

Abstract
Organic molecules with heavy main-group elements frequently form supramolecular links to electron-rich centres. One particular case of such interactions is halogen bonding. Most studies of this phenomenon have been concerned with either dimers or infinitely extended structures (polymers and lattices) but well-defined cyclic structures remain elusive. Here we present oligomeric aggregates of heterocycles that are linked by chalcogen-centered interactions and behave as genuine macrocyclic species. The molecules of 3-methyl-5-phenyl-1,2-tellurazole 2-oxide assemble a variety of supramolecular aggregates that includes cyclic tetramers and hexamers, as well as a helical polymer. In all these aggregates, the building blocks are connected by Te…O–N bridges. Nuclear magnetic resonance spectroscopic experiments demonstrate that the two types of annular aggregates are persistent in solution. These self-assembled structures form coordination complexes with transition-metal ions, act as fullerene receptors and host small molecules in a crystal.
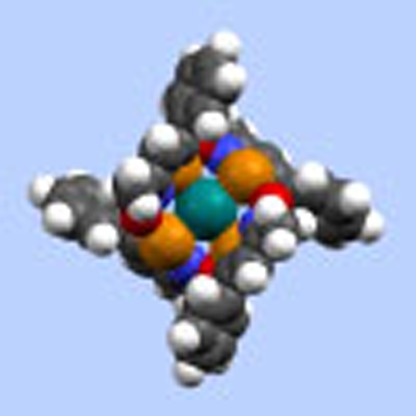 Similarly to halogen bonding, the heavier chalcogens are capable of forming supramolecular links with electron rich sites. Here, the authors show that these forces can allow the formation of well-defined cyclic structures that are stable in solution and are capable of forming host-guest complexes.
Similarly to halogen bonding, the heavier chalcogens are capable of forming supramolecular links with electron rich sites. Here, the authors show that these forces can allow the formation of well-defined cyclic structures that are stable in solution and are capable of forming host-guest complexes.
One of the most remarkable developments in supramolecular chemistry in the last two decades is the evolution of halogen bonding1,2 from being an intriguing structural feature to becoming a powerful tool in crystal engineering3,4,5,6, which is also applicable to systems and processes as diverse as luminescent7 and non-linear optical8 materials, photo-patterning of surfaces9, the assembly of fractal patterns from molecular building blocks10, supramolecular gelation11, organocatalysis12, macromolecular alignment at macroscopic scale13, anion recognition14, transmembrane anion transport15,16 and mimicking the activity of the deiodinase enzyme17.
Our current understanding attributes such interactions to the depletion of electron density in a region of space surrounding the nucleus of a halogen atom in a molecule (termed a sigma hole)11, which consequently is attracted to lone-pairs and π clouds of electrons. This electrostatic factor is supplemented by polarization of the electron density—which in a strong case would be modelled by the mixing of electron donor and acceptor orbitals—and electron correlation manifested in the London dispersion force. Those stabilizing contributions are countered by the Pauli repulsion and the balance results in interaction energies of about 5–70 kJ mol−1 and preference for a linear R-X…:B geometry (X is a halogen atom and B a Lewis base)18,19,20. The conditions that give rise to each of those stabilizing factors are common to molecules that contain heavy elements from other p-block families18,21,22,23,24,25. While it is long-recognized26,27,28 that intra- and intermolecular short interatomic contacts are pervasive in structural main-group chemistry, terms such chalcogen and pnictogen bonding have been recently suggested by analogy to the halogen case. There are indeed common traits to all these interactions; for instance, the trends in ratio of interatomic distance to sum of van der Waals radii denote a correlation of interaction strength with the mass of the p-block element and enhancement by electronegative substituents29,30,31. However, one important difference is that atoms of group-16 (refs 32, 33) and 15 (refs 34, 35) elements can engage in up to two and three concurrent supramolecular interactions, respectively36,37.
The potential of chalcogen-centered interactions in supramolecular chemistry is illustrated by two well-studied molecular families: the dichalcogena alkynes, which consistently crystalize in tubular structures assembled by chalcogen–chalcogen interactions38,39,40, and the 1,2,5-chalcogenadiazoles, in which two pairs of antiparallel chalcogen–nitrogen interactions per molecule tend to build infinite ribbons. The latter structures are of interest because of their charge transport properties41 and, through moderate steric repulsion, can be distorted to induce non-linear optical properties or chromotropism42,43,44. The auto-association of chalcogenadiazoles is amenable to combination with metal-ion coordination and hydrogen bonding45,46. In spite of their charge, N-alkylated chalcogenadiazolium cations do associate in the solid state and, according to electrochemical data, the tellurium derivatives may also be associated in solution47.
Large assemblies (gels, supramolecular polymers and crystals) provide tangible demonstration of the power of these supramolecular interactions but comparatively less has been investigated at the other end of the size spectrum: small, discrete aggregates of a few molecules held together by supramolecular bonds. In the halogen-bonding case, I…N interactions have been employed in the construction of molecular capsules48 from a pair of complementary molecules; the assembly of such structures in solution was recently demonstrated49,50.
In the chalcogen case, the crystal of 3-methyl-5-(1,1-dimethylethyl)-1,2-tellurazole 2-oxide (1a)51 features a tetramer spontaneously assembled by short Te…O interactions, its arrangement is specially intriguing because it evokes a macrocycle. However, until now it has been unclear whether such an aggregate would be stable enough to function in the same way as do molecules from the vast category of supramolecular building blocks that encompasses crown ethers, polyazacycloalkanes, tetrapyrroles, phthalocyanines, calixarenes, cyclodextrins, cucurbiturils and cyclophanes. If so, building macrocycles by the spontaneous addition of molecular building blocks would be a uniquely convenient approach because, in general, the synthesis of macrocycles is laborious and low-yielding—even when template methods are used—with notable exceptions such as the recently synthesized macrocyclic cyanostar52. We have investigated in detail the stability of the cyclic aggregates of iso-tellurazole N-oxides in solution and probed their chemical behaviour. Here we report that indeed these assemblies are persistent in solution and display properties of actual macrocyclic molecules in their ability to coordinate transition-metal ions, form adducts with fullerenes and host small species in the solid state.
