

Abstract
While ab initio modeling of protein structures is not routine, certain types of proteins are more straightforward to model than others. Proteins with short repetitive sequences typically exhibit repetitive structures. These repetitive sequences can be more amenable to modeling if some information is known about the predominant secondary structure or other key features of the protein sequence. We have successfully built models of a number of repetitive structures with novel folds using knowledge of the consensus sequence within the sequence repeat and an understanding of the likely secondary structures that these may adopt. Our methods for achieving this success are reviewed here.
Keywords: protein modeling, repetitive protein, antifreeze protein, Beta‐solenoid, tandem repeat, molecular dynamics
Abbreviations
- AFGP
antifreeze glycoprotein
- AFP
antifreeze protein
- HMM
hidden Markov models
- IBP
ice‐binding protein
- IBS
ice‐binding site
- INP
ice‐nucleating protein
- MD
molecular dynamics
- NMR
nuclear magnetic residence
- RMSD
root‐mean‐square deviation
- RMSF
root‐mean‐square fluctuation
Introduction to Repetitive Protein Structures
In order to relate structure to function it is preferable to determine atomic‐resolution structures by experimental means (X‐ray crystallography or NMR), but in the event that these methods are not successful, and there are no structures available for homology modeling, it is possible to conduct ab initio modeling. Then those predicted structures can be used as a guide for site‐directed mutagenesis experiments to confirm the protein fold and reveal information about those amino acids that are essential for function. One focus of our work has been to understand the structure and function of ice‐binding proteins (IBPs).1 Our ability to model structures in silico has been aided by the repetitive nature of many of the IBPs, which is consistent with their binding to a crystalline lattice.
IBPs are produced in many organisms that survive in icy environments. They protect the organism from freezing damage by binding to ice.1 Their adsorption to the ice surface makes it more difficult for water to join the ice crystal. In organisms that cannot survive freezing they function as antifreeze proteins (AFPs) to depress the freezing point of a solution and prevent the ice from growing further. In organisms that can tolerate freezing their role is to stop the recrystallization of ice into bigger more damaging crystals. IBPs have one contiguous surface that binds to ice, referred to as the ice‐binding site (IBS). Our current view of an IBS is that it organizes surface waters into an ice like pattern in order to bind particular planes of ice. IBSs are typically flat, hydrophobic, regular and extensive compared to the other surfaces of the molecule. Often they are rich in small hydrophobic residues such as Thr and Ala.
The overall structures of IBPs are remarkably different, as might be expected for proteins that have independently evolved from different progenitor genes.1 Although these proteins have convergently evolved to serve the same function of binding to ice, even their IBSs can vary as befits proteins that bind to different planes of ice with somewhat different surface structures. But they can also be remarkably similar like the two parallel ranks of Thr seen on the IBSs of beetle2 and moth3 AFPs.
Many of the IBPs with known structures have been determined experimentally to have sequences that are composed of multiple short repeats (3–19 residues in length) that in turn form repeating structural motifs. These structural motifs include α‐helices and polyproline type II helices, but the majority of repetitive IBPs form a β‐solenoid structure.1 In a repetitive β‐solenoid each sequence repeat forms a coil that consists of one or more β‐strands connected by loops. These coils then form a solenoid structure in which the equivalent β‐strands of each coil form hydrogen‐bonds to the neighboring strands to form β‐sheets. Single β‐solenoid structures have been observed with one to four β‐sheets.4
Examples of three repetitive ice‐binding protein structures and their sequence patterns are shown in Figure 1. Type I AFP is a simple, alanine‐rich, short α‐helical AFP found in some fishes.5 This AFP has an 11‐amino‐acid repeating motif that forms three helical turns with a slightly different periodicity (3.7 residues/turn) from the classic α‐helix (3.6 residues/turn). The best conserved positions are a Thr and two Ala [colored red and blue, respectively, in Fig. 1(A)]. These residues are presented in the structure adjacent to each other on the same side of the helix to form the ice‐binding surface.6
Figure 1.
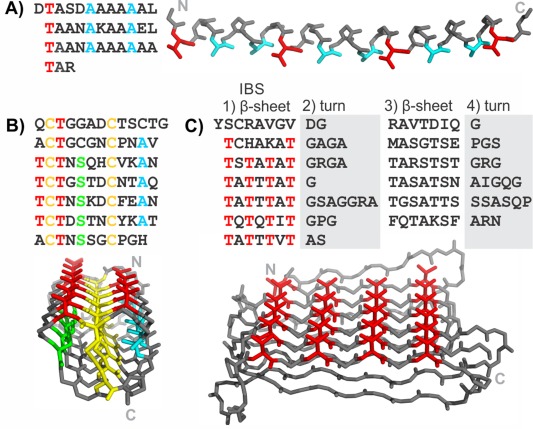
Sequences and structures of some repetitive ice‐binding proteins. Antifreeze proteins from A) Pseudopleuronectes americanus (type I AFP), B) T. molitor, and C) Rhagium inquisitor. Key repetitive amino acids are colored: Thr (red), Ala (cyan), Cys (yellow), Ser (green). The N and C termini are labeled on the structures by N and C, respectively.
The AFP from the mealworm beetle, Tenebrio molitor (TmAFP) has a 12‐residue repeating motif of TCTxSxxCxxAx [Fig. 1(B)]. The structure is a very tightly wound β‐solenoid that is stabilized by disulfide bonds connecting opposite sides in each coil of the solenoid. The other residues that point into the solenoid core are regularly spaced Ser and Ala on either side of the disulfide ladder, which both have short side‐chains that fill the respective halves of the divided core. The regularly spaced Thr of the TXT motifs are the ice‐binding residues and point outward from the solenoid on the same surface making up a narrow, untwisted β‐sheet that is the IBS. The sequence diverges from the regular pattern at the N and C termini to allow for helix‐capping.
