

Abstract
Mutations in gene encoding for bone morphogenetic protein type 2 receptor (BMPR-2) have been reported in pulmonary arterial hypertension (PAH), but their functional relevance remains incompletely understood.
BMP receptors expression was evaluated in human lungs and in cultured pulmonary artery smooth muscle cells (PASMCs) isolated from 19 idiopathic PAH patients and 9 heritable PAH patients with demonstrated BMPR-2 mutations. BMP4-treated PASMCs were assessed for Smad and p38MAPK signaling associated to mitosis and apoptosis.
Lung tissue and PASMCs from heritable PAH patients presented with decreased BMPR-2 expression and variable increases in BMPR-1A and BMPR-1B expressions, while a less important decreased BMPR-2 expression was observed in PASMCs from idiopathic PAH patients. Heritable PAH PASMCs showed no increased phosphorylation of Smad1/5/8 in the presence of BMP4, which actually activated the p38MAPK pathway. Individual responses varied from one mutation to another. PASMCs from PAH patients presented with an in vitro proliferative pattern, which could be inhibited by BMP4 in idiopathic PAH, not in heritable PAH. PASMCs from idiopathic PAH and more so from heritable presented an inhibition of BMP4-induced apoptosis.
Most heterogenous BMPR-2 mutations are associated with defective Smad signaling compensed for by an activation of p38MAPK signaling, accounting for PASMC proliferation and deficient apoptosis.
INTRODUCTION
Pulmonary arterial hypertension (PAH) is an uncommon disease with a poor prognosis and mysterious pathobiology, characterized by a progressive increase in pulmonary vascular resistance and eventual right ventricular failure (1). Mutations of bone morphogenetic protein receptor-2 (BMPR-2), a member of the TGF-β receptor family have been reported in a high proportion of patients with the heritable form of the disease, and in 10 to 30 % of patients with sporadic idiopathic PAH (2). To date, more then 200 distinct BMPR-2 mutations have been described, widely dispersed across the gene, with the majority predicting premature truncation of the transcript (3). BMPR signaling involves heterodimerization of two transmembrane serine/threonine-kinase receptor chains, the constitutively active BMPR-2 and the corresponding type 1 receptor, BMPR-1A/ALK3 or BMPR-1B/ALK6 (4,5). With interaction of a ligand, for example BMP4, the activated kinase domain of BMPR-2 phosphorylates the corresponding BMPR-1, which in turn initiates intracellular signaling through the phosphorylation of a set of BMP restricted Smad proteins (Smad1/5/8). Subsequently, these phosphorylated Smads associate with Smad4, translocate to the nucleus and then modulate the transcription of target genes. Alternative Smad-independent signaling pathways involving mitogen activated protein kinase (MAPK), including ERK1/2, JNK and p38MAPK have been reported to be activated by BMP ligands (6). The resulting imbalance is believed to be the cause of a proliferation of pulmonary artery smooth muscle cells (PASMCs) as a major component of pulmonary arteriolar remodeling in PAH (1).
It is of interest that the histopathology and clinical picture of PAH with or without BMPR-2 mutations appear similar, except for an earlier age of onset, more severe hemodynamic compromise at diagnosis, and less common reversibility at vasodilator testing (7,8). Therefore, the functional consequences of BMPR-2 mutations remain incompletely understood, but it may be hypothesized that their phenotypic impact may vary with type of mutation or interaction with alternative signaling pathways.
We have previously reported that PASMCs from idiopathic PAH patients present with an in vitro proliferative phenotype (9,10). In the present study, we investigate the effects of BMP4 on Smad and p38MAPK signaling associated with mitosis and apoptosis in cultured PASMCs isolated from idiopathic PAH patients without detected mutations and from heritable PAH patients with mutations. The results are in keeping with the notion of a crucial role for BMP/Smad signaling in the prevention of abnormal growth and apoptosis of PASMCs that is lost in most but not all types of mutations.
METHODS
Tissue samples
Lung tissue and pulmonary arteries were sampled at lung transplantation and sequenced to screen BMPR-2 mutations. After confirmatory cross-check with medical records, patients with PAH were segregated in two groups, according the presence or the absence of mutations. These 2 groups were respectively called heritable PAH (n=9) and idiopathic PAH (n=19) patients. Pulmonary specimens were also sampled in control subjects (n=10) at lobectomy or pneumonectomy for a suspected localized lung tumor. These control subjects did not bear any BMPR-2 mutations or polymorphisms.
All PAH patients were in New York Heart Association functional class III or IV and were treated with iv epoprostenol. In the control subjects, transthoracic echocardiography was performed preoperatively to rule out pulmonary hypertension, and pulmonary arteries were sampled at a distance from tumor areas. The study was approved by the local Institutional Review Board.
Screening for mutations in the gene encoding the BMPR-2 receptor
Mutations in the BMPR-2 gene in lung specimens from patients with PAH (n=28) were screened as previously described (11,12). Briefly, the entire protein-coding region (sequence corresponding to exons 1–13 of the BMPR-2 gene) was amplified from genomic DNA samples by polymerase chain reaction with specific primers. PCR products were then separated by electrophoresis in a 1% agarose gel and purified using the QIAquick PCR purification kit (QIAGEN, Courtaboeuf, France). Amplified and purified fragments were sequenced wih a dye-terminator cycle-sequencing system (ABI PRISM 377, Perkin-Elmer Applied Biosystems).
Culture of human pulmonary artery smooth muscle cells (PASMCs) and pulmonary microvascular endothelial cells (PECs)
Human PASMCs were cultured from explants of pulmonary arteries (1.5 to 10 mm in diameter) derived from previously described patient groups transplanted for heritable and idiopathic PAH, but also from controls. PASMCs were cultured in 10% FCS/DMEM and used between passages 3 and 6, as previously described (13). The phenotype of cultured PASMCs was assessed for expression of muscle-specific contractile and cytoskeletal proteins, including smooth muscle α-actin (α-SMA), desmin, and vinculin (13).
Human PECs were obtained first by Dispase I (Roche Diagnostics, Penzbeg, Germany) digestion and after by immunomagnetic purification with anti-platelet endothelial cell adhesion molecule-1 (CD31) monoclonal antibody-labeled Dynabeads (Dynal Biotech, Compiegne, France) of a fragment of lung tissue isolated from heritable and idiopathic PAH patients and controls, as previously described (9,14). To characterize the endothelial phenotype, PECs were labeled with acetylated low-density lipoprotein coupled to a fluorescent carbocyanine dye (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate [Dil-Ac-LDL]; Tebu, Le Perray en Yvelines, France) and stained with antibodies against the endothelial cell-specific lectin Ulex europaeus agglutinin-1 (UEA-1; Sigma-Aldrich, Ayrshire, UK) (15). Experiments were also performed with monoclonal antibodies against desmin and vimentin (Dako, Glostrup, Denmark). Cells with positive staining for Dil-Ac-LDL and UEA-1 and negative staining for desmin and vimentin were taken as endothelial cells and constituted more than 95% of our PEC cultures. PECs were used between passages 3 and 6 (9,14).