Results
Preparation and structural studies
Improvements from literature procedures51,53 afford the iso-tellurazole N-oxides 1a and 1b in good yields; these products are remarkably stable in air and tolerant of moisture. Depending on the solvent used for crystallization, 3-methyl-5-phenyl-iso-tellurazole N-oxide (1b) crystallizes in a remarkable variety of polymorphs, all the structures in Fig. 1 were identified by single-crystal X-ray diffraction. In each case, the morphology of the whole sample indicates that only one phase is reproducibly obtained. The unit cells of several polymorphs include solvent molecules and, while the solvent likely influences packing efficiency, the molecules of 1b are all associated to each other only. The aggregates observed in the crystals (Fig. 2a) include an infinite spiral chain (1b∞) and cyclic tetra- (1b4) and hexamers (1b6). In every case, consistently with the σ-hole/donor–acceptor model, the oxygen atom of one molecule is bound to the tellurium atom of another, always trans to the nitrogen atom and to distances that span 2.171(3) to 2.242(1) Å. These are comparable to the 2.299(2) Å measured in 1a4 (ref. 51) and are slightly longer than the 2.122(1) Å of the axial bonds in β-TeO2 (ref. 54). The Te–N distances (2.197(2) to 2.258(2) Å) are slightly longer than those measured for other single bonds between these elements. In each crystal, the geometry around the chalcogen approximates a T-shape, with N–Te–O†, N–Te–C and O–N–Te average angles of 165.3°, 76.0° and 126.9°, respectively. Individual iso-tellurazole heterocyles are planar, with bond distances consistent with localized single and double bonds. Adjacent iso-tellurazole rings tend to lay perpendicular to each other (Fig. 2b) with inter-planar angles ranging from 60° to 83°.
Figure 1. Summary of supramolecular species derived from 1b.

Supramolecular species formed by auto-association of 1b, alone or in combination with a Pd(II) salt or C60.
Figure 2. Crystallographically characterized aggregates of 1b.

(a) ORTEPs of the aggregates observed in the crystal structures of 3-methyl-5-phenyl-isotellurazole-N-oxide, 1b. While 1b∞ is contained in 3(1b)·(C6H6), the macrocyclic tetramer crystallizes in a solvent-free polymorph and the hexamer forms the co-crystal 12(1b)·(CH2Cl2). (b) Detail of the structure of 1b∞ displaying the relative orientations of the iso-tellurazole planes and the labelling sequence used in the discussion. Displacement ellipsoids are plotted at 75% probability in all cases. For clarity, hydrogen atoms are omitted, the phenyl and methyl groups are portrayed using a wireframe representation and are partially hidden in b.
Crystallization from benzene yields a phase of composition 3(1b)·(C6H6), which is built by infinite spiral chains (1b∞) coiling in alternating directions along b with a periodicity of 3. However, the P212121 space group contains no ternary screw axis, thus the unit cell contains three crystallographically distinct molecules. The macrocyclic tetramer 1b4 is formed in non-solvated crystals obtained from CHCl3 or by layering acetonitrile over a CH2Cl2 solution. Its structure resembles that of 1a4 but there are important differences: the structure of the phenyl derivative approaches a chair conformation, has Ci symmetry and is built by two crystallographically independent molecules while the geometry of the t-Bu aggregate corresponds to a boat conformer, belongs to the S4 point group and the four constituting 1a molecules are all related by symmetry51. There are two distinct trans-annular Te–Te distances in 1b4, 5.5895(2) and 5.3043(2) Å. Crystallization from THF and a hexanes/CH2Cl2 mixture produces crystals of compositions 3(1b)·(C4H8O) and 12(1b)·(CH2Cl2), respectively. Both phases contain macrocyclic hexamers. In the latter case, the crystallization solvent occupies voids external to two crystallographically distinct macrocycles, each built from three molecular units that are unique by symmetry. Packing distorts the macrocyle, thus there are three different trans-annular Te–Te distances for each ring: 7.117(1), 7.300(1) and 7.544(1) Å in one case and 7.151(1), 7.227(1) and 7.692(1) Å in the other. As the macrocycles in this crystal stack along b (Fig. 3a) a methyl group of one hexamer extends towards the cavity of the neighbouring macrocycle (Fig. 3b). In contrast, the crystal that contains THF packs in a hexagonal lattice (Fig. 3c); the macrocyclic aggregate is built by six equivalent molecular units of 1b and the trans-annular Te–Te distances are all 7.638(2) Å. The macrocycles pack in a layer and a second cavity is flanked by the phenyl groups. Vertical stacking of the layers in an ABA sequence alternates the two types of cavity-forming tubular channels (Fig. 3d). THF molecules disordered in three different orientations sit in the macrocycle cavities, above and below the plane defined by the chalcogens. Although these channels are small, the crystals slowly loose solvent and become opaque.
Figure 3. Detail of the crystalline structures that feature the hexamer 1b6.

From the crystal of composition 12(1b)·(CH2Cl2): (a) stacking of the macrocycles, (b) interaction of the methyl group with the cavity of a neighbouring ring. From the crystal of composition 3(1b)·(C4H8O): (c) packing of a layer in the (0,0,1) plane, (d) detail of the crystal structure, highlighting the channels, as well as the location and three orientations of the THF molecules. Panels c and d are simplified for clarity; after ruling out twinning, larger unit cells and less symmetric space groups, the best approximate model features three distinct orientations for the phenyl groups but only one orientation for the C3NTe ring. The THF molecules were treated as rigid groups.
Evidence of persistent auto-association in solution
In spite of the large molecular mass, the electrospray mass spectrum (Supplementary Fig. 1) displays isotopic patterns characteristic of aggregates [1bn-H]+ (n=1–7). Given the apparent strength of the Te…O supramolecular interactions, it became of interest to probe the aggregation of iso-tellurazole oxides in solution with nuclear magnetic resonance (NMR) spectroscopy. At room temperature, the 125Te NMR spectrum of 1b, measured in CH2Cl2, displays only a single line. However, a second line appears on cooling and grows in intensity at the expense of the first (Fig. 4a). These changes are fully reverted when room temperature is restored and are paralleled by those in the 1H-NMR spectrum, albeit with some differences. For example, the lines of the methyl resonances coalesce at 230 K in the 500 MHz spectrum. Furthermore, the relative intensities of the methyl lines are dependent on concentration (Fig. 4b). At low temperature, negative nuclear Overhauser effect (NOE) is observed between the 1H nuclei of the methyl and phenyl or t-butyl groups of 1b (Fig. 4c) and 1a (Supplementary Fig. 2), respectively. The separation between the 1H nuclei of these pendant groups in the individual molecules of 1 is too large for NOE (>5 Å). However, the crystal structures show that the Te…O interactions bring the substituents of neighbouring molecules to shorter distances (<3 Å).
Figure 4. NMR Investigations of the auto-association of 1 in solution.

NMR (500 MHz) spectra of 1b solutions in CDCl3: (a) 125Te and 1H at 74 mmol l−1 and variable temperature; (b) 1H at 190 K as a function of concentration; *denotes the resonance of a trace amount of H2O. (c) 1H-NMR and 1D-NOESY spectra of 1b in CD2Cl2 at 190 K; the over-imposed structure portrays the van der Waals surfaces of the methyl and phenyl groups of each molecule in a pair modelled with coordinates extracted from the crystal structures of 1b4; ‡denotes the resonance of residual CHDCl2. 1H-NMR (600MHz) from the equimolar mixture of 1a and 1b: (d) methyl resonances above room temperature, (e) methyl and t-butyl resonances at low temperature. (f) evolution of selected methyl 1H-NMR lines as a function of composition (molar fraction of 1a) of the mixtures, dashed lines correspond to the calculated abundance of each type of aggregate.