An unrelated AFP was identified in the inquisitor beetle (RiAFP).7 Its sequence is not as regular as the two presented previously, but careful analysis of the sequence allowed for trends to be observed and successful modeling of the structure.8 The motif can be separated into four parts [Fig. 1(C)]. The first part is the most regular. It has alternating polar and non‐polar residues indicative of β‐sheets, and polar residues, which point outward, that are mostly Thr and make up the AFP's IBS. The second and fourth parts of the motif have several Gly, Ser, and Pro that are often involved in turns. The third part of the motif has again alternating polar and non‐polar residues indicative of another β‐sheet, of a similar length to the first but less regular. The structure, as predicted by modeling, is a flat β‐solenoid with two β‐sheets stabilized by interdigitating small hydrophobic residues in its extremely narrow core.9
Introduction to Modeling
When modeling a protein structure from its sequence, the most common method is homology modeling, in which the sequence of the unknown structure is aligned with sequences of proteins of known structure.10 The known structures are used as templates to which the sequence of the target is restrained. This requires sufficient sequence similarity to identify the homologous structures. A recent advance in homology modeling is the use of Hidden Markov Models (HMM) to find more distant relatives of a target sequence.11 Despite the added sensitivity of detecting distant homologs that this method provides, the evolution of ice‐binding proteins has occurred relatively recently so it is rare to find true homologs in any databases.1 In the case of the repetitive IBPs, their sequences have likely evolved through duplication events. The only repetitive AFP whose evolution has been explained to date is that of the antifreeze glycoprotein (AFGP), which arose from multiple duplication events of a small region within the trypsinogen gene.12
In the absence of a homologous structure, we perform “intuition‐guided” modeling. We make use of predicted secondary structure and similarity to other known structures, along with any information that might be known about functionally important residues. For example, when the targets are ice‐binding proteins, we would expect there to be a flat, relatively hydrophobic face to serve as the IBS. A common structural feature that allows the generation of such a surface is a flat β‐sheet and indeed, repeats that contain β‐strands are very common in ice‐binding proteins. Furthermore, a common feature among several AFPs is a TXT sequence motif in which the two Thr residues are pointing outward and the “X” residue is a hydrophobic residue or a disulfide bonded Cys that point inward to form part of the hydrophobic core of the protein. In this case the TXT motif is likely to be part of a β‐strand. Another common feature that aids in the modeling is that the tandem repeats found in ice‐binding proteins are typically short sequences that together fold into a single domain, rather than longer sequences that each fold into a domain that is then repeated.
General Outline of the Building Process
Identifying the repeating sequence
The first step in modeling a repetitive protein is identifying the repeating sequence motif. Given the short repeats that are commonly observed in IBPs (3–19 residues) and their tandem arrangement the repeats can usually be observed through visual inspection of the sequence. They may also be detected using repeat detection algorithms such as T‐REKS, XSTREAM and REPTITIA,13, 14, 15 and by dot‐matrix plots.16 Examples of dot‐matrix plots of the ice‐nucleating protein from Pseudomonas borealis (PbINP), of T. molitor AFP (TmAFP), and Marinomonas primoryensis IBP (MpAFP region IV) show extensive repetition in these sequences (Fig. 2). Dot‐matrix analysis shows that PbINP has a 16‐residue repeating motif that continues for ∼800 residues in the central section flanked by N‐ and C‐terminal regions devoid of repeats [Fig. 2(A)]. A higher order 48‐residue repeat is visible in the expansion of the red boxed area [Fig. 2(B)]. Dot‐matrix analyses of TmAFP and MpAFP indicate their 12‐ and 19‐residue repeating motifs, respectively [Fig. 2(C,D)]. The lack of repetition in the very N‐terminal regions (and to a lesser extent the C‐terminal regions is due to formation of capping structures essential for the stability of β‐solenoids.17
Figure 2.
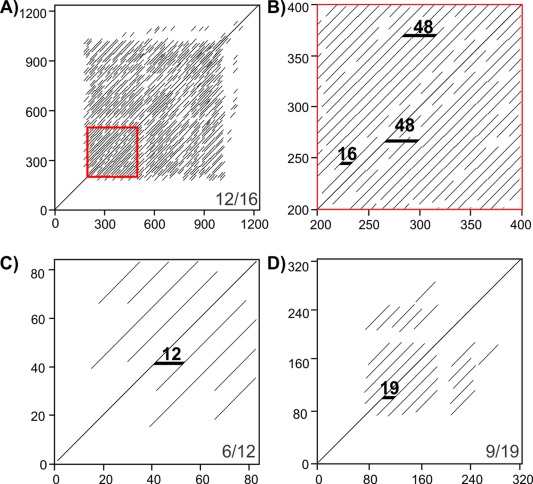
Dot‐matrix analyses of repetitive proteins. A) and B) P. borealis ice‐nucleating protein, C) T. molitor antifreeze protein, and D) M. primoryensis ice‐binding protein (region IV). B is an enlargement of the red box in A. Residue numbers in the sequence are indicated on the vertical and horizontal axes. Bottom right corners of A, C, and D indicate dot‐matrix analysis parameters (matches per window size). Black horizontal markers in B, C, and D indicate sequence length between matched segments.
We also typically run secondary structure predictions. The repeating sequence pattern is examined to look for trends, such as alternating hydrophobic and hydrophilic residues that may indicate the presence of a β‐strand and the presence of Gly or Pro residues that may indicate the locations of tight turns. For the shorter repeat lengths these may not result in any observed secondary structure pattern. Positions within the repeat that are not well conserved from one repeat to another may also be present in the connecting loops or they may indicate the regions of the molecule that are not functionally important. The natural isoform variation of the sequences within a family of ice‐binding proteins can also aid in the identification of the residues that make up a sequence repeat as well as of functionally important residues. For examples, variants of type I AFP are known that differ by exactly 11 residues corresponding to one repeat [Fig. 1(A)].18 Spruce budworm AFP (SbwAFP) has a well‐characterized isoform (501) that is 30 residues longer than most, with the extra sequence making exactly two 15‐residue repeats that each makes a coil of the β‐solenoid structure.19
While detection of repeats is often not especially difficult, finding the correct “frame” of the repeats is important for establishing the repeating structure, or coil. Frequently the repeats near the amino‐ and carboxy‐termini of the domain are less like those in the middle of the domain, so definition of the actual repeat boundaries can be more difficult than the initial identification of a repeating sequence. The N‐ and C‐terminal repeats may show less identity to the central repeats due to fewer steric restrictions and also because they may have a role in capping the repeat structure as mentioned earlier. Use of a regular expression to describe the repeat pattern can be useful for establishing the limits of the domains. Capping is often necessary to satisfy the hydrogen‐bonding requirements of the backbone and prevent end‐to‐end associations that would lead to amyloid formation and deposition. In the case of β‐helical structures, the cap is often observed to be amphipathic in order to bury the hydrophobic core,20 while particular residues are frequently found near the N‐ (often Asn and Pro) and C‐termini (often Gly) of α‐helices.21
Building the initial model
While there may be no known homologs to a repetitive ice‐binding protein sequence we often observe that portions of the repeat sequence show similarity to known structures or motifs. Combining that information with patterns of hydrophobicity and hydrophilicity as well as secondary structure predictions can help define a conformation for a single repeat or coil. We describe specific examples of how this is achieved below.