RNA extraction and cDNA preparation
Total RNA was prepared from snap-frozen human lung tissue samples (weight 100 mg) by homogenization according to the method of Chomczynski and Sacchi (16), using TRIzol reagent (Invitrogen, Cergy-Pontoise, France). Total RNA was extracted from growth-arrested primary cultures of human PASMCs and PECs using Qiagen RNeasy Mini kit (QIAGEN S.A., Courtabeouf, France), according to the manufacturer’s instructions. RNA concentration was determined by standard spectrophotometric techniques and the RNA integrity was assessed by visual inspection of ethidium bromide-stained denaturing agarose gels. First-strand cDNA synthesis was carried out using SuperScript II Reverse Transcriptase System (Life Technologies, Inc.), as previously described (9,14).
Real-time quantitative polymerase chain reaction (RTQ-PCR)
RTQ-PCR primers were designed using the computer program Primer3 (Primer Express Software, Applied Biosystems) for human BMPR-1A, BMPR-1B, BMPR-2, Bax, Bcl2 mRNA and 18s rRNA, as housekeeping gene. To avoid inappropriate amplification of residual genomic DNA, intron-spanning primers were selected. RTQ-PCR was performed in triplicate on an ABI PRISM 7000 (Applied Biosystems, Foster City, CA), in mixtures of 12.5μl Sybr Green PCR Master Mix (Applied Biosystems, Warrington, UK), 300 nM (each) primer and 5μl of diluted template DNA in a total volume of 25μl. Signal detection and analysis of results were performed with ABI PRISM 7000 sequence detection software (Applied Biosystems). Relative quantification was achieved with the comparative 2−ΔΔCt method by normalization with 18s ribosomal RNA. For assays of Bax and Bcl2 mRNAs, PASMCs were seeded and synchronized. The cells were then exposed to BMP4 (100 ng/mL) for 4 hours and then used for mRNA extraction and RTQ-PCR.
Protein extraction and BMPR-1A, BMPR-1B and BMPR-2 Western Blotting
Proteins were extracted from snap-frozen tissue samples (weight 100 mg) by homogenization in an appropriate amount of homogenizing buffer [Complete Mini Protease Inhibitor Cocktail (Roche Diagnostics, Mannheim, Germany) in PBS and 0.1% Triton X-100]. The homogenates were centrifuged at 4°C and the supernatants were collected. After determination of the protein concentration using the method of Bradford (17), 40 μg of protein from each lung sample were resuspended in 3x Laemmli’s buffer, boiled for 5 minutes, and separated on 10% acrylamide gel by electrophoresis. Proteins were electrophoretically transferred to a nitrocellulose membrane (Sigma-Aldrich, Ayrshire, UK) for 1h at room temperature. After blocking with 5% BSA in 1x Tween (T)-TBS (10 mM Tris-HCl, pH 8.0; 150 mM NaCl; and 0.1% Tween 20) for 2 h at room temperature, the membrane was washed three times with T-TBS at room temperature for 5 min. The membrane was incubated with goat anti-human BMPR-1A, BMPR-1B or BMPR-2 antibody (1:500) (R&D systems, Mineapolis, MN) at 4°C overnight with rocking. Then the membrane was washed three times for 5 min and incubated with the secondary antibody (rabbit antigoat IgG conjugated with horseradish peroxidase; Dako, Glostrup, Denmark; 1:2,000) for 1 h at room temperature. Immunoreactive bands were detected using the enhanced chemiluminescence Western blotting analysis system (Amersham Pharmacia Biosciences, Buckinghamshire, UK) and quantified by laser densitometry. Relative quantification was performed by normalization with β-actin (Sigma-Aldrich CO, St Louis, USA).
Immunoblotting for BMP Signaling Pathways
PASMCs were plated in fresh 10% FCS/DMEM medium for 24 hours, and then quiesced for 48 hours in serum-free medium. BMP4 (100 ng/mL) or vehicle was then added to the cells for 20 minutes. Protein was harvested by washing cells in cold PBS and by scraping in 300 μL of 1x sample loading buffer [Tris-HCl (pH 7.4), NaCl, NaF, sodium pyrophosphate (all at 25 mM), sodium vanadate (1mM), EDTA, EGTA (both at 2,5mM), phenylmethylsulfonyl fluoride (1mM), aprotinine, leupeptine (both at 5μg/mL), SDS, deoxycholate and NP-40 (all at 0,50%)] on ice. The samples were then stored at −20°C. After determination of the protein concentration, using the method of Bradford (17), samples (20μg) were resuspended in 3x Laemmli buffer, boiled at 95°C for 5 minutes and electrophoresed on acrylamide gels (10%). Immunoblotting assays were performed as described above with monoclonal mouse anti-human phospho-p38MAPK(Thr180/Tyr182) (1:1000, Cell Signalling Technology Inc, Danvers, MA), polyclonal rabbit anti-human phospho-Smad1 (Ser463/465)/Smad5(Ser463/465)/Smad8(Ser426/428) (1:1000, Cell Signalling Technology Inc, Danvers, MA) and polyclonal goat anti-human total Smad 1/5/8 (1:1000, Santa-Cruz Biotechnology, Santa Cruz, CA). Relative quantification was performed by normalization with total Smad1/5/8 for phospho-Smad1/5/8 and β-actin (Sigma-Aldrich, France) for p38MAPK. A treatment of 100 ng/mL during 20 minutes was chosen based on preliminary studies of BMP4 concentrations in relation to the capacity to activate downstream signaling pathways and to inhibate growth-promoting activity of mitogenic agents on PASMCs (18,19).
PASMC Proliferation Assays
The growth of human cultured PASMCs was determined by [3H]thymidine incorporation, representing DNA synthesis. Briefly, PASMCs were seeded in 24-well plates in 10% FCS/DMEM at a density of 5 × 104 cells/well and allowed to adhere for 24 hours. The medium was then removed and the cells subjected to growth arrest by incubation with serum-free DMEM. After 48 hours, the medium was replaced with fresh DMEM completed with 10% FCS or PDGF (10 ng/mL) in the presence or absence of BMP4 (100ng/mL). PASMC proliferation was also assessed in response to 10% FCS and PDGF (10 ng/mL) alone. For each condition, [3H]thymidine (0.6 μCi/ml) was added to each well. After incubation for 24 hours, the cells were washed twice with PBS, treated with ice-cold 10% trichloroacetic acid and neutralized with 0,1N NaOH (0,5 ml/well). [3H]thymidine incorporation into DNA was counted and reported as counts per minute per well.