NMR scrambling experiments
Mixtures of 1a and 1b were investigated by 1H-NMR to further probe the structure of their aggregates in solution. At room temperature, the spectrum of the 1:1 mixture displays a very broad band (1.9–1.6 p.p.m.) for the methyl resonances, this feature is more clearly discernible at 323 K (Fig. 4d), these observations are indicative of intermolecular association and exchange. The spectrum with the best resolution of resonances from this mixture was acquired at 190 K using a 600 MHz instrument; in these conditions 14 lines are observed for the methyl and 6 for the t-butyl resonances (Fig. 4e). The methyl lines in the 1H-NMR spectra of mixtures of variable composition but constant total concentration were classified by their behaviour as a function of molar fraction (Fig. 4f). It was possible in this way to identify the patterns characteristic of cyclic tetramers: resonances that continuously increase or decrease in intensity following a fourth-degree polynomial trajectory for the homogeneous aggregates; lines with one maximum each at molar fractions 0.25 or 0.75 that correspond to the mixed tetramers of 1:3 and 3:1 composition; as well as lines with a maximum at 0.5 that correspond to the 2:2 stoichiometry. Lines with a more complex behaviour would result from the superposition of more than one resonance.
As this interpretation attributes the resonance at 1.70 p.p.m. to the methyl protons of the tetramer 1b4, it became possible to explain the effect of concentration on the 1H-NMR spectrum of 1b (Fig. 4b) as the result of further aggregation (equation (1)). The stoichiometric coefficient (n) and equilibrium constant (K, equation (2)) of the process were determined from the intensities (I) of the lines (relative to the resonance of the residual proton on the solvent) as a function of concentration (equation (3), Supplementary Fig. 3a). The fitted stoichiometric coefficient n=1.53±0.02 implies that the resonance at 1.56 p.p.m. belongs to the methyl protons of the hexamer. At 190 K, the equilibrium constant is therefore K=0.28±0.01 dm1.5 mol−0.5; the corresponding van't Hoff analysis (Supplementary Fig. 3b) yielded ΔH=−16±1 kJ mol−1 and ΔS=−88±6 J mol−1K−1. However, diffusion-ordered spectroscopy (DOSY) experiments were unable to provide the hydrodynamic radii and mass of the aggregates because the magnetization decay during diffusion did not follow simple Gaussian profiles, which likely is a consequence of the equilibrium between tetramer and hexamer.
 |
 |
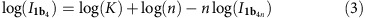 |
Computational modelling
The relative stabilities of the oligomeric structures formed by aggregation of iso-tellurazole N-oxides in solution was assessed with dispersion- and gradient-corrected relativistic density-functional-theory (DFT) gas-phase calculations. For computational expediency, the models were based on the molecular building block with R=Me, 1c. Calculations for the individual molecule were also used to build a map of electrostatic potential (Fig. 5a), which demonstrate the occurrence of two σ holes on the Te atom opposite to the C and N atoms, the latter hole being the most prominent. The LUMO of 1c has a predominant contribution from the σ*Te–N orbital (Fig. 5b). The plot of the electron localization function in the molecular plane (Fig. 5c) features two dips in the space surrounding the chalcogen. These data confirm that the molecules of 1 are predisposed to associate through contacts between the electrophilic (O) and nucleophilic (Te) regions and emphasize that the most favourable position for attachment of a Lewis base to is opposite to the nitrogen atom.
Figure 5. The aggregation of 1 originates in its electronic properties.

(a) Two views of the map of electrostatic potential for a molecule of 1c; (b) LUMO of 1c; (c) contour plot of the electron localization function in the iso-tellurazole plane.
There is only one published53 crystallographic determination of an iso-selenazole N-oxide (3-methyl-phenyl-, 2b), which features a centro-symmetric dimer formed by a pair of antiparallel Se…O interactions. The analogous structure for 1c2, could only be optimized by imposing symmetry constraints. In their absence, the geometry converges to a dimer bridged by only one Te…O interaction (d=2.40 Å) in which the two heterocycles define an inter-planar angle of 87.6°, which is consistent with all the observed structures of the aggregates of 1a and 1b. The structures of the cyclic 1c4 tetramers (chair and boat conformations) and the hexamer 1c6 were also optimized, in each case vibrational calculations return all real frequencies confirming that all are minima in the potential-energy surface. Structures of hypothetical trimer and pentamer cyclic aggregates could not be satisfactorily optimized, the preferred nearly perpendicular orientation of the iso-tellurazole rings imposes a preference for an even number of molecules in a cyclic aggregate. Periodic calculations used to optimize the infinite chain 1c∞, based on the structure observed in the crystal of 3(1b)·(C6H6). The results of these calculations are summarized in Table 1 as thermodynamic parameters for aggregation equilibria.
Table 1. Calculated (PBE-D3) thermodynamic parameters of aggregation in gas phase.
| Equilibrium | ΔH per Te…O interaction (kJ mol−1) | ΔS per Te…O interaction (J mol−1 K−1) |
|---|---|---|
| 2 1c⇌1c2 | −68.7 | −185 |
| 4 1c⇌1c4 (chair) | −68.6 | −158 |
| 4 1c⇌1c4 (boat) | −69.3 | −140 |
| 6 1c⇌1c6 | −75.7 | −171 |
| ∞ 1c⇌1c∞ | −82.1 | −291 |
Coordination of a transition-metal ion
Mixing [Pd(NC-CH3)4](BF4)2 with 1b dissolved in a CH2Cl2/acetonitrile mixture yields a dark brown mixture, its visible absorption spectrum features a well-defined shoulder at 500 nm. Job's continuous variations method showed that this spectrum is due to a complex of 1:4 stoichiometry (Fig. 1), the composition of which was confirmed by a structural determination from crystals grown by slow diffusion of an acetonitrile solution of the metal salt into a CH2Cl2 solution of 1b. Figure 6a,b displays two views of the structure of the coordination complex in the crystal of [Pd(1b4)](BF4)2·2(CH2Cl2)2. The crystal structure features the tetrameric aggregate of 1b in the boat conformation while the metal centre displays a square planar coordination geometry with Pd–Te distances (2.5804(4)Å) that are comparable to those measured in complexes of anionic tellurium ligands55; the Te–Pd–Te trans bond angles of 172.38(2)° denote a slight pseudo D2d distortion. This crystal structure features metal depletion due to partial occupation of the coordination sites. CH2Cl2 molecules replaced the tetrafluoroborate anions in proportion to the missing metal ions. After refinement, the final ratios of occupancies from two crystals grown in separate batches, 0.863(7) and 0.797(5), are different as expected for independently prepared samples.