Once an initial conformation for a single repeat (coil) has been built as described in specific examples below, that repeat is then “copied and pasted” in a graphics program such as PyMOL22 (Fig. 3). The copy of the first coil is translated such that it can make favorable interactions with the initial coil such as backbone‐to‐backbone hydrogen bonding, side chain salt bridging and aromatic group stacking. The process is then repeated for subsequent coils. The conformations of the terminal residues of each coil are altered to allow connection of each coil to its neighbors. While performing this process for β‐solenoid structures we typically make models for both left‐ or right‐handed versions of the solenoid. In the case of α‐helical IBPs, one may need to adjust the phi and psi angles away from the standard α‐helical angles in order to achieve the correct helical repeat. While building the complete structure, one must correct the sequence of each coil to match the sequence of the protein and be aware that repeats often have additional residues in them that form bulges or loops that can be accommodated into the model.
Figure 3.
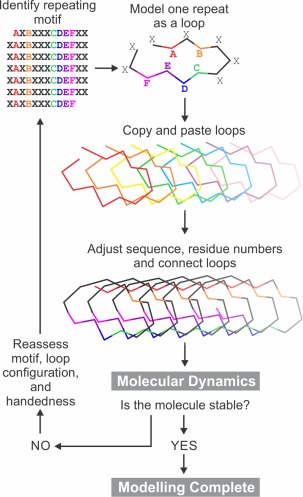
General overview of the method used for structural modeling of proteins with repetitive sequences. A–F are residues that show repeating patterns. X signifies non‐repetitive residues.
Testing of Models
Once a model has been constructed, it must be tested for stability and its ability to predict the functional features of the IBP. Typically a model in explicit solvent would be subjected to energy minimization and then a molecular dynamics (MD) simulation of at least 20 ns duration. This length of simulation is usually sufficient to determine if the structure is in a stable conformation. Here, we are defining stability as the maintenance of structural conformation from the starting model over the simulation time. The resulting trajectory can be analyzed visually using a program such as VMD23 or PyMOL22 as well as by plotting various calculated quantities, such as the root‐mean‐square deviation (RMSD) from the starting structure and the root‐mean‐square fluctuation (RMSF). The RMSD plot will always show an early jump as the initial structure relaxes, but if it quickly levels off to a nearly constant RMSD value this indicates a stable structure (Fig. 4). If instead it shows a steady rise in RMSD, or at a later time point shows a large jump in RMSD, this indicates that the initial structure contained significant strain. Simple energy minimization is not likely to reveal this situation.
Figure 4.
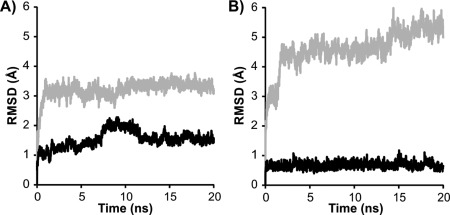
Assessing model stability by molecular dynamics. Root‐mean‐square deviation (RMSD) is plotted as a function of time during 20 ns molecular dynamics simulations of A) L. perenne ice‐binding protein at 4°C and B) midge antifreeze protein at 23°C. RMSDs for right‐ and left‐handed models are shown in gray and black, respectively.
Initial analysis of LpAFP models suggested that both right‐ and left‐handed LpAFP models had similar stability.24 An analysis performed more recently suggested slightly more stability for the left‐handed form [Fig. 4(A)], which agrees with the crystal structure.25 MD analysis of the midge AFP model clearly showed that the left‐handed solenoid was much more stable than the right handed version [Fig. 4(B)]. The RMSD of the left handed model increased slightly at the beginning of the simulation then remained unchanged through the simulation. The right handed model increased drastically at the start of the simulation indicating strain in the model then continuously increased over the 20 ns simulation indicating that model was unstable.
An RMSF plot will indicate those areas of the structure that fluctuate the most and while it usually shows that the termini fluctuate, it can help to identify areas that may have been built in a strained conformation. Using the time of that transition one can use VMD to identify what structural transition occurred at the same time and then rebuild the structure of the coils to match the energetically more favorable conformation.
In the event that the MD simulation indicates an unstable structure, due to a steadily rising RMSD plot, the initial structure can be compared with one late in the simulation to see if a more stable conformation can be used for further modeling. For example, in the case of a β‐solenoid structure, there may be some peptide bonds that were initially built in the wrong conformation and need to be flipped for every coil to arrive at a lower energy structure while still maintaining the overall coil shape.
Once a stable structure of an IBP has been obtained it can be assessed for its ability to predict the ice‐binding site. Within VMD, the volmap function can be used to calculate the water density. It has been observed that the ice‐binding site will tend to order the bound waters into an ice‐like array.26 In addition, the model can be used to design mutagenesis experiments to test the role of specific amino acid side chains on the function of the IBP. If there are multiple sequences available (isoforms or orthologs) then the pattern of sequence conservation or variation can be mapped onto the model.
These basic principles are illustrated here by following a number of case histories where new IBP structures and even new folds have been determined by modeling from repetitive IBP sequences.
MpAFP and Its Ice‐Binding Domain RIV
The AFP domain (RIV) in the giant ice adhesin of M. primoryensis (MpAFP) is a good example where a model was predicted well ahead of solving the structure and with sufficient accuracy to guide mutagenesis experiments that both validated the model and identified the AFP's ice‐binding site.27 The antifreeze activity of this Gram‐negative Antarctic bacterium (MpAFP) was found to be calcium‐dependent.28 Sequencing of tryptic peptides from ice‐affinity and gel‐purified MpAFP produced some matches to the repeating regions of RTX proteins.27 Since the RTX repeats require calcium for folding into a β‐solenoid structure, these two features drew our attention to this small region that comprises only ∼2% of the 1.5‐MDa bacterial protein. Several AFPs were known to be β‐solenoids29, 30 and an RTX‐type structure would fit with calcium dependency. Initial expression of constructs from this region of MpAFP in Escherichia coli confirmed that RIV had antifreeze activity, although the constructs showed a tendency to aggregate and were not amenable to crystallization.