Apoptosis Assays
Apoptosis evaluation was performed by flow cytometry analysis of the DNA content by propidium iodide incorporation and RTQ-PCR analysis of Bax/Bcl2 ratio. For flow cytometry analysis of DNA content, PASMCs were seeded and treated for 24 h with fresh serum-free DMEM in presence or absence of BMP4 (100ng/mL). Culture medium was removed and saved. Cells were trypsinized and returned to the medium they had grown in and then centrifuged. Cells were then washed twice in ice-cold phosphate-buffered saline (PBS) and stored at 4°C in 75% ethanol. Fixed cells were centrifuged, washed with PBS and incubated with 200 μl Rnase I (1 mg/mL, Invitrogen, Cergy-Pontoise, France) and 200 μl of propidium iodide (1mg/mL; Sigma, France). Cells were incubated at room temperature for one hour in the dark. Samples were analyzed by flow cytometry. The red fluorescence of single events was recorded using a laser beam at 488 nm excitation λ with 610 nm as emission λ, to measure the DNA index. For Bax/Bcl2 ratio determination, PASMCs were seeded, synchronized and treated for 4 hours as described above. mRNA extraction, cDNA synthesis and RTQ-PCR were performed to determinate the expression of pro-apopototic Bax and anti-apoptotic Bcl2 genes compared to 18s as housekeeping gene, as described above.
Statistical Analyses
All data are reported as mean ± SEM. Effects of BMPR-2 mutations and BMP4 treatment were analysed by repeated-measures ANOVA. When the F-ratio of the ANOVA reached a critical value of p<0.05, nonparametric Mann-Withney tests were used to compare specific situations (20). A linear squared regression analysis was used to calculate correlations between pulmonary vascular resistance and contents (mRNA and protein) of the investigated BMP signaling molecules (20).
RESULTS
Clinical and Hemodynamic Characteristics of PAH Patients
There were no differences between PAH patients with and without BMPR-2 mutations in age (41 ± 2 vs 39 ± 3 years), female to male sex ratio (10/9 versus 5/4), mean pulmonary artery pressure (62 ± 2 vs 63 ± 3 mmHg), pulmonary vascular resistance (20 ± 1 vs 20 ± 2 U.m−2) and cardiac index (2.19 ± 0.09 vs 2.41 ± 0.25 L.min−1.m−2). None of patients presented with reversibility at vasodilator testing.
Identification and description of BMPR-2 mutations (Figure 1)
Figure 1.
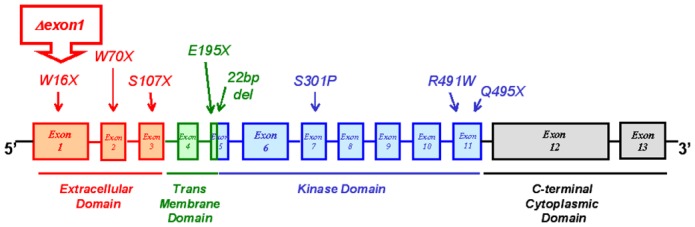
Genetic characteristics of patients. Schematic representation of BMPR-2 functional domains, demonstrating the range of BMPR-2 mutations studied in this study and indicating the nature of amino acid substitution or nonsense mutations (X).
Germline mutations in the 13 exons encoding BMPR-2 were identified in the 9 heritable PAH patients. Three heterozygous nonsense mutations were identified in exons 1 (W16X), 2 (W70X) and 3 (S107X), which encode part of the extracellular domain of BMPR-2. One mutation consisted in total deletion of exon 1 (Δexon1). Two mutations were found in exon 5, which encodes the transmembrane domain of BMPR-2: one heterozygous nonsense mutation (E195X) and one by loss of 22bp (22bp del). Two heterozygous missense and one nonsense mutations were identified respectively in exons 7 (S301P) and 11 (R491W and Q495X), which encode parts of the kinase domain of BMPR-2. No ALK-1 mutations was found in the PAH patients with and without mutations.
Pulmonary and cellular expression of BMPR’s (Figures 2–4)
Figure 2.
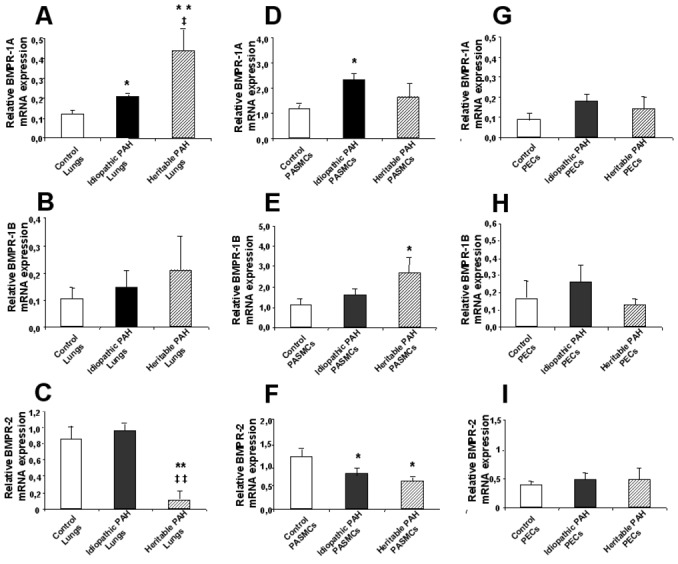
Relative BMPR-1A (A, D, G), BMPR-1B (B, E, H) and BMPR-2 (C, F, I) mRNA expression. Whole lung tissue samples (A, B, C), cultured pulmonary artery smooth muscle cells (PASMCs; D, E, F) and pulmonary microvascular endothelial cells (PECs; G, H, I) from controls (white bars), PAH patients without (black bars; idiopathic PAH) and with BMPR-2 mutations (dashed bars; heritable PAH) were assessed by RTQ-PCR as described in METHODS. Results are expressed as mean ± S.E.M. Statistical differences are assessed by Mann-Withney’s test: *P<0.05, ** P<0,001 vs control conditions; ‡P<0.05, ‡‡ P<0.001 idiopathic PAH vs heritable PAH conditions.
Figure 4.