Figure 6. Detail of the crystal structures of the derivatives of the macrocyclic tetramer 1b4.

(a,b) ORTEP perspectives of the [Pd(1b4)]2+ complex along (0,1,0) and along (2,1,0), respectively. (c) ORTEP of the crystal structure of 1b4·C60. All displacement ellipsoids are shown at 75% probability. (d) Space-filling depiction of molecular packing the same crystal.
C60 adduct
Mixing 1b with C60 in chloroform immediately yields a solid that is not soluble enough for spectroscopic investigations. However, slow diffusion of C60 into a solution of 1b produces crystals of composition 4(1b)·C60 (Fig. 1). Along the b-axis, the crystal structure features stacks of alternating fullerene molecules and distorted boat conformers of the 1b4 aggregate (Fig. 6c). Compared with the boat 1a4, in the adduct crystal the iso-tellurazole heterocycles are tilted towards the meridional plane of the macrocycle to maximize their contact with the fullerene. The stack is not symmetrical, there are two distinct distances between the centroids defined by the 4 tellurium atoms of each 1b4 aggregate and the fullerene molecule, the closest macrocycle engaged in two short Te…C contacts (3.457(4) Å, cf., the sum of van der Waals radii 3.76 Å) with the fullerene. The C60 molecule is slightly distorted, it features three crystallographically distinct diameter values (6.952(5), 6.9393(5) and 6.9223(5) Å), which evokes a Jahn–Teller distortion that would result from electron transfer into the t1u LUMO56. However, there are no significant changes in the bond distances and angles of 1b4 in this structure and the material is diamagnetic. As compared with the other crystalline phases in this report, the main differences are in the torsion angles only. Along c, the C60 molecules are organized in a columnar arrangement with even C…C spacing of 3.496 Å (Fig. 6d). The distance between C60 centroids is 10.533(2) Å, longer than the 10.008 Å observed in the crystal of pure C60 (ref. 57), and may be determined by the size of the macrocycle.
Discussion
The variety of supramolecular structures obtained from 1b and the ease with which the crystallization conditions select the aggregate and polymorph clearly indicate that these assemblies undergo reversible dissociation in solution. On the other hand, the short Te…O distances in the crystals structures and the results of mass spectrometry, NMR spectroscopy and DFT calculations show that the interaction between the tellurium and oxygen atoms is very strong.
The observation of aggregates in the electrospray mass spectrum 1b is remarkable; supramolecular dimers assembled by Te…N interactions have been observed in mass spectra acquired from the laser-ablation plume of benzotelluradiazoles58 but the detection of oligomers with aggregation numbers 3–7 is unprecedented for organo-tellurium molecules.
Multinuclear NMR spectroscopic experiments demonstrate that the annular tetra- and hexamers are persistent and exist in equilibrium in solution. Direct observation of σ-hole interactions in solution is usually difficult59,60 but encapsulation of halogen-bonded adducts in cavitands61,62 has been helpful and in some cases can be monitored by spectroscopic methods63. Earlier observations of broadening in the 1H-NMR spectrum of 1a at low temperature hinted at the existence of a dynamic process but in that instance the 125Te resonance could not be located and the nature of the process could not be conclusively established. The observation of NOE is one of the strongest evidences of the association of iso-tellurazole N-oxide molecules in solution at low temperature. Such spin cross-relaxation is only observable when the distance between the interacting nuclei is <5 Å, which is not possible within individual molecules of 1a or 1b. Moreover, that the NOE is negative indicates the zero quantum path is dominant in these systems, such situation is characteristic of restricted mobility due to large molecular weights, high viscosity and—arguably—cyclic structures. Provided there is no significant difference in the association energies of these molecules, the combination of 1a and 1b in solution would result in an even distribution of mixed structures that could be identified by their NMR spectra. For instance, an equimolar mixture that only forms centro-symmetric dimers would yield three different structures and display four lines from the methyl protons and two from the t-butyl groups; more complex patterns would arise from the dimers (Me: 8, t-Bu: 4), the tetramers (Me: 16, t-Bu: 8) and hexamers (Me: 41, t-Bu: 26). Of course, those are the maximum number of lines that would arise in each case; whether each of those lines could actually be observed would depend on the actual separation of their resonance frequencies, as well as on the dispersion and resolution provided by the instrument. The experimental result from the 1:1 mixture (Me: 14, t-Bu: 6; Fig. 4e) points to the tetramers, the size of these aggregates is confirmed by the observation of the mixed 1:3 and 2:2 macrocycles in the continuous variations experiments (Fig. 4f). Furthermore, the study of the concentration dependency of the 1H-NMR of 1a at low temperature is consistent with the equilibrium between tetramers and hexamers in solution (Supplementary Fig. 3).
The thermodynamic parameters calculated with DFT-D3 for the model compound 1c are indeed favourable for supramolecular association; however, their magnitudes are taken with caution as it has been argued that this method overestimates the binding energies of this type of interaction due to delocalization error. Also, solvation is likely to have an important role but these calculations do not account for it nor the effect of packing in a crystalline lattice. The calculations indicate that the binding energies of the Te…O interactions are nearly additive, there is little strain in the annular structures and only a small energetic difference between the two tetramer conformations. By enthalpy alone the hexamer would be the most stable cyclic structure, although entropy favours the smaller aggregates and individual molecules. Even more enthalpically favourable would be the infinite polymer chain but its formation naturally imposes the highest entropic cost.
The annular aggregates of iso-tellurazole N-oxides not only are persistent in solution but also display properties of actual macrocycles. The crystal structures of the hexamers already showcase their ability to host small molecules and suggest the construction of rotaxanes and inclusion compounds. Here we further demonstrate that they form coordination complexes and act as fullerene receptors.
The cyclic arrangement of chalcogen atoms and the trans-annular Te–Te distances of 5.0–5.6 Å suggest that the tetramers would be suitable to host transition-metal ions, this is indeed the case with Pd(II). The formation of the macrocyclic complex [Pd(1b4)]2+ is particularly significant; while the reversibility of the Te…O interactions favours the discreet oligomeric aggregates as the predominant species in solution, the lack of kinetic stabilization could compromise the structural integrity of the macrocycle. Iso-tellurazole oxides are potentially ambidentate ligands, coordination by oxygen would likely compete with the Te…O interactions. Such complication is possible even with soft metal ions; pyridine oxides, for example, easily coordinate palladium(II)64. As a macrocyclic ligand, the tetramer will enable the study of metal ions in a uniquely soft coordination sphere, which is difficult to achieve using more traditional approaches. Indeed, metal complexes of telluracrown ethers are difficult to obtain because their Te–C bonds are very reactive65.
