree of a plasmin-mediated process. Boonstra et al. had previously shown that TUCA could suppress PAI-1 activity allowing for uninhibited tissue plasminogen activation of plasminogen causing fibrinolysis [1]. The receptor-mediated effects of TUCA warrant further investigation, as it may involve previously unrecognized receptors and pathways.
In multiple rat studies, acute liver injury increased systemic TUCA levels above control levels [6,7]. In our animal study, plasma from 30 minutes of systemic shock also contained increased TUCA levels. These animal models suggest that both an ischemic insult and mechanical damage to the liver can release TUCA locally and systemically. This correlates to clinical outcomes in which the volume of liver removed (tissue injury)[19,20], pringle time (local ischemia)[19,21], and blood loss (systemic ischemia)[22] are associated with an increased risk of post-operative complications. While TUCA levels have not been clinically measured at the cut edge of a liver, the concentration is likely to be markedly elevated. The gastric contents from patients with pathologic reflux have been measured to have TUCA concentrations 1000 times greater than plasma levels [3]. This is likely is a lower level estimate of TUCA concentrations from intrahepatic bile which is not diluted by gastric contents. However, TXA does partially suppress both the loss of clot strength and increase in fibrinolysis, therefore it may be a potential topical therapeutic for difficult to control liver bleeding.
This study is limited to an in vitro analysis on healthy volunteer blood. Because of the testing environment, the blood contained normal levels of all liver synthesized proteins and blood components, which is not the case for many trauma patients. Additionally, the testing environment only looked at the interaction of the one metabolite. There can be other metabolite interactions that are not being assessed during our study, as well as cellular and organ specific effects. TUCA has previously been implicated in causing impairment of fibrinolysis in a rat acute pancreatitis model [23]. However these effects were not appreciated until 3 hours after injection with evidence of inflammation of the pancreas and systemic levels of TUCA were not analyzed. This observation may have been an adaptive response to inflammation, which has previously been shown to shutdown fibrinolysis [24]. It would not be unexpected that TUCA has tissue dependent effects on fibrinolysis.
In conclusion, our results have shown that taurocholic acid has the ability to cause platelet dysfunction, in addition to increased fibrinolysis. TUCA has the potential to create post-operative complications in trauma and liver patients by impaired hemostasis and containment of bile, in addition to causing systemic impairment of coagulation. With the use of rapid output metabolomics our understanding of the effect of many previously unmeasured small biologic molecules will likely change our understanding of the bodies response to surgical procedures and shock. TUCA provides an example of a small highly active biological molecule that previously received minimal attention on its impact on coagulation.
Acknowledgments
This study was supported in part by National Institute of General Medical Sciences grants: T32-GM008315, P50-GM049222 1, UM 1HL120877 and CCTSI supported in part by Colorado CTSA Grant UL1 TR001082 from NCATS/NIH.
Footnotes
Presented at the 10th Annual Academic Surgical Congress in Las Vegas, NV, February 4, 2015
All listed authors made significant contributions to the manuscript. G.W. and H.M. were integral in all parts of the study, including the study design, data collection, manuscript drafting, and in finalizing critical manuscript revisions. E.M. and A.B. assisted in formulating the study design and the preliminary manuscript draft, and in completing critical revisions. E.G. and A.D. performed data collection and interpretation, in addition to completing work on the draft of the manuscript. S.D. and S.Z. performed data collection and completed critical manuscript revisions for submission.
None of the authors have any conflicts of interest to disclose.
5. Disclosure
The authors have no personal conflicts of interest or proprietary interests of products or concepts mentioned in the article.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIGMS, NHLBI or National Institutes of Health.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boonstra EA, Adelmeijer J, Verkade HJ, de Boer MT, Porte RJ, et al. Fibrinolytic proteins in human bile accelerate lysis of plasma clots and induce breakdown of fibrin sealants. Ann Surg. 2012;256:306–312. doi: 10.1097/SLA.0b013e31824f9e7e. [DOI] [PubMed] [Google Scholar]
- 2.Burkard I, von Eckardstein A, Rentsch KM. Differentiated quantification of human bile acids in serum by high-performance liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;826:147–159. doi: 10.1016/j.jchromb.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 3.Iftikhar SY, Ledingham S, Steele RJ, Evans DF, Lendrum K, et al. Bile reflux in columnar-lined Barrett’s oesophagus. Ann R Coll Surg Engl. 1993;75:411–416. [PMC free article] [PubMed] [Google Scholar]