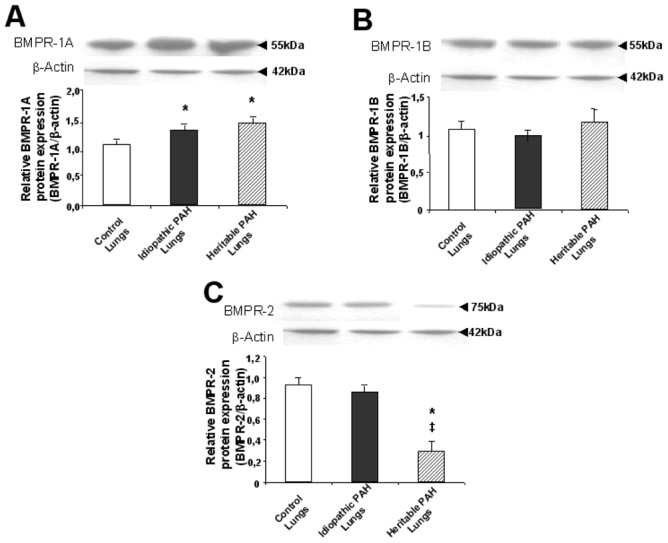
Relative BMPR-1A (A), BMPR-1B (B) and BMPR-2 (C) protein expression in whole lung tissue samples from controls (white bars), PAH patients without (black bars; idiopathic PAH) and with BMPR-2 mutations (dashed bars; heritable PAH) were assessed by Western blotting as described in METHODS. Results are expressed as mean ± S.E.M. Statistical differences are assessed by Mann-Withney’s test: *P<0.05 vs control conditions, ‡P<0.05 idiopathic PAH vs heritable PAH conditions.
The expression of BMPR-1A was increased in lung tissue from both heritable and idiopathic patients (mARN and protein), but only in PASMCs from idiopathic PAH patients, and was not different from controls in PECs from heritable and idiopathic PAH patients (mRNA). The expression of BMPR-1B was increased in PASMCs from heritable PAH patients only (mRNA). The expression of BMPR-2 was decreased in lung tissue from heritable PAH only (mRNA and protein), and in PASMCs from both heritable and idiopathic PAH, and was not different from controls in PECs (mRNA). However, as indicated by relatively large SEM’s, the increased expressions of BMPR-1A and BMPR-1B, and decreased expressions of BMPR-2 in PASMCs varied greatly from one mutation to another, with no consistent pattern. No correlation was found between pulmonary vascular resistance and lung expressions of BMPR-1A, BMPR-1B and BMPR-2 proteins.
Differencial effects of BMP4 on Smad and p38MAPK signaling (Figures 5 and 6)
Figure 5.
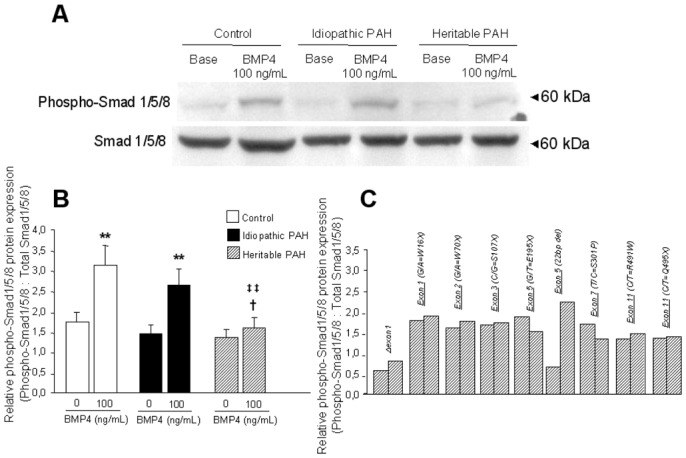
Smad 1/5/8 phosphorylation induced by BMP4. 90% confluent PASMCs isolated from controls (n=10), PAH patients without (idiopathic PAH; n=10) and without BMPR-2 mutations (heritable PAH; n=9) were stimulated with 100 ng/mL BMP4 for 20 minutes, followed by lysis for total protein. (A) Representative Western blots for phospho-Smad 1/5/8 and for total Smad 1/5/8 to show equal loading. (B) Densitometry of phospho-Smad 1/5/8 and total Smad 1/5/8 bands from Western blots of control (white bars), idiopathic PAH (black bars) and heritable PAH (dashed bars) PASMCs. Results are presented as relative protein expression ratio of phospho-Smad1/5/8 and total Smad1/5/8 band intensity. Data are presented as mean ±SEM. ** P<0,001 vs basal conditions in nonstimulated PASMCs; ‡‡ P<0.001 vs values for PASMCs isolated from controls stimulated by BMP4; †P<0.05 vs values for PASMCs isolated from idiopathic PAH patients stimulated by BMP4. (C) Relative densitometry ratio of phospho-Smad 1/5/8 in PASMCs isolated from definied naturally occuring BMPR-2 mutated patients (Δexon1, G/A=W16X, G/A=W70X, C/G=S107X, G/T=E195X, 22bp del, T/C=S301P, C/T=R491W, C/T=Q495X).
Figure 6.
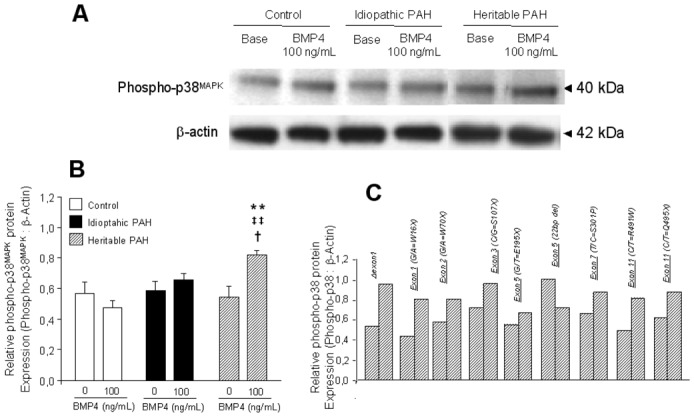
p38MAPK phosphorylation induced by BMP4. 90% confluent PASMCs isolated from controls (n=10), PAH patients without (idiopathic PAH; n=10) and with BMPR-2 mutations (heritable PAH, n=9) were stimulated with 100 ng/mL BMP4 for 20 minutes, followed by lysis for total protein. (A) Representative Western blots for phospho-p38 and for β-actin to show equal loading. (B) Densitometry of phospho-p38 and β-actin bands from Western blots of control (white bars), idiopathic PAH (black bars) and heritable PAH (dashed bars) PASMCs. Results are presented as relative protein expression ratio of phospho-p38 and β-actin band intensity. Data are presented as mean ±SEM. ** P<0,001 vs basal conditions in nonstimulated PASMCs; ‡‡ P<0.001 vs values for PASMCs isolated from controls stimulated by BMP4; †P<0.05 vs values for PASMCs isolated from idiopathic PAH patients stimulated by BMP4. (C) Relative densitometry ratio of phospho-p38MAPK in PASMCs isolated from definied naturally occuring BMPR-2 mutated patients (Δexon1, G/A=W16X, G/A=W70X, C/G=S107X, G/T=E195X, 22bp del, T/C=S301P, C/T=R491W, C/T=Q495X).