Fullerenes form adducts with a variety of macrocyclic and polycyclic molecules in solution and are amenable to structural characterization by X-ray diffraction66. In the case of 1b, poor solubility restricted the study of the product of reaction with C60 to the crystallographic determination but the ability of the tetramer 1b4 to bind the fullerene receptor is well-demonstrated. The fullerene adduct is an intriguing material in its own right; its columnar arrangement of C60 molecules could facilitate charge transport, which calls for further investigations of applications in photovoltaics and molecular semiconductors.
As shown here, iso-tellurazole N-oxides have an unparalleled ability to spontaneously assemble functional macrocycles and thus hold great promise as supramolecular building blocks.
Methods
Experimental
The manipulation of air-sensitive materials was carried out in a glove box or using standards Schlenk techniques under an atmosphere of UHP argon (Praxair). Photosensitive materials were handled under a red LED illumination source. Elemental tellurium (CERAC), DMF (EMD), sodium hydroxide (EMD), acetic anhydride (Sigma-Aldrich), boron trifluoride diethyl etherate (Sigma-Aldrich), Boron trifluoride diethyl etherate (Sigma-Aldrich), hydroxylamine-O-sulfonic acid (Sigma-Aldrich), sodium borohydride (Sigma-Aldrich), tetrakis(acetonitrile)palladium(II) tetrafluoroborate (Sigma-Aldrich), Fullerene C60 (Sigma-Aldrich), dimethylcarbamoyl chloride (Alfa Aesar), phenylacetylene (Alfa Aesar), t-butylacetylene (Alfa Aesar), Chloroform (Caledon), dichloromethane (Caledon), diethyl ether (Caledon), ethyl acetate (Caledon), methanol (Caledon), Sodium sulphate (Caledon), toluene (Caledon), n-butyllithium (Acros Organics), Silica gel 60 (VWR) and sodium carbonate (VWR) were used as received from the commercial suppliers without further purification. Solvents for used synthesis were dehydrated within an Innovative Technologies solvent purification system (THF, acetonitrile) or by reflux with an appropriate dehydrating agent (Methanol over magnesium). 4-Phenylbut-3-yn-2-one and 5,5-dimethyl-hex-3-yn-2-one were prepared by literature methods. All NMR spectra were acquired in solution with a deuterated solvent. Spectra were obtained using Bruker AVANCE 500 MHz (Bruker 5-mm Broad Band Inverse probe) or Bruker AVANCE 600 MHz (Bruker 5-mm BROAD BAND OBSERVE probe) Spectrometers at 287.5 K unless otherwise indicated. Variable temperature spectra were acquired using either a cold or ambient temperature gas flow with a BV-T 2000 variable temperature controller. The sample temperature in the spectrometers was calibrated with a chemical-shift thermometer consisting of a 4% solution of methanol in methanol-d4. The 1H, 13C and 125Te spectra were processed using Bruker TopSpin 2.1 or 3.2 software packages. The 1H and 13C spectra were referenced to tetramethyl silane using the deuterated solvent signal as a secondary reference. The 125Te chemical shifts are reported respect to the room-temperature resonance of TeMe2 (δ=0.00 p.p.m.) but were measured using a secondary reference of diphenyl ditelluride in CD2Cl2 (δ=420.36 p.p.m.). Electrospray ionization mass spectra were acquired in positive ion mode on a Waters/Micromass Quattro Ultima Global ToF mass spectrometer operating in W Mode. Pure samples were dissolved in dichloromethane followed by dilution with methanol. High resolution Mass spectra were obtained in a Waters Global and Ultima (ES Q-TOF) Mass Spectrometer (capillary=3.20 V, cone=100 V, source temp=80 °C and resolving power=10,000). Infrared vibrational spectra were acquired in a Bio-Rad FTS-40 FT-IR spectrometer or a Thermo Scientific Nicolet 6700 FT-IR spectrometer. Melting points were determined with Uni-Melt Thomas Hoover capillary melting point apparatus and are reported uncorrected. Combustion elemental analyses were carried out by Guelph Analytical Laboratories (Guelph, Ontario, Canada).
Synthesis overview
The iso-tellurazole N-oxides were prepared by a method (Supplementary Fig. 4) that includes the trans addition of an in situ-generated tellurocarbamic acid to an ynone. The resulting enone undergoes condensation with hydoxylamine-O-sulfonic acid to introduce the nitrogen atom and the heterocycle is closed by hydrolysis of the intermediate product. The process yields DMF and sulfuric acid as by-products that are separated in an aqueous workup.
Bis-(N,N-dimethylcarbamoyl)-ditelluride
Sodium hydrogen telluride was prepared in situ from elemental tellurium (1.58 g), anhydrous sodium borohydride (2.33 g, 5 eq.) and anhydrous deoxygenated DMF (70 ml) at 95 °C under argon in a single-piece glass vessel. Shortly after heating started, the tellurium began to dissolve into a dark red–purple solution. After about 1 h, all the tellurium was consumed and the mixture became a light yellow suspension. The NaTeH dispersion was cooled to room temperature with a water bath and dimethylcarbamoyl chloride (3.97 g, 3 eq.) was added by cannula under argon; the reaction mixture was then stirred at 95 °C. The light yellow slurry was removed from the heat and cooled to room temperature in a water bath. Argon was removed with vacuum, and oxygen was introduced into the apparatus at 1 atm and the slurry became dark brown after 1 h. About 700 ml of distilled water was added into the mixture with stirring, the brown slurry turned black and was extracted repeatedly with 70 ml of diethyl ether until the aqueous solution was no longer yellow. The yellow organic solution was washed with aqueous sodium carbonate followed by distilled water, then dehydrated with Na2SO4.The organic fraction was concentrated under vacuum until a dark yellow solid began to precipitate at room temperature. The mixture was placed in a freezer (−20 °C) to promote crystallization of the pure ditelluride. Yield: 68%; mp: 105–110 °C (decomposed); 1H-NMR (500 MHz, CDCl3): δ 3.11 (s, 3H), 3.08 (s, 3H) (cf. ref. 66 3.11 (s, 3H), 3.08 (s, 3H)); 13C NMR (500 MHz, CDCl3): δ 145.4, 40.6, 36.1; IR (KBr): 1,660, 1,353, 1,249, 1,076, 872, 665 cm−1.