Initial modeling of MpAFP_RIV using RTX proteins as the template
The central region of MpAFP_RIV contains eleven tandem repeats of a 19‐aa sequence [Fig. 5(C)]; each beginning with the consensus xGTGNDxUx where x can be any residue and U is typically a large hydrophobic residue, and with two repeats extended by two residues. This consensus sequence is similar to the 9‐aa Ca2+‐binding sequence repeats GGxGxDxUx from the repeats‐in‐toxin (RTX) proteins [Fig. 5(A)], which are a family of secreted proteins with a wide range of biological functions produced by Gram‐negative bacteria. Each RTX repeat binds one Ca2+ as it curls around the ion and coordinates it with an inward pointing Asp and backbone carbonyl groups. The 9‐aa RTX repeats typically appear in tandem pairs, forming symmetrical 18‐aa coils that bind two Ca2+ ions inside both bends of the β‐roll that are shared with neighboring coils [Fig. 5(B)].
Figure 5.
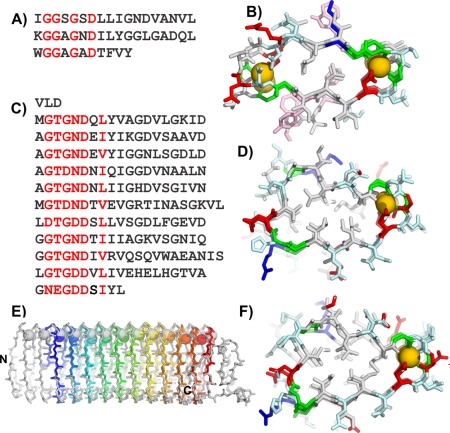
Modeling the M. primoryensis ice‐binding protein (region IV) (MpAFP_RIV). A) Sequence and B) crystal structure of the repetitive β‐roll domain of P. aeruginosa alkaline protease. C) Sequence and D) modeled structure of the repetitive domain of the MpAFP_RIV. E) Peptide backbone (sticks) and calcium ion (spheres) alignment of MpAFP_RIV crystal structure (gray) and model (rainbow). F) MpAFP_RIV crystal structure. For A and C red letters indicate most repetitive residues. For B, D, and F, acidic residues are in red, basic residues are in blue, polar residues are in light cyan, aromatic residues are in light pink, and Gly are green, all other non‐polar residues are in gray. Calcium ions are gold spheres.
The alkaline protease RTX protein produced by the Gram‐negative bacterium Pseudomonas aeruginosa [Fig. 5(A)], had a known crystal structure [Fig. 5(B)], which we used as the template to model MpAFP_RIV. Each of the 19‐aa repeats of MpAFP_RIV was initially modeled as one coil of a right‐handed β‐solenoid with Ca2+ bound internally down both sides of the structure. However, MpAFP_RIV only contains one canonical RTX repeat (xGTGNDxUx) in each of the 19‐aa repeats followed by the 10‐aa sequence of UGGxUxGxUx. In contrast, each β‐helical loop of alkaline protease is comprised of two tandem RTX repeats (18 residues). When this initial model of MpAFP_RIV was subjected to molecular‐dynamics, it became apparent that the structure was not able to support the second row of Ca2+ binding down the solenoid. The canonical RTX repeat (xGTGNDxUx) in each of the 19‐aa coils held its Ca2+ but the following 10‐aa repeat (UGGxUxGxUx) did not retain the second Ca2+ [Fig. 5(D)]. This 10‐residue segment has a large hydrophobic residue in place of a canonical glycine residue, and the aspartate residue that is essential for binding a Ca2+ is replaced by a glycine. Thus the backbone conformation was adjusted such that this non‐Ca2+‐binding side of the β‐helix was stabilized by a conserved hydrophobic core [Fig. 5(D)].
Final model of the MpAFP_RIV
The 322‐aa MpAFP_RIV model folds as a right‐handed β‐helix containing 11 Ca2+‐binding loops [Fig. 5(D)]. In contrast to the symmetrical cross‐section of alkaline protease with Ca2+ bound at both ends, MpAFP_RIV has a more triangular cross‐section with Ca2+ ions aligned down one side. Circular dichroism analyses confirmed that these Ca2+ are needed for the proper folding of the protein.27 MpAFP_RIV is also stabilized on the opposite side of the loops by a conserved hydrophobic core throughout the length of the model.
The final model of MpAFP_RIV revealed a new potential IBS comprised of two parallel rows of outward pointing Thr and Asn/Asp residues. To validate the model and the putative IBS, numerous mutations were made at positions around the coils and one toward the middle of the AFP. As expected, the insertion of an Arg into the core in place of a Val spoiled the fold of the protein and knocked out its antifreeze activity.27 When Thr and Asn/Asp on the IBS were replaced by Tyr, to disrupt the IBS due to steric hindrance and/or a disturbance in the anchored clathrate water pattern their activities were at least 50% lower than those of the wild type. CD analyses showed that all the IBS mutants folded identically to the wild type in the presence of Ca2+. In contrast, the tyrosine mutants on other surfaces of MpAFP had negligible effects on the protein's antifreeze activity.
Comparison of the MpAFP_RIV's model and its crystal structure
The 1.7‐Å crystal structure of MpAFP_RIV was solved several years later once the solenoid capping sequences were in place. The structure has a very similar overall fold to the model and is also a right‐handed helix [Fig. 5(E,F)]. It forms a β‐solenoid with 13 Ca2+ bound down one side of the structure, and it produced an RMSD of 1.64 Å compared to the initial model. The structure confirmed that the IBS of MpAFP_ RIV was correctly modeled; displaying two parallel rows of Thr and Asp/Asn residues that are key to ice binding. The crystal structure also revealed distinct capping structures for the β‐helix that were not modeled. At its N terminus, MpAFP_RIV is stabilized by several large hydrophobic residues such as Phe and Tyr, whereas the C terminus is capped by a short α‐helix. These features and an internal loop were not included in the original model due to a lack of sequence similarity to the template structure.