BMP4 (100ng/mL, 20 minutes) induced the activation (phosphorylation) of Smad1/5/8 in PASMCs isolated from idiopathic PAH patients and controls, indicating that the transmission of BMP signaling was intact in these cells. In contrast, no BMP4-induced phosphorylation of Smad1/5/8 was observed in PASMCs from heritable PAH, with the exception of the 22 bp del mutation. BMP4 activated p38MAPK signaling in PASMCs from heritable PAH patients (with the exception of the PASMC with 22bp del mutation) but not from idiopathic PAH patients or controls.
Effects of BMP4 on PASMC proliferation induced by serum (10%) and PDGF (10 ng/mL) treatment (Figure 7)
Figure 7.
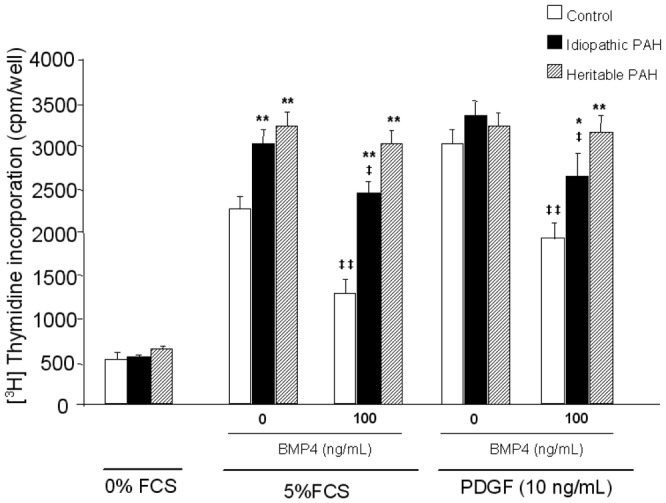
Basal [3H] thymidine incorporation in PASMCs derived from controls (white bars; n=10), PAH patients without (black bars; idiopathic PAH, n=19) and with BMPR-2 mutations (dashed bars; heritable PAH, n=9) in response to incubation with or without BMP4 (100 ng/mL), in presence of FCS 10% or PDGF (10 ng/mL). Data are presented as mean ±SEM. * P<0.01, ** P<0.001 vs PASMCs isolated from controls; ‡ P<0.05, ‡‡ P<0.001 vs basal conditions in BMP4-nonstimulated PASMCs.
PASMCs isolated from heritable and idiopathic PAH exhibited an increased proliferation, as assessed by [3H]-thymidine incorporation, in the presence of serum but not PDGF. The addition of BMP4 induced an inhibition of [3H]-thymidine incorporation in both serum- and PDGF-treated PASMCs from idiopathic PAH and controls, but not of PASMCs from heritable PAH patients. The absence of BMP4-induced inhibition of proliferation was observed on PASMCs from all the heritable PAH patients, excepted in the patient with the 22bp del mutation.
Effects of BMP4 on PASMC apoptosis (Figure 8)
Figure 8.
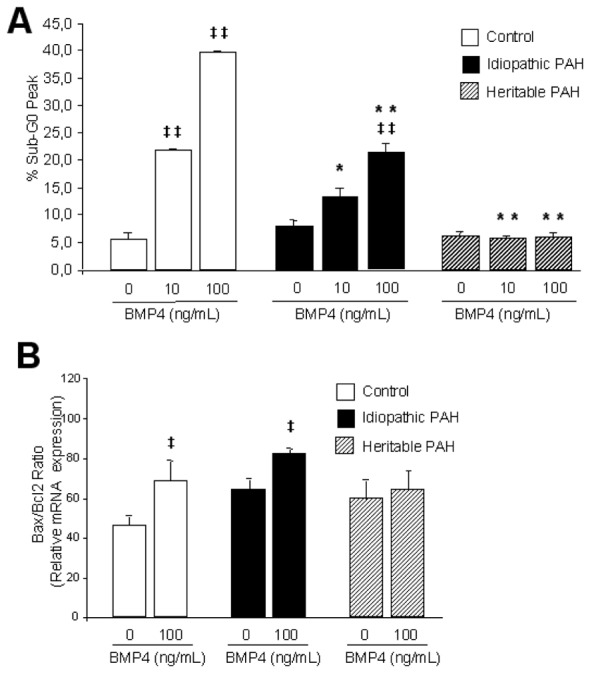
BMP4-induced apoptosis of PASMCs isolated from controls (n=10), PAH patients without (idiopathic PAH, n=10) and with BMPR-2 mutations (heritable PAH, n=9). (A) Flow cytometric analysis of propidium iodide stained control (white bars), idiopathic PAH (black bars) and heritable PAH (dashed bars) PASMCs treated with 10 and 100 ng/mL BMP4 to evaluate the distribution of cells in different phases of cell cycle. Apoptotic cells are evaluated as % sub-G0/G1 phase of cell population. (B) Apoptotic index (Bax/Bcl-2 ratio) of BMP4 (100 ng/mL)-treated PASMCs isolated from controls (white bars), PAH patients without (idiopathic PAH, black bars) and with BMPR-2 mutations (heritable PAH, dashed bars). PASMCs were seeded, synchronized for 48 hours and treated with BMP4 (100 ng/mL). Relative mRNA expression (Bax, Bcl-2) ratio was determined by RTQ-PCR. Data are presented as mean ±SEM. * P<0.05, ** P<0.001 vs similarly treated PASMCs isolated from controls; ‡ P<0.05, ‡‡ P<0.001 vs basal conditions in BMP4-nonstimulated PASMCs.
Flow cytometry analysis showed that BMP4 (10 and 100 ng/mL, 24 hours) increased apoptotic rates in PASMCs from idiopathic PAH patients and controls but with less extent in PASMCs from idiopathic PAH and not from heritable PAH patients (Figure 8A). The Bax/Bcl-2 pro-apoptotic ratio was increased by BMP4 at 100 ng/ml in PASMCs from controls and idiopathic PAH, not from heritable PAH patients (Figure 8B).
DISCUSSION
The present results show that (1) lung tissue and PASMCs, not PECs, from heritable PAH patients present with decreased expressions of BMPR-2 and variable increases in the expressions of BMPR1-A and BMPR-1B, while only a (relatively less important) decreased expression in BMPR-2 is observed in PASMCs from idiopathic PAH patients, (2) PASMCs from heritable PAH patients show no BMP4-induced Smad 1/5/8 phosphorylation but BMP4-induced activation of the p38MAPK pathway, (3) PASMCs from PAH patients present with an in vitro proliferative and anti-apoptotic pattern, that can be inhibited by BMP4 in idiopathic PAH but not in heritable PAH and (4) individual responses vary from one mutation to another, with in particular PASMCs from PAH patients with the 22bp del mutation showing no difference as compared to PASMCs from idiopathic PAH patients without identified mutations. The present work confirmed previous studies about BMPR-2 signaling in PAH and presented for the first time a large in vitro comparison of PASMCs with and without naturally occuring BMPR-2 mutations.