(Z)-4-[(dimethylamino)carbonyltelluro]-4-phenyl-3-buten-2-one
Based on the procedure from the study by Shimada et al.53, bis(N,N-dimethylcarbamoyl)-ditelluride (1.54 g, 3.86 mmol) was dissolved in 35 ml of anhydrous DMF under argon; the solution was dark yellow. Anhydrous NaBH4 (0.321 g, 8.49 mmol) was dissolved in anhydrous methanol (18 ml) then added dropwise into the ditelluride solution while maintaining the temperature between −50 and −78 °C. The mixture was stirred at 0 °C for 30 min, the evolution of gas was observed and the colour of the mixture became dark red. The 4-phenylbut-3-yn-2-one (1.95 g, 13.51 mmol) was added to the reaction at 0 °C dropwise. The solution turned light yellow and stirring continued for 3 h. The reaction was quenched with 5 ml of distilled water followed by extraction with toluene in 50 ml portions from a 500 ml brine solution. The organic solution was washed with distilled water, dried with Na2SO4 and evaporated under high vacuum at 35 °C. The organic residue was purified by silica gel column chromatography with CH2Cl2:ethyl acetate (95:5% v/v). The solvent from eluate was evaporated and the product was a yellow solid. Yield: 72%; mp: 70–71 °C; 1H-NMR (500 MHz, CD2Cl2): δ 7.35–7.65 (m, 5H), 2.77 (s, 3H), 2.60 (s, 3H), 2.34 (s, 3H) (cf. ref. 53 7.33–7.41 (m, 5H), 2.81 (s, 3H), 2.60 (s, 3H), 2.56 (s, 3H)); 13C-DEPTq NMR (500 MHz, CD2Cl2): δ 197.2, 160.7, 156.6, 143.9, 131, 129.17, 128.9, 128.2, 41.7, 33.9, 30.1.
(Z)-4-[(dimethylamino)carbonyltelluro]-4-t-butyl-3-buten-2-one
This derivative was synthesized in a similar way and obtained as a yellow solid. Yield: 37%; 1H-NMR (500 MHz, CDCl3): δ 6.81 (s, 1H), 3.00 (s, 6H), 2.28 (s, 3H), 1.25 (s, 9H); 13C-DEPTq NMR (500 MHz, CD2Cl2): δ 199.6, 156.9, 151.6, 133.4, 41.4, 35.9, 31.2, 30.7.
3-methyl-5-phenyl-1,2-tellurazole N-oxide, R=Ph (1b)
This compound was synthesized following the method described by Kübel51 with some modifications. The tellurocarbamate (1.42 g, 4.13 mmol) was refluxed with hydroxylamine-O-sulfonic acid (2.05 g, 18.16 mmol) for 1 h in anhydrous methanol (90 ml). The product was extracted with chloroform, washed with distilled water, dehydrated and dried under vacuum. The crude product was dissolved again in methylene chloride and was deposited on a layer of silica (2 cm). Impurities were eluted with methylene chloride through the silica. The pure product was then eluted with a CH2Cl2/methanol solution (50:50% v/v) and the solvent was removed under vacuum. The product was obtained as a pale yellow solid. Yield: >95%; mp: 207–211 °C (decomposed); 1H-NMR (500 MHz, CD2Cl2): δ 7.26–7.42 (m, 5H), 7.10 (s, 1H), 1.77 (s, 3H); 13C NMR (500 MHz, CD2Cl2): δ 157.9, 152.6, 140.8, 129.9, 128.2, 128.0, 127.7, 15.9; 125Te NMR (500 MHz, CD2Cl2): δ 1,595.2; IR (KBr): 3,050, 3,022, 2,918, 1,571, 1,493, 1,468, 1,443, 1,373, 1,343, 1,222, 1,109, 1,028, 927, 908, 869, 832, 759, 713, 696, 617, 584, 534 cm−1; HRMS (m/z): [M−H]+ calcd. for C10H10NOTe, 289.7961; found, 289.9831.
3-methyl-5-t-butyl-1,2-tellurazole N-oxide, R=t-Bu (1a)
This compound was synthesized in a similar way. Yield: 80%; mp: 180–185 °C (decomposed); 1H-NMR (500 MHz, CDCl3): δ 6.96 (s, 1H), 2.17 (s, 3H), 1.42 (s, 9H). 13C-DEPTq NMR (125.8 MHz, CD2Cl2): δ 168.9, 156.4, 122.7, 41.5, 32.1, 16.0; IR (KBr): 2,953, 2,912, 2,865, 1,565, 1,466, 1,424, 1,389, 1,370, 1,361, 1,337, 1,243, 1,231, 1,202, 1,125, 1,030, 1,001, 967, 896, 842, 828, 794, 760, 756, 697 cm−1; HRMS (m/z): [M-H]+ calcd. for C8H14ON129Te, 270.0138; found, 270.0122.
[Pd(1b 4 )](BF4)2
A solution of isotellurazole-N-oxide in anhydrous dichloromethane (0.031 g, 0.108 mmol) was added dropwise to a solution of tetrakis(acetonitrile)palladium(II) tetrafluoroborate in anhydrous acetonitrile (0.012 g, 0.027 mmol). The solution turned from light yellow to deep red and a reddish–brown solid precipitated. The mixture was stirred under nitrogen for a day and the solid was filtered off, washed with dichloromethane and dried under vacuum. Yield: 98%; mp: 190–191 °C; 1H-NMR (500 MHz, CD3CN): δ 7.49-7.42 (m, 6H), 2.11 (s, 3H); 13C NMR (500 MHz, DMSO-d6): δ 138.4, 131.9, 129.2, 129.0, 128.7, 128.4, 128.1, 15.7; Not soluble enough for 125Te NMR; IR (KBr): 1,615, 1,575, 1,490, 1,442, 1,384, 1,219, 1,113, 1,084, 1,062, 928, 866, 760, 696, 614, 573, 533 cm−1; analysis (calcd., found for C40H36N4O4B2F8Te4Pd): C (33.66, 33.48), H (2.54, 2.28), N (3.93, 4.08). Slow diffusion in long tube yielded instead single crystals of idealized composition [Pd(1b4)](BF4)2.(CH2Cl2)2 in the mixing zone and crystals of pure 1b4 at the bottom. The former loose the crystallization solvent under vacuum.
[1b 4 ]C60
A concentrated solution of 1b (0.048 g, 0.167 mmol) was dissolved in chloroform. The layer of 1b solution was allowed to diffuse with a layer concentrated solution of fullerene (0.030 g, 0.041 mmol) in tetrachloroethane that was filtered through an activated neutral alumina. Crystals suitable for X-ray diffraction were obtained by slow diffusion with the two layers of solution until the growth of crystals reached equilibrium. Yield: 76.9%; the material did not melt or appear to decompose up to 280 °C; [1b4]C60 is not soluble enough to acquire meaningful 1H, 13C and 125Te NMR spectra. IR (KBr): 1,572, 1,491, 1,468, 1,428, 1,372, 1,221, 1,182, 1,107, 1,028, 927, 908, 869, 833, 755, 715, 694, 616, 577, 527 cm−1; analysis (calcd., found for C100H36N4O4Te4): C (64.30, 64.16), H (1.94, 1.77 ), N (3.00, 2.75).