Modeling the Ryegrass Antifreeze Protein
The 118‐residue sequence of the Lolium perenne antifreeze protein (LpAFP), has a 7‐residue repeating motif of xxNxVxG with the occasional insertion between x and G to give an 8‐residue repeat [Fig. 6(A)].31 Fourier transform infrared spectrophotometry results indicated a high content of solvent‐exposed β‐sheet. The repetitive nature of the sequence and evidence of β‐structure content guided the modeling of LpAFP to a β‐solenoid design.
Figure 6.
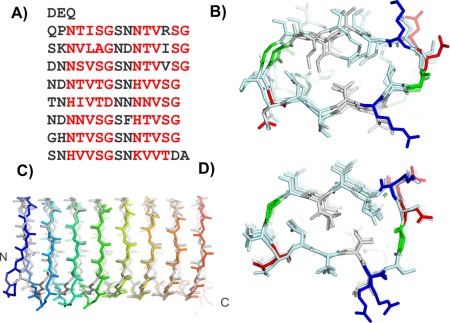
Modeling L. perenne antifreeze protein (LpAFP). A) Sequence and B) modeled structure of LpAFP. C) Peptide backbone alignment of LpAFP crystal structure (gray) and model (rainbow). D) LpAFP crystal structure. Sequence and structures are colored the same as in Figure 5.
Initial modeling of LpAFP
The first β‐helical model was built with a triangular cross section where a 7‐residue repeat made up each side of the triangle.24 The conserved Val of the repeat pointed inside the helix. However, their small side chains did not fill the core to form strong hydrophobic interactions that would stabilize the protein fold. LpAFP was remodeled with two repeating motifs making up one loop of the solenoid, a structure loosely based on the β‐roll domain of the alkaline protease from P. aeruginosa.32 Again, the conserved Val pointed inward but the new arrangement brought apposing Val side chains into a Val‐stacked ladder that built a much stronger hydrophobic core [Fig. 6(B)]. In this new model, the conserved Asn residues were located at the turns connecting the β‐strands and pointed internally to form two Asn ladders. These reinforced the fold by forming hydrogen bonds to the neighboring coil while contributing the β‐C atoms' methylene group to the hydrophobic core. On one side of the helix (b‐side), three additional Ser residues on consecutive loops resulted in a bulge while the opposing side was flatter (a‐side). Both a‐ and b‐ sides were considered potential ice‐binding surfaces. The model was built as both a right‐ and left‐handed helix, however, only the right‐handed model was deposited into the protein data bank (PDB 1I3B).
Testing the LpAFP model
Mutations of predicted outward‐pointing residues supported the LpAFP model. As predicted by the model, none of these mutations disrupted the LpAFP fold or caused solubility problems. When these mutants were assayed for antifreeze activity, only those with changes on the a‐side resulted in decreased antifreeze activity. This series of experiments identified the a‐side as the IBS and eliminated the b‐side from contention.33 Based on the model, putative internal residues were selected for substitution by Met for seleno‐methionine labeling. Although this strategy was not used for solving the phase problem with native LpAFP crystals, the Met substitutions were well tolerated and did little to alter the antifreeze activity of the protein, thus confirming the basic fold.
Comparison of the LpAFP model with the crystal structure
The accuracy of the model was established by the crystal structure solved 11 years later.25 Although the LpAFP solenoid proved to be a left‐handed helix, there was little difference in the energetics of coiling in either direction [Fig. 4(A)]. In all other details the model was spot on [Fig. 6(C)]. It predicted the 14–15 residue coils, internal Asn ladders, which occasional substitutions by His, apposed Val hydrophobic core and a flat a‐side β‐sheet [Fig. 6(D)]. Access to the LpAFP model allowed us to identify the ice‐binding site long before the structure was solved and accelerate analysis of what constitutes an IBS.
Modeling the Midge Antifreeze Protein
A new antifreeze protein has recently been discovered in the midge.34 The AFP is 79 residues in length and has a 10‐residue repeating motif (xxCxGxYCxG), which in turn contains two 5‐residue motifs (xxCxG) [Fig. 7(A)]. This sequence pattern was reminiscent of the T. molitor antifreeze protein (TmAFP) sequence, which has a 12‐residue repeating motif each of which contains the common TXT motif and Cys residues at every 6 positions.30 The 12‐residue repeat of TmAFP forms one coil of its helix.2 The structure is stabilized by disulfide bonds that bridge together opposing sides of the helix and internal water molecules that form hydrogen bonds with the main chain backbone.
Figure 7.
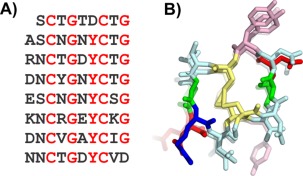
Modeling the midge antifreeze protein. A) Sequence and B) modeled structure of midge antifreeze protein. Sequence and structure are colored the same as in Figure 5. Additionally, Cys are yellow in B.
Modeling of the midge AFP using TmAFP as a template
The midge AFP model was built with the TmAFP structure as a template by aligning the Cys residues of the midge AFP's 10‐residue repeat with the Cys residues of the TmAFP 12‐residue repeat. Two residues per coil of the TmAFP were deleted (one on either side of the internal disulfide bridge) and the remaining residues were changed to the midge AFP sequence. The final model was a helix with eight 10‐residue coils.34 The helix was built in both right‐ and left‐handed orientations, but during molecular dynamics, only the left‐handed helix was stable [Fig. 4(B)].
Testing the midge AFP model
The midge AFP is the tightest disulfide‐bridged solenoid that we are aware of. Secondary structure analysis of the resulting model structure does not find any β‐content in the helix, likely due to the tight winding of the coil and so it cannot be referred to as a β‐helix. The tight solenoid has several stabilizing features besides the disulfide core. Down one side of the helix there is a row of 7 external pi‐stacked tyrosine residues, and several acid and base residues in adjacent loops have the potential to form salt bridges. Circular dichroism (CD) analysis has provided experimental evidence in favor of the proposed model.34 Additionally, other midge AFP isoforms have been discovered by reverse transcription PCR that are shorter by exactly 10‐residue sections providing further evidence that the one‐loop unit of the helix is 10 residues long (Basu et al., in preparation).