Even though BMPR-2 mutations are identified in the majority of heritable PAH patients, and carry a significant risk of developing the disease in asymptomatic carriers, two large clinical studies have failed to disclose differences in clinical presentation, hemodynamics and histopathology between heritable or idiopathic PAH patients, except for an earlier onset of the disease, more compromised hemodynamics, and maybe a less frequent reversibility at vasodilator testing in heritable PAH (7,8). These observations suggest heterogenous functional consequences of the various hitherto reported over 200 BMPR-2 mutations, and also interactions with coexisting signaling abnormalities. In the present study, there was no difference in clinical presentation of heritable and idiopathic PAH, but this is probably related to minor phenotypic differences, individual variability and small sample size.
Our results confirm that the expression of BMPR-2 is decreased in idiopathic PAH, and much more so in heritable PAH (21). The relative magnitudes of lung tissue and isolated PASMC and PEC expressions were suggestive of a predominant PASMC location of BMPR-2. This is in contrast with predominant endothelial cell location previously reported in normal and in lungs from PAH patients (21). However, recent studies on human PASMCs and PECs showed relatively high levels of BMPR-2 on both cell types, but very low expressions of BMPR-1A and -1B in PECs consistent with a lack of BMP4 responsiveness (22). In the present study, the expressions of BMPR-2 and of BMPR-1A and BMPR-1B were much lower in PECs as compared to PASMCs with no apparent impact of BMPR-2 mutations, so that further analysis focused on PASMCs. Moreover, in heritable PAH patients, the level of BMPR-2 expression was lower than predicted by the state of haploinsufficiency and the process of nonsense mediated decay secondary to the presence of nonsense mutations. These observations are in keeping with the notion that some additional environmental and/or genetic factors may be responsible to further reduce BMPR-2 expression.
Decreased expressions of BMPR-2 have also been reported to occur in experimental animal models of pulmonary hypertension such as induced by chronic systemic-to-pulmonary shunting (23), hypoxic exposure (24) and monocrotaline (25). In the latter study, decreased BMPR-2 expression was described in both lung tissue and PASMCs (25). Suprisingly, exclusive overexpression of BMPR-2 by gene therapy did not ameliorate monocrotaline-induced PAH in rats (26), indicating that reconstitution of the receptor was unable to restore BMP signaling and, thus, did not prevent disease onset or progression. The present results suggest that decreased expression associated to mutations in BMPR-2 seem to be crucial to explain the pro-proliferative and anti-apoptotic effects in PASMCs.
In the present study, the expressions of BMPR-1A and BMPR-1B tended to increase, though quite variably, with significance for the BMPR-1A in lung tissue of both heritable and idiopathic PAH patients, but only for BMPR-1A on PASMCs from idiopathic PAH patients and BMPR-1B in PASMCs from heritable PAH patients. The expression of BMPR-1A was decreased in a study on patients with various causes of severe pulmonary hypertension, including PAH, but also mitral stenosis and thromboembolic pulmonary hypertension, and this was explained in relation to an overexpression of angiopoietin-1 (27). The expression of BMPR-2 was unaltered in that same study (27). We previously reported RTQ-PCR and Western Blotting of unchanged expression of angiopoietin–1 in whole lung tissue and in PASMCs from PAH patients (14). In monocrotaline-induced pulmonary hypertension, the expressions of BMPR-2 and BMPR-1B were decreased, while the expression of BMPR-1A remained unchanged (25). An increased expression of BMPR-1B has been reported in one idiopathic PAH patient (28). The reasons for these discrepant results are unclear. Since BMPR-1A/BMPR-2 is the main BMP4 responsive receptor complex allowing for Smad 1/5/8 stimulation (22), the overexpression of BMPR-1A in the PASMCs of the idiopathic PAH patients could be some how related to the normal BMP4-responsiveness of these cells. As for the overexpression of BMPR-1B in heritable PAH, this could be speculated to be related to alternative p38MAPK signaling activation.
Of the 9 mutations identified in the present study, the W16X, R491X, Q495X have been previously reported (11,29,30). The 6 other mutations (Δexon1, W70X, S107X, E195X, 22bp del, S301P) across all four domains of the receptor are all novel including one total deletion of exon, three nonsense, one partial deletion and one missense mutation. Previous studies showed that missense mutations in the ligand binding domain by cysteine substitutions impair BMP signaling by mutant receptor mislocalisation in the cytosol (31). Moreover non-cysteine substitutions localise to the cell surface but also exhibit defects in BMP signaling activity (31). In contrast mutations in the cytoplasmic C-terminal domain only moderately inhibit Smad-signaling (31,32). Therefore, in the present study, all reported mutations would be susceptible to be deleterious by changing the protein sequence at important functional sites of the receptor and the associated protein functions.
Mutations of BMPR-2 (including R491X) heterogeneously interfer with BMP donwstream signaling, but all of them activate proliferative pathways (24). We previously reported that PASMCs isolated from idiopathic PAH patients present with enhanced growth responses to serum, but not to PDGF (10). In the present study, PASMCs with or without BMPR-2 mutations did not behave differently in this respect, but BMP4-induced growth inhibition and increased apoptosis was markedly more important in PASMCs with BMPR-2 mutations. It is of interest that all the BMPR-2 mutations identified in the present study, excepted the 22bp deletion in the transmembrane domain, responded homogeneously to these effects of BMP4, supporting the notion that the majority of BMPR-2 mutations are functionally linked. Along the same line, decreased BMPR-2 expression in monocrotaline-induced pulmonary hypertension has been reported to be associated with decreased phosphorylation of Smad1 and a decrease in BMP-induced apoptosis of PASMCs (25).
In the present study, the application of BMP4 was associated in PASMCs with BMPR-2 mutations with a decreased activation (phosphorylation) of Smad 1/5/8 together with an increased activation (phosphorylation) of the p38MAPK pathway. We selected a dose of BMP4 of 100ng/mL on the basis of available literature (18, 19, 32) and preliminary testing showing maximum efficacy of 100ng/mL as compared to 10ng/mL and 1ng/mL in discriminating PASMCs with and without BMPR-2 mutations. However, a recent study showed maximum efficacy at a lower dose of 10 ng/mL of BMP4 (33). The reasons for these discrepancies are not clear, and therefore the absence of complete dose-response curves may be a limitation to our findings. In monocrotaline-induced pulmonary hypertension, decreased BMPR-2 and phospho-Smad1 occurred without change in p38MAPK signaling in PASMCs (25). In hypoxia-induced pulmonary hypertension, downregulation of BMPR-2 did not affect Smad 1/5/8 phosphorylation, and was associated with a decreased p38MAPK signaling (24). Even though part of these differences may be related to model specificities and PASMCs versus whole lung measurements, the present data confirm previous report (31,32) that most mutations of BMPR-2 are associated with more profound changes in downstream signaling and are associated with increased p38MAPK signaling as a cause of increased PASMC proliferation.