Scrambling experiment
The NMR samples were prepared by mixing 1b with 1a in 10:0, 9:1, 8:2, 7:3, 6:4, 5:5, 4:6, 3:7, 2:8, 1:9, 0:10 molar ratios while maintaining a total amount of 8.717 × 10−3 mmol. Each sample was dissolved in 0.7 ml of deuterated methylene chloride, yielding a total concentration 12.6 mmol l−1. 1H-NMR spectra were acquired at both 179.9 and 287.5 K using a Bruker Avance 600 MHz spectrometer and are provided as Supplementary Fig. 5
Single-crystal X-ray diffraction
Single crystals were grown under the following conditions: 3(1b)·(C6H6): slow evaporation from a benzene solution. 1b: slow evaporation from a CH2Cl2 solution. 12(1b)·(CH2Cl2): slow evaporation from a concentrated solution in a mixture of or CH2CH2/pentane (90:10% v/v). 3(1b)·(C4H8O): slow evaporation from a THF solution. [Pd(1b4)](BF4)2(CH2Cl2)2: diffusion of a solution of 1b in CH2Cl2 into [Pd(CH3CN)4])](BF4)2 in acetonitrile. (1b4)·C60: diffusion of a 1b solution in CHCl3 into saturated C60 in tetrachloroethane. All crystals were mounted on a MiTeGen Micromounts with Paratone-n oil. Crystals were mounted on nylon loops (Hampton, CA) or MiTeGen Micromounts (Ithica, NY) with Paratone-n oil. A Bruker APEX2 diffractometer was used to collect data at 100 K with Mo-Kα radiation (λ=0.71073 Å). A CCD area detector was used and equipped with a low-temperature accessory Oxford cryostream. Solution and refinement procedures are presented in the Supplementary Methods and specific details are compiled in Supplementary Table 1. Selected distances and angles are provided in Supplementary Table 2.
Computational
All DFT calculations were performed using the ADF/BAND DFT package (versions 2013 and 2014). Details of the method are provided in the Supplementary Methods. Coordinates of all optimized structures are provided in Supplementary Tables 3–8.
Additional information
Accession codes: The X-ray crystallographic coordinates for all structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 1414076-1414081 and 1415229. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre (www.ccdc.cam.ac.uk/data_request/cif).
How to cite this article: Ho, P. C. et al. Supramolecular macrocycles reversibly assembled by Te…O chalcogen bonding. Nat. Commun. 7:11299 doi: 10.1038/ncomms11299 (2016).
Supplementary Material
Supplementary Figures 1-5, Supplementary Tables 1-8, Supplementary Methods and Supplementary References
Acknowledgments
The support of the Natural Sciences and Engineering Research Council Canada (I.V.-B.—DG, L.M.L.—PSD, J.S.—USRA) and McMaster University (P.C.H.—Summer Work Program) is gratefully acknowledged. We thank Professor Goward and Drs Berno and Krachkovskiy (McMaster) for their valuable advice on the NMR experiments. Portions of this work were made possible by the facilities of the Shared Hierarchical Academic Research Computing Network (SHARCNET: www.sharcnet.ca) and Compute/Calcul Canada.
Footnotes
Author contributions P.C.H. optimized the synthetic methods, isolated 3(1b)·(C6H6), 12(1b)·(CH2Cl2), [Pd(1b4)](BF4)2·2(CH2Cl2)2 and 4(1b)C60, performed the NOESY and scrambling NMR experiments, analysed the data and assisted writing the manuscript. J.S. isolated 3(1b)·(C4H8O), analysed the structural data and assisted with the scrambling and other experiments. J.K. and P.J.W.E. performed the synthesis and spectroscopic characterization of 1a and DFT calculations. P.S. and C.G. performed the synthesis and preliminary spectroscopic characterization of 1b. L.M.L., H.J. and J.F.B. screened samples for X-ray crystallography, acquired diffraction data, solved and refined the structures. D.R.M. performed synthetic experiments and some DFT calculations. I.V.-B. designed the experiments, performed some DFT calculations and wrote the manuscript.
References
- Desiraju G. R. et al. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 85, 1711–1713 (2013). [Google Scholar]
- Pedireddi V. R. et al. The nature of halogen halogen interactions and the crystal structure of 1,3,5,7-tetraiodoadamantane. J. Chem. Soc., Perkin Trans. 2, 2353–2360 (1994). [Google Scholar]
- Rissanen K. Halogen bonded supramolecular complexes and networks. CrystEngComm. 10, 1107–1113 (2008). [Google Scholar]
- Metrangolo P., Meyer F., Pilati T., Resnati G. & Terraneo G. Halogen bonding in supramolecular chemistry. Angew. Chem. Int. Ed. 47, 6114–6127 (2008). [DOI] [PubMed] [Google Scholar]
- Metrangolo P. & Resnati G. Chemistry: halogen versus hydrogen. Science 321, 918–919 (2008). [DOI] [PubMed] [Google Scholar]
- Raatikainen K. & Rissanen K. Breathing molecular crystals: halogen- and hydrogen-bonded porous molecular crystals with solvent induced adaptation of the nanosized channels. Chem. Sci. 3, 1235–1239 (2012). [Google Scholar]
- Bolton O., Lee K., Kim H.-J., Lin K. Y. & Kim J. Activating efficient phosphorescence from purely organic materials by crystal design. Nat. Chem. 3, 205–210 (2011). [DOI] [PubMed] [Google Scholar]
- Virkki M. et al. Halogen bonding enhances nonlinear optical response in poled supramolecular polymers. J. Mater. Chem. C 3, 3003–3006 (2015). [Google Scholar]
- Saccone M. et al. Supramolecular hierarchy among halogen and hydrogen bond donors in light-induced surface patterning. J. Mater. Chem. C 3, 759–768 (2015). [Google Scholar]
- Shang J. et al. Assembling molecular Sierpiński triangle fractals. Nat. Chem. 7, 389–393 (2015). [DOI] [PubMed] [Google Scholar]
- Meazza L. et al. Halogen-bonding-triggered supramolecular gel formation. Nat. Chem. 5, 42–47 (2013). [DOI] [PubMed] [Google Scholar]
- Kniep F. et al. Organocatalysis by neutral multidentate halogen-bond donors. Angew. Chem. Int. Ed. 52, 7028–7032 (2013). [DOI] [PubMed] [Google Scholar]