- 4.Sato S, Yamamoto H, Mukaisho K, Saito S, Hattori T, et al. Continuous taurocholic acid exposure promotes esophageal squamous cell carcinoma progression due to reduced cell loss resulting from enhanced vascular development. PLoS One. 2014;9:e88831. doi: 10.1371/journal.pone.0088831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rainer PP, Primessnig U, Harenkamp S, Doleschal B, Wallner M, et al. Bile acids induce arrhythmias in human atrial myocardium--implications for altered serum bile acid composition in patients with atrial fibrillation. Heart. 2013;99:1685–1692. doi: 10.1136/heartjnl-2013-304163. [DOI] [PubMed] [Google Scholar]
- 6.Luo L, Schomaker S, Houle C, Aubrecht J, Colangelo JL. Evaluation of serum bile acid profiles as biomarkers of liver injury in rodents. Toxicol Sci. 2014;137:12–25. doi: 10.1093/toxsci/kft221. [DOI] [PubMed] [Google Scholar]
- 7.Buness A, Roth A, Herrmann A, Schmitz O, Kamp H, et al. Identification of metabolites, clinical chemistry markers and transcripts associated with hepatotoxicity. PLoS One. 2014;9:e97249. doi: 10.1371/journal.pone.0097249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffman M, Monroe DM., 3rd A cell-based model of hemostasis. Thromb Haemost. 2001;85:958–965. [PubMed] [Google Scholar]
- 9.Maloney SF, Brass LF, Diamond SL. P2Y12 or P2Y1 inhibitors reduce platelet deposition in a microfluidic model of thrombosis while apyrase lacks efficacy under flow conditions. Integr Biol (Camb) 2010;2:183–192. doi: 10.1039/b919728a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clasquin MF, Melamud E, Rabinowitz JD. LC-MS data processing with MAVEN: a metabolomic analysis and visualization engine. Curr Protoc Bioinformatics. 2012;Chapter 14(Unit14):11. doi: 10.1002/0471250953.bi1411s37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shiao YJ, Chen JC, Wang CN, Wang CT. The mode of action of primary bile salts on human platelets. Biochim Biophys Acta. 1993;1146:282–293. doi: 10.1016/0005-2736(93)90367-9. [DOI] [PubMed] [Google Scholar]
- 12.Koseoglu SS, Meyer AF, Kim D, Meyer BM, Wang Y, et al. Analytical Characterization of the Role of Phospholipids in Platelet Adhesion and Secretion. Anal Chem. 2014 doi: 10.1021/ac502293p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dopico AM, Walsh JV, Jr, Singer JJ. Natural bile acids and synthetic analogues modulate large conductance Ca2+-activated K+ (BKCa) channel activity in smooth muscle cells. J Gen Physiol. 2002;119:251–273. doi: 10.1085/jgp.20028537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee HK, Lee KH, Cho ES. Bile Acid Inhibition of N-type Calcium Channel Currents from Sympathetic Ganglion Neurons. Korean J Physiol Pharmacol. 2012;16:25–30. doi: 10.4196/kjpp.2012.16.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dolan AT, Diamond SL. Systems modeling of Ca(2+) homeostasis and mobilization in platelets mediated by IP3 and store-operated Ca(2+) entry. Biophys J. 2014;106:2049–2060. doi: 10.1016/j.bpj.2014.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salooja N, Perry DJ. Thrombelastography. Blood Coagul Fibrinolysis. 2001;12:327–337. doi: 10.1097/00001721-200107000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Kratzer S, Irl H, Mattusch C, Burge M, Kurz J, et al. Tranexamic acid impairs gamma-aminobutyric acid receptor type A-mediated synaptic transmission in the murine amygdala: a potential mechanism for drug-induced seizures? Anesthesiology. 2014;120:639–649. doi: 10.1097/ALN.0000000000000103. [DOI] [PubMed] [Google Scholar]
- 18.Weber CF, Gorlinger K, Byhahn C, Moritz A, Hanke AA, et al. Tranexamic acid partially improves platelet function in patients treated with dual antiplatelet therapy. Eur J Anaesthesiol. 2011;28:57–62. doi: 10.1097/EJA.0b013e32834050ab. [DOI] [PubMed] [Google Scholar]
- 19.Benzoni E, Lorenzin D, Favero A, Adani G, Baccarani U, et al. Liver resection for hepatocellular carcinoma: a multivariate analysis of factors associated with improved prognosis. The role of clinical, pathological and surgical related factors. Tumori. 2007;93:264–268. doi: 10.1177/030089160709300306. [DOI] [PubMed] [Google Scholar]
- 20.Shirabe K, Shimada M, Gion T, Hasegawa H, Takenaka K, et al. Postoperative liver failure after major hepatic resection for hepatocellular carcinoma in the modern era with special reference to remnant liver volume. Journal of the American College of Surgeons. 1999;188:304–309. doi: 10.1016/s1072-7515(98)00301-9. [DOI] [PubMed] [Google Scholar]
- 21.Boyko VV, Pisetska ME, Tyshchenko OM, Skoryi DI, Kozlova TV, et al. Role of ischemic preconditioning in hepatic ischemia-reperfusion injury. Hepatobiliary Surg Nutr. 2014;3:179–184. doi: 10.3978/j.issn.2304-3881.2014.06.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Si-Yuan F, Yee LW, Yuan Y, Sheng-Xian Y, Zheng-Guang W, et al. Pringle manoeuvre versus selective hepatic vascular exclusion in partial hepatectomy for tumours adjacent to the hepatocaval junction: a randomized comparative study. Int J Surg. 2014;12:768–773. doi: 10.1016/j.ijsu.2014.05.068. [DOI] [PubMed] [Google Scholar]
- 23.Rydzewska G, Kosidlo S, Gabryelewicz A, Rydzewski A. Tissue plasminogen activator, plasminogen activator inhibitor, and other parameters of fibrinolysis in the early stages of taurocholate acute pancreatitis in rats. Int J Pancreatol. 1992;11:161–168. doi: 10.1007/BF02924181. [DOI] [PubMed] [Google Scholar]
- 24.Ostrowski SR, Berg RM, Windelov NA, Meyer MA, Plovsing RR, et al. Discrepant fibrinolytic response in plasma and whole blood during experimental endotoxemia in healthy volunteers. PLoS One. 2013;8:e59368. doi: 10.1371/journal.pone.0059368. [DOI] [PMC free article] [PubMed] [Google Scholar]