The pi‐stacked tyrosine side chains make a flat, relatively hydrophobic surface that fits all the requirements for an ice‐binding surface. It is the predicted ice‐binding surface, and if this prediction turns out to be true, it will be the first tyrosine‐rich ice‐binding surface to be discovered. The model has provided a tool for intelligent design to confirm the proposed ice‐binding surface by mutagenesis of residues on and off the ice‐binding surface. The model can also be used in molecular dynamics to study the potential for water molecule ordering by the predicted ice‐binding surface.
Modeling the Snow Flea Antifreeze Protein
Compared to the AFPs found in other organisms, the AFP that was discovered in the snow flea, Hypogastrura harveyi Folsom exhibits a unique amino acid composition and sequence.35 The 6.5‐kDa isoform, whose structure was ultimately determined by crystallography,36 contains more than 45% glycine residues. The sequence contained a Gly‐X‐Y repeat in which X was also often Gly and it contained just four Cys that were distributed throughout the sequence. While the 3‐residue repeat was reminiscent of the 3‐residue repeat in collagen, the snow flea AFP only contained 5 proline residues, which periodically interrupted the Gly‐X‐Y repeats. Examination of the sequence showed that there was an underlying pattern that divided the sequence up into six segments of about 12 to 15 residues [Fig. 8(A)]. Those six segments could be described as having patterns Goi, Goo, GGi, GGo, GGi and GAP, respectively, where “o” represents a small residue and “i” represents a large, hydrophilic residue.
Figure 8.
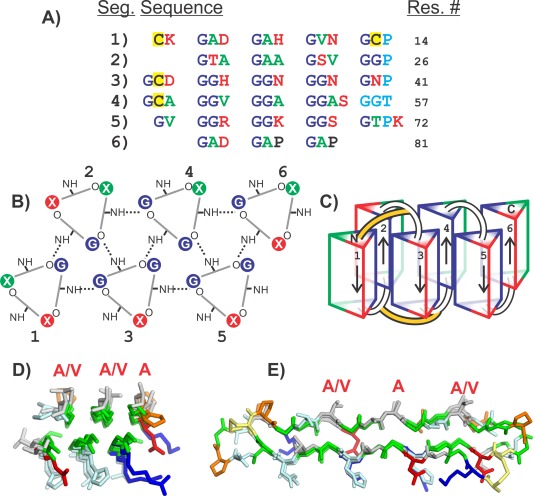
Modeling the snow flea antifreeze protein (sfAFP). A) SfAFP sequence separated into six segments. Green letters are residues with small non‐polar side chains (Ala, Val). Red letters are charged or hydrophilic residues (Arg, Lys, Asp, Asn, Ser, Thr, His). Dark blue letters are Gly. Cyan letters are the residues that disturb the PPII helix. Yellow backgrounds are Cys. B) Top view of PPII helices showing hydrogen bonding pattern between coils. Segment numbers are labeled 1–6. Hydrogen bonds are shown as dotted lines. Residue positions are colored the same as in A. C) Side view of PPII bundles showing direction of each segment and disulfide bonding positions (yellow connections). D) Top view and E) side view of crystal structure of SfAFP. Structures in D and E are colored the same as in Figure 5. Additionally Cys are yellow and Pro are orange. Residues labeled with red letters are on the ice‐binding site.
Modeling of the SfAFP as a bundle of polyproline‐II helices
Given that the 3‐residue repeat was suggestive of the polyproline‐II helix, we expected that the structure must be held together by backbone‐backbone hydrogen bonds similar to that seen in the collagen triple helix. The interspersed prolines (and a break in the 3‐residue repeat between segments 4 and 5) suggested that the structure might be composed of six segments of polyproline type II helix. Loops at the breaks in the repeat pattern would allow those helices to fold back on one another to form a globular structure.
The resulting model was one in which the segments 1, 3, and 5 formed one face of the molecule and segments 2, 4, and 6 formed the other and were antiparallel to the helices of the first face37 [Fig. 8(B)]. Those segments with the Gly‐Gly‐Y repeat were less solvent exposed and were hydrogen‐bonded to three or four neighboring helices, while those with only one Gly within the Gly‐X‐Y repeat were more solvent exposed and hydrogen‐bonded to two or three neighbors. The model also resulted in one face of the molecule being formed of mostly small hydrophobic side chains (mostly Ala and Val) while the other face contained large hydrophilic side chains). In addition, the four cysteine residues were positioned to form disulfide bonds between the N‐terminal ends of segments 1 and 3 and between the C‐termini of segments 1 and 3 [Fig. 8(C)].
Subsequent to our modeling of the snowflea AFP, Pentelute et al.36 determined the crystal structure of the protein. They were able to crystallize a racemic mixture of a chemically synthesized version of the protein [Fig. 8(D,E)]. The RMSD for fitting our model to the crystal structure is 1.85 Å for 69 of the 81 α‐carbons.
Modeling the P. borealis Ice Nucleation Protein
The bacterial ice nucleation protein (INP) from P. borealis is distinct from the proteins described above because, as its name suggests, it promotes the formation of ice crystals rather than inhibiting their growth. Like many AFPs, the INP contains a highly repetitive sequence with a series of tandem repeats of 16 amino acids in length [Figs. 2(A) and 9(C)]. What distinguishes it from AFPs is its size. While AFPs are generally small (molecular weights of 3 kDa to 35 kDa), the PbINP has a molecular weight of 123 kDa. The sequence can be divided up into three regions: an N‐terminal region of about 163 residues, a C‐terminal region of 41 residues and a central region containing 64 of the 16‐residue repeats.38
Figure 9.
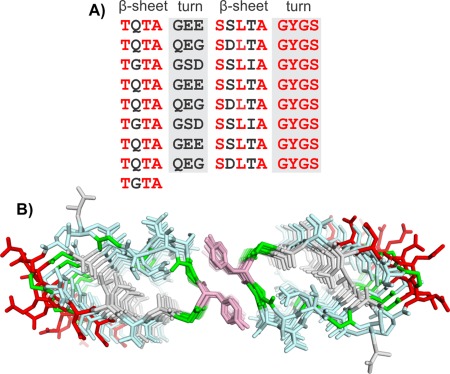
Modeling P. borealis ice‐nucleating protein (PbINP). A) Sequence and B) modeled structure of PbINP. Sequence and structure are colored the same as in Figure 5.