The present results support the notion of altered BMPR-2/Smad signaling as a cause of increased proliferation of PASMCs playing an important role in the remodeling of pulmonary resistance vessels in PAH.
Figure 3.
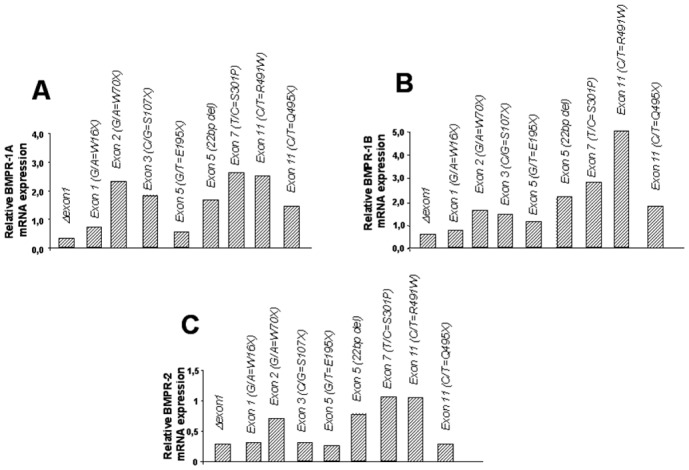
Relative BMPR-1A (A), BMPR-1B (B) and BMPR-2 (C) mRNA expression in PASMCs isolated from heritable PAH patients with naturally occuring BMPR-2 mutations (Δexon1, G/A=W16X, G/A=W70X, C/G=S107X, G/T=E195X, 22bp del, T/C=S301P, C/T=R491W, C/T=Q495X, G/A=W70X, C/G=S107X, G/T=E195X, 22bp del, T/C=S301P, C/T=R491W, C/T=Q495X). Results are assessed by RTQPCR as described in METHODS.
Acknowledgments
This study was supported by grants from the “Institut National de la Santé et de la Recherche Médicale”, the “Ministère de la Recherche”, the “Institut des Maladies Rares”, the “Délégation à la Recherche Clinique de l’Assistance Publique – Hôpitaux de Paris”, the Belgian Foundation for Cardiac Surgery and from the “Fonds de la Recherche Scientifique Médicale” (grant n° 3.4551.05). This research project received financial support from the European Commission under the 6th Framework Programme (Contracts n° LSHM-CT-2005-018275 and LSHM-CT-2005-018724, PULMOTENSION). This publication reflects only the authors’ views, and under no circumstances is the European Community liable for any use that may be made of the information it contains.
Footnotes
CONFLICT OF INTEREST DISCLOSURES
The authors have no conflict of interests to disclose.
References
- 1.Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–1665. doi: 10.1056/NEJMra035488. [DOI] [PubMed] [Google Scholar]
- 2.Newman JH, Trembath RC, Morse JA, Grunig E, Loyd JE, Adnot S, Coccolo F, Ventura C, Phillips JA, Knowles JA, Janssen B, Eickelberg O, Eddahibi S, Herve P, Nichols WC, Elliott G. Genetic basis of pulmonary arterial hypertension: current understanding and future directions. J Am Coll Cardiol. 2004;43:33S–39S. doi: 10.1016/j.jacc.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 3.Machado RD, Aldred MA, James V, Harrison RE, Patel B, Schwalbe EC, Gruenig E, Janssen B, Koehler R, Seeger W, Eickelberg O, Olschewski H, Elliott CG, Glissmeyer E, Carlquist J, Kim M, Torbicki A, Fijalkowska A, Szewczyk G, Parma J, Abramowicz MJ, Galie N, Morisaki H, Kyotani S, Nakanishi N, Morisaki T, Humbert M, Simonneau G, Sitbon O, Soubrier F, Coulet F, Morrell NW, Trembath RC. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum Mutat. 2006;27:121–132. doi: 10.1002/humu.20285. [DOI] [PubMed] [Google Scholar]
- 4.Liu F, Ventura F, Doody J, Massague J. Human type II receptor for bone morphogenic proteins (BMPs): extension of the two-kinase receptor model to the BMPs. Mol Cell Biol. 1995;15:3479–3486. doi: 10.1128/mcb.15.7.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moore RK, Otsuka F, Shimasaki S. Molecular basis of bone morphogenetic protein-15 signaling in granulosa cells. J Biol Chem. 2003;278:304–310. doi: 10.1074/jbc.M207362200. [DOI] [PubMed] [Google Scholar]
- 6.Ono M, Sawa Y, Mizuno S, Fukushima N, Ichikawa H, Bessho K, Nakamura T, Matsuda H. Hepatocyte growth factor suppresses vascular medial hyperplasia and matrix accumulation in advanced pulmonary hypertension of rats. Circulation. 2004;110:2896–2902. doi: 10.1161/01.CIR.0000146342.30470.30. [DOI] [PubMed] [Google Scholar]
- 7.Elliott CG, Glissmeyer EW, Havlena GT, Carlquist J, McKinney JT, Rich S, McGoon MD, Scholand MB, Kim M, Jensen RL, Schmidt JW, Ward K. Relationship of BMPR2 mutations to vasoreactivity in pulmonary arterial hypertension. Circulation. 2006;113:2509–2515. doi: 10.1161/CIRCULATIONAHA.105.601930. [DOI] [PubMed] [Google Scholar]
- 8.Sztrymf B, Coulet F, Girerd B, Yaici A, Jais X, Sitbon O, Montani D, Souza R, Simonneau G, Soubrier F, Humbert M. Clinical Outcomes of Pulmonary Arterial Hypertension in Carriers of BMPR2 Mutation. Am J Respir Crit Care Med. 2008;177:1377–1383. doi: 10.1164/rccm.200712-1807OC. [DOI] [PubMed] [Google Scholar]
- 9.Eddahibi S, Guignabert C, Barlier-Mur AM, Dewachter L, Fadel E, Dartevelle P, Humbert M, Simonneau G, Hanoun N, Saurini F, Hamon M, Adnot S. Cross talk between endothelial and smooth muscle cells in pulmonary hypertension: critical role for serotonin-induced smooth muscle hyperplasia. Circulation. 2006;113:1857–1864. doi: 10.1161/CIRCULATIONAHA.105.591321. [DOI] [PubMed] [Google Scholar]
- 10.Marcos E, Fadel E, Sanchez O, Humbert M, Dartevelle P, Simonneau G, Hamon M, Adnot S, Eddahibi S. Serotonin-induced smooth muscle hyperplasia in various forms of human pulmonary hypertension. Circ Res. 2004;94:1263–1270. doi: 10.1161/01.RES.0000126847.27660.69. [DOI] [PubMed] [Google Scholar]
- 11.Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet Sep. 2000;67:737–744. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet. 2000;26:81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 13.Eddahibi S, Humbert M, Fadel E, Raffestin B, Darmon M, Capron F, Simonneau G, Dartevelle P, Hamon M, Adnot S. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest. 2001;108:1141–1150. doi: 10.1172/JCI12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dewachter L, Adnot S, Fadel E, Humbert M, Maitre B, Barlier-Mur AM, Simonneau G, Hamon M, Naeije R, Eddahibi S. Angiopoietin/Tie2 Pathway Influences Smooth Muscle Hyperplasia in Idiopathic Pulmonary Hypertension. Am J Respir Crit Care Med. 2006;174:1025–1033. doi: 10.1164/rccm.200602-304OC. [DOI] [PubMed] [Google Scholar]