Modeling the centre region of the PbINP
Examination of the central repeats showed that each could be broken down into 4‐residue segments.39 Two of those segments contained at least one Thr residue (consensus sequences TQTA and SLTA) and were predicted to form short β‐strands with the sidechains of the Thr and Ser residues pointing outward and those of the Gln, Leu, and Ala residues pointing inward [Fig. 9(B)]. The other two segments are rich in Gly and Ser and are expected to form turns that connect the two β‐strands.
The similarity of this architecture to the RTX proteins again led us to use the alkaline protease (PDB 1KAP)32 as a template structure for building PbINP [Fig. 9(A,B)]. The Ca2+‐binding loops were replaced by the 4‐residue Gly‐ and Ser‐rich sequences, which were manually built to connect the two β‐strands. The resulting β‐solenoid structure contained a hydrophobic core made up of the two Ala residues and one Leu residue per repeat but also contained inward pointing Ser and Gln residues that are able to form a hydrogen‐bonded ladder. The Ser side chains form hydrogen bonds to neighboring backbone amides, while the Gln side chains form hydrogen bonds to each other. In the sequence repeat pattern, every third Gln is typically replaced by a Gly resulting in a 48 amino acid repeating motif [Fig. 2(B)]. This appears to provide some space in the interior that may be occupied by an internal water, but may also help the β‐solenoid to maintain a flat surface.
Modeling suggests PbINP multimerizes through the Tyr ladder
Analysis of the ice‐nucleation ability of several bacterial species has suggested that ice nucleation at elevated temperatures (−2°C) would require a nucleation site much larger than a single protein molecule.40 Thus it is expected that INPs function as multimers. The row of stacked Tyr side chains of PbINP was suggestive of other structures in which Tyr ladders promote dimerization.41 Two molecules of the β‐solenoid model of PbINP were, therefore, placed in a parallel arrangement with the two Tyr ladders adjacent to each other. A molecular dynamics simulation showed that this resulted in a close interaction of the two monomers with the Tyr side chains of each monomer forming a hydrogen‐bonded network with the Ser side chains of the opposing monomer and with the exclusion of water molecules. The resulting dimer was stable over 10‐ns simulations even at temperatures as high as 310 K,39 while simulations of the monomer resulted in unraveling of the C terminus of the β‐solenoid structure.
Conclusions
We are not yet at a point where the structures of proteins can be reliably modeled ab initio using only the knowledge of their sequences. The use of additional information, such as the discovery of repetitive sequences and predicted secondary structure, can allow the successful model building. These models can then serve as a guide for further biochemical experiments to test their validity. In the event that a predicted model is sufficiently close to the true structure and X‐ray diffraction data of native crystals are available, that model could potentially allow the determination of reasonable phases by the molecular replacement method.
In 2009, the application of a search algorithm specifically developed to locate proteins with tandemly‐repeating sequences in the Swiss‐Prot database found that 9.5% of total proteins were repetitive.13 Assuming the proportion of tandem repeating proteins in the Swiss‐Prot database has not changed, their number today would be >50,000. This is equivalent to half the number of protein structures in the Protein Databank. Thus, applications of the methods outlined in this review could substantially add to the wealth of structural information in the database.
Acknowledgment
The authors thank Dr. Laurie Graham for valuable discussions on dot matrix analyses.
References
- 1. Davies PL (2014) Ice‐binding proteins: a remarkable diversity of structures for stopping and starting ice growth. Trends Biochem Sci 39:548–555. [DOI] [PubMed] [Google Scholar]
- 2. Liou YC, Tocilj A, Davies PL, Jia ZC (2000) Mimicry of ice structure by surface hydroxyls and water of a beta‐helix antifreeze protein. Nature 406:322–324. [DOI] [PubMed] [Google Scholar]
- 3. Leinala EK, Davies PL, Jia ZC (2002) Crystal structure of beta‐helical antifreeze protein points to a general ice binding model. Structure 10:619–627. [DOI] [PubMed] [Google Scholar]
- 4. Kajava AV, Steven AC (2006) Beta‐rolls, beta‐helices, and other beta‐solenoid proteins. Adv Protein Chem 73:55–96. [DOI] [PubMed] [Google Scholar]
- 5. Graham LA, Hobbs RS, Fletcher GL, Davies PL (2013) Helical antifreeze proteins have independently evolved in fishes on four occasions. PLoS One 8:e81285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baardsnes J, Kondejewski LH, Hodges RS, Chao H, Kay C, Davies PL (1999) New ice‐binding face for type I antifreeze protein. FEBS Lett 463:87–91. [DOI] [PubMed] [Google Scholar]
- 7. Kristiansen E, Ramlov H, Hojrup P, Pedersen SA, Hagen L, Zachariassen KE (2011) Structural characteristics of a novel antifreeze protein from the longhorn beetle Rhagium inquisitor. Insect Biochem Mol Biol 41:109–117. [DOI] [PubMed] [Google Scholar]
- 8. Lin FH, Davies PL, Graham LA (2011) The thr‐ and ala‐rich hyperactive antifreeze protein from inchworm folds as a flat silk‐like beta‐helix. Biochemistry 50:4467–4478. [DOI] [PubMed] [Google Scholar]
- 9. Hakim A, Nguyen JB, Basu K, Zhu DF, Thakral D, Davies PL, Isaacs FJ, Modis Y, Meng W (2013) Crystal structure of an insect antifreeze protein and its implications for ice binding. J Biol Chem 288:12295–12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234:779–815. [DOI] [PubMed] [Google Scholar]
- 11. Kelley LA, Sternberg MJ (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4:363–371. [DOI] [PubMed] [Google Scholar]
- 12. Chen L, DeVries AL, Cheng CH (1997) Evolution of antifreeze glycoprotein gene from a trypsinogen gene in Antarctic notothenioid fish. Proc Natl Acad Sci USA 94:3811–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jorda J, Kajava AV (2009) T‐REKS: identification of Tandem REpeats in sequences with a K‐meanS based algorithm. Bioinformatics 25:2632–2638. [DOI] [PubMed] [Google Scholar]
- 14. Marsella L, Sirocco F, Trovato A, Seno F, Tosatto SC (2009) REPETITA: detection and discrimination of the periodicity of protein solenoid repeats by discrete Fourier transform. Bioinformatics 25:i289–i295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Newman AM, Cooper JB (2007) XSTREAM: a practical algorithm for identification and architecture modeling of tandem repeats in protein sequences. BMC Bioinformatics 8:382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maizel JV Jr, Lenk RP (1981) Enhanced graphic matrix analysis of nucleic acid and protein sequences. Proc Natl Acad Sci USA 78:7665–7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bryan AW, Starner‐Kreinbrink JL, Hosur R, Clark PL, Berger B (2011) Structure‐based prediction reveals capping motifs that inhibit β‐helix aggregation. Proc Natl Acad Sci USA 108:11099–11104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chao H, Hodges RS, Kay CM, Gauthier SY, Davies PL (1996) A natural variant of type I antifreeze protein with four ice‐binding repeats is a particularly potent antifreeze. Protein Sci 5:1150–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leinala EK, Davies PL, Doucet D, Tyshenko MG, Walker VK, Jia ZC (2002) A beta‐helical antifreeze protein isoform with increased activity ‐ Structural and functional insights. J Biol Chem 277:33349–33352. [DOI] [PubMed] [Google Scholar]
- 20. Richardson JS, Richardson DC (2002) Natural beta‐sheet proteins use negative design to avoid edge‐to‐edge aggregation. Proc Natl Acad Sci USA 99:2754–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Richardson JS, Richardson DC (1988) Amino‐acid preferences for specific locations at the ends of alpha‐helices. Science 240:1648–1652. [DOI] [PubMed] [Google Scholar]
- 22. DeLano WL (2003) The PyMOL Molecular Graphics System. Available at: http://www.pymol.org. Accessed February 2016.
- 23. Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38, 27–28. [DOI] [PubMed] [Google Scholar]
- 24. Kuiper MJ, Davies PL, Walker VK (2001) A theoretical model of a plant antifreeze protein from Lolium perenne. Biophys J 81:3560–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Middleton AJ, Marshall CB, Faucher F, Bar‐Dolev M, Braslavsky I, Campbell RL, Walker VK, Davies PL (2012) Antifreeze protein from freeze‐tolerant grass has a beta‐roll fold with an irregularly structured ice‐binding site. J Mol Biol 416:713–724. [DOI] [PubMed] [Google Scholar]
- 26. Garnham CP, Campbell RL, Davies PL (2011) Anchored clathrate waters bind antifreeze proteins to ice. Proc Natl Acad Sci USA 108:7363–7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garnham CP, Gilbert JA, Hartman CP, Campbell RL, Laybourn‐Parry J, Davies PL (2008) A Ca2+‐dependent bacterial antifreeze protein domain has a novel beta‐helical ice‐binding fold. Biochem J 411:171–180. [DOI] [PubMed] [Google Scholar]
- 28. Gilbert JA, Davies PL, Laybourn‐Parry J (2005) A hyperactive, Ca2+‐dependent antifreeze protein in an Antarctic bacterium. FEMS Microbiol Lett 245:67–72. [DOI] [PubMed] [Google Scholar]
- 29. Graether SP, Kuiper MJ, Gagne SM, Walker VK, Jia ZC, Sykes BD, Davies PL (2000) Beta‐helix structure and ice‐binding properties of a hyperactive antifreeze protein from an insect. Nature 406:325–328. [DOI] [PubMed] [Google Scholar]
- 30. Liou YC, Thibault P, Walker VK, Davies PL, Graham LA (1999) A complex family of highly heterogeneous and internally repetitive hyperactive antifreeze proteins from the beetle Tenebrio molitor. Biochemistry 38:11415–11424. [DOI] [PubMed] [Google Scholar]
- 31. Sidebottom C, Buckley S, Pudney P, Twigg S, Jarman C, Holt C, Telford J, McArthur A, Worrall D, Hubbard R, Lillford P (2000) Phytochemistry ‐ heat‐stable antifreeze protein from grass. Nature 406:256‐256. [DOI] [PubMed] [Google Scholar]
- 32. Baumann U, Wu S, Flaherty KM, McKay DB (1993) Three‐dimensional structure of the alkaline protease of Pseudomonas aeruginosa: a two‐domain protein with a calcium binding parallel beta roll motif. EMBO J 12:3357–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Middleton AJ, Brown AM, Davies PL, Walker VK (2009) Identification of the ice‐binding face of a plant antifreeze protein. FEBS Lett 583:815–819. [DOI] [PubMed] [Google Scholar]
- 34. Basu K, Graham LA, Campbell RL, Davies PL (2015) Flies expand the repertoire of protein structures that bind ice. Proc Natl Acad Sci USA 112:737–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Graham LA, Davies PL (2005) Glycine‐rich antifreeze proteins from snow fleas. Science 310:461‐461. [DOI] [PubMed] [Google Scholar]
- 36. Pentelute BL, Gates ZP, Tereshko V, Dashnau JL, Vanderkooi JM, Kossiakoff AA, Kent SB (2008) X‐ray structure of snow flea antifreeze protein determined by racemic crystallization of synthetic protein enantiomers. J Am Chem Soc 130:9695–9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lin FH, Graham LA, Campbell RL, Davies PL (2007) Structural modeling of snow flea antifreeze protein. Biophys J 92:1717–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu ZQ, Qin L, Walker VK (2009) Characterization and recombinant expression of a divergent ice nucleation protein from 'Pseudomonas borealis'. Microbiology 155:1164–1169. [DOI] [PubMed] [Google Scholar]
- 39. Garnham CP, Campbell RL, Walker VK, Davies PL (2011) Novel dimeric beta‐helical model of an ice nucleation protein with bridged active sites. BMC Struct Biol 11:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Govindarajan AG, Lindow SE (1988) Size of bacterial ice‐nucleation sites measured in situ by radiation inactivation analysis. Proc Natl Acad Sci USA 85:1334–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Biancalana M, Makabe K, Koide S (2010) Minimalist design of water‐soluble cross‐beta architecture. Proc Natl Acad Sci USA 107:3469–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]