- 15.Hewett PW, Murray JC. Immunomagnetic purification of human microvessel endothelial cells using Dynabeads coated with monoclonal antibodies to PECAM-1. Eur J Cell Biol. 1993;62:451–454. [PubMed] [Google Scholar]
- 16.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 17.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 18.Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, Trembath RC. Circulation. 2001;104:790–795. doi: 10.1161/hc3201.094152. [DOI] [PubMed] [Google Scholar]
- 19.Sobolewski A, Rudarakanchana N, Upton P, Yang J, Crilley TK, Trembath RC, Morrell NW. Hum Mol Genet. 2008;17:3180–3190. doi: 10.1093/hmg/ddn214. [DOI] [PubMed] [Google Scholar]
- 20.Winer BJ, Brow DR, Michels KM. Statistical Principles in Experimental Design. 3. New York: MacGraw-Hill; 1991. pp. 220–283. [Google Scholar]
- 21.Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105:1672–1678. doi: 10.1161/01.cir.0000012754.72951.3d. [DOI] [PubMed] [Google Scholar]
- 22.Upton PD, Long L, Trembath RC, Morrell NW. Functional characterization of bone morphogenetic protein binding sites and Smad1/5 activation in human vascular cells. Mol Pharmacol. 2008;73:539–552. doi: 10.1124/mol.107.041673. [DOI] [PubMed] [Google Scholar]
- 23.Rondelet B, Kerbaul F, Van Beneden R, Motte S, Fesler P, Hubloue I, Remmelink M, Brimioulle S, Salmon I, Ketelslegers JM, Naeije R. Signaling molecules in overcirculation-induced pulmonary hypertension in piglets: effects of sildenafil therapy. Circulation. 2004;110:2220–2225. doi: 10.1161/01.CIR.0000143836.40431.F5. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi H, Goto N, Kojima Y, Tsuda Y, Morio Y, Muramatsu M, Fukuchi Y. Down-regulation of type II bone morphogenetic protein receptor in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2005;290:L450–458. doi: 10.1152/ajplung.00206.2005. [DOI] [PubMed] [Google Scholar]
- 25.Morty RE, Nejman B, Kwapiszewska G, Hecker M, Zakrzewicz A, Kouri FM, Peters DM, Dumitrascu R, Seeger W, Knaus P, Schermuly RT, Eickelberg O. Dysregulated bone morphogenetic protein signaling in monocrotaline-induced pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol. 2007;27:1072–1078. doi: 10.1161/ATVBAHA.107.141200. [DOI] [PubMed] [Google Scholar]
- 26.McMurtry MS, Moudgil R, Hashimoto K, et al. Overexpression of human bone morphogenetic protein receptor II does not ameliorate monocrotaline pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2006;292:L872–878. doi: 10.1152/ajplung.00309.2006. [DOI] [PubMed] [Google Scholar]
- 27.Du L, Sullivan CC, Chu D, Cho AJ, Kido M, Wolf PL, Yuan JX, Deutsch R, Jamieson SW, Thistlethwaite PA. Signaling molecules in nonfamilial pulmonary hypertension. N Engl J Med. 2003;348:500–509. doi: 10.1056/NEJMoa021650. [DOI] [PubMed] [Google Scholar]
- 28.Takeda M, Otsuka F, Nakamura K, Inagaki K, Suzuki J, Miura D, Fujio H, Matsubara H, Date H, Ohe T, Makino H. Characterization of the bone morphogenetic protein (BMP) system in human pulmonary arterial smooth muscle cells isolated from a sporadic case of primary pulmonary hypertension: roles of BMP type IB receptor (activin receptor-like kinase-6) in the mitotic action. Endocrinology. 2004;145:4344–4354. doi: 10.1210/en.2004-0234. [DOI] [PubMed] [Google Scholar]
- 29.Koehler R, Grunig E, Pauciulo MW, Hoeper MM, Olschewski H, Wilkens H, Halank M, Winkler J, Ewert R, Bremer H, Kreuscher S, Janssen B, Nichols WC. Low frequency of BMPR2 mutations in a German cohort of patients with sporadic idiopathic pulmonary arterial hypertension. J Med Genet. 2004;41:e127. doi: 10.1136/jmg.2004.023101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, Wheeler L, Phillips JA, Newman J, Williams D, Galiè N, Manes A, McNeil K, Yacoub M, Mikhail G, Rogers P, Corris P, Humbert M, Donnai D, Martensson G, Tranebjaerg L, Loyd JE, Trembath RC, Nichols WC. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet. 2001;68:92–102. doi: 10.1086/316947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R, Patel D, Trembath RC, Morrell NW. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2002;11:1517–1525. doi: 10.1093/hmg/11.13.1517. [DOI] [PubMed] [Google Scholar]
- 32.Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, Atkinson C, Chen H, Trembath RC, Morrell NW. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res. 2005;96:1053–1063. doi: 10.1161/01.RES.0000166926.54293.68. [DOI] [PubMed] [Google Scholar]
- 33.Yang J, Davies RJ, Southwood M, Long L, Yang X, Sobolewski A, Upton PD, Trembath RC, Morrell NW. Mutations in bone morphogenetic protein type II receptor cause dysregulation of Id gene expression in pulmonary artery smooth muscle cells: implications for familial pulmonary arterial hypertension. Circ Res. 2008;102:1212–1221. doi: 10.1161/CIRCRESAHA.108.173567. [DOI] [PubMed] [Google Scholar]