- Houbenov N. et al. Halogen-bonded mesogens direct polymer self-assemblies up to millimetre length scale. Nat. Commun. 5, 4043 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langton M. J., Robinson S. W., Marques I., Félix V. & Beer P. D. Halogen bonding in water results in enhanced anion recognition in acyclic and rotaxane hosts. Nat. Chem. 6, 1039–1043 (2014). [DOI] [PubMed] [Google Scholar]
- Jentzsch A. V. et al. Transmembrane anion transport mediated by halogen-bond donors. Nat. Commun. 3, 905 (2012). [DOI] [PubMed] [Google Scholar]
- Jentzsch A. V. & Matile S. Transmembrane halogen-bonding cascades. J. Am. Chem. Soc. 135, 5302–5303 (2013). [DOI] [PubMed] [Google Scholar]
- Manna D. & Mugesh G. Regioselective deiodination of thyroxine by iodothyronine deiodinase mimics: an unusual mechanistic pathway involving cooperative chalcogen and halogen bonding. J. Am. Chem. Soc. 134, 4269–4279 (2012). [DOI] [PubMed] [Google Scholar]
- Politzer P., Murray J. S. & Clark T. Halogen bonding and other σ-hole interactions: a perspective. Phys. Chem. Chem. Phys. 15, 11178–11189 (2013). [DOI] [PubMed] [Google Scholar]
- Rosokha S. V., Stern C. L. & Ritzert J. T. Experimental and computational probes of the nature of halogen bonding: complexes of bromine-containing molecules with bromide anions. Chem. Eur. J. 19, 8774–8788 (2013). [DOI] [PubMed] [Google Scholar]
- Ramasubbu N., Parthasarathy R. & Murray-Rust P. Angular preferences of intermolecular forces around halogen centers: preferred directions of approach of electrophiles and nucleophiles around carbon-halogen bond. J. Am. Chem. Soc. 108, 4308–4314 (2002). [Google Scholar]
- Brezgunova M. E. et al. Chalcogen bonding: experimental and theoretical determinations from electron density analysis. geometrical preferences driven by electrophilic–nucleophilic interactions. Crystal Growth Des. 13, 3283–3289 (2013). [Google Scholar]
- Sarkar S., Pavan M. S. & Row T. N. G. Experimental validation of ‘pnicogen bonding' in nitrogen by charge density analysis. Phys. Chem. Chem. Phys. 17, 2330–2334 (2014). [DOI] [PubMed] [Google Scholar]
- Scheiner S. Sensitivity of noncovalent bonds to intermolecular separation: hydrogen, halogen, chalcogen, and pnicogen bonds. CrystEngComm. 15, 3119–3124 (2013). [Google Scholar]
- Bauza A., Quiñonero D., Deyà P. M. & Frontera A. Halogen bonding versus chalcogen and pnicogen bonding: a combined Cambridge structural database and theoretical study. CrystEngComm. 15, 3137–3144 (2013). [Google Scholar]
- Zahn S., Frank R., Hey Hawkins E. & Kirchner B. Pnicogen bonds: a new molecular linker? Chem. Eur. J. 17, 6034–6038 (2011). [DOI] [PubMed] [Google Scholar]
- Alcock N. W. Secondary bonding to nonmetallic elements. Adv. Inorg. Chem. Radiochem. 15, 1–58 (1972). [Google Scholar]
- Landrum G. A. & Hoffmann R. Secondary bonding between chalcogens or pnicogens and halogens. Angew. Chem. Int. Ed. 37, 1887–1890 (1998). [Google Scholar]
- Rosenfield R. E., Parthasarathy R. & Dunitz J. D. Directional preferences of nonbonded atomic contacts with divalent sulfur. 1. Electrophiles and nucleophiles. J. Am. Chem. Soc. 99, 4860–4862 (1977). [Google Scholar]
- de Paul N, Nziko V. & Scheiner S. Intramolecular S…O chalcogen bond as stabilizing factor in geometry of substituted phenyl-SF3 molecules. J. Org. Chem. 80, 2356–2363 (2015). [DOI] [PubMed] [Google Scholar]
- Chaudhary P. et al. Lewis acid behavior of SF4: synthesis, characterization and computational study of adducts of SF4 with pyridine and pyridine derivatives. Chem. Eur. J. 21, 6247–6256 (2015). [DOI] [PubMed] [Google Scholar]
- Alikhani E., Fuster F., Madebene B. & Grabowski S. J. Topological reaction sites—very strong chalcogen bonds. Phys. Chem. Chem. Phys. 16, 2430–2442 (2014). [DOI] [PubMed] [Google Scholar]
- Chivers T. & Laitinen R. S. Tellurium: a maverick among the chalcogens. Chem. Soc. Rev. 44, 1725–1739 (2015). [DOI] [PubMed] [Google Scholar]
- Tiekink E. R. T. & Zukerman-Schpector J. Stereochemical activity of lone pairs of electrons and supramolecular aggregation patterns based on secondary interactions involving tellurium in its 1,1-dithiolate structures. Coord. Chem. Rev. 254, 46–76 (2010). [Google Scholar]
- Politzer P., Murray J., Janjić G. & Zarić S. σ-hole interactions of covalently-bonded nitrogen, phosphorus and arsenic: a survey of crystal structures. Crystals 4, 12–31 (2014). [Google Scholar]
- Setiawan D., Kraka E. & Cremer D. Strength of the pnicogen bond in complexes involving group Va elements N, P and As. J. Phys. Chem. A 119, 1642–1656 (2014). [DOI] [PubMed] [Google Scholar]
- Cozzolino A. F., Elder P. J. W. & Vargas-Baca I. A survey of tellurium-centered secondary-bonding supramolecular synthons. Coord. Chem. Rev. 255, 1426–1438 (2011). [Google Scholar]
- Politzer P., Murray J. S. & Clark T. Halogen bonding: an electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 12, 7748–7757 (2010). [DOI] [PubMed] [Google Scholar]
- Werz D. B. et al. Self-organization of chalcogen-containing cyclic alkynes and alkenes to yield columnar structures. Org. Lett. 4, 339–342 (2002). [DOI] [PubMed] [Google Scholar]
- Gleiter R. & Werz D. B. Elastic cycles as flexible hosts: how tubes built by cyclic chalcogenaalkynes individually host their guests. Chem. Lett. 34, 126–131 (2005). [Google Scholar]
- Lari A., Gleiter R. & Rominger F. Supramolecular organization based on van der Waals forces: syntheses and solid state structures of isomeric [6.6]cyclophanes with 2,5-diselenahex-3-yne bridges. Eur. J. Org. Chem. 2009, 2267–2274 (2009). [Google Scholar]
- Pushkarevsky N. A. et al. First charge-transfer complexes between tetrathiafulvalene and 1,2,5-chalcogenadiazole derivatives: Design, synthesis, crystal structures, electronic and electrical properties. Synth. Metals 162, 2267–2276 (2012). [Google Scholar]
- Cozzolino A. F., Vargas-Baca I., Mansour S. & Mahmoudkhani A. H. The nature of the supramolecular association of 1,2,5-chalcogenadiazoles. J. Am. Chem. Soc. 127, 3184–3190 (2005). [DOI] [PubMed] [Google Scholar]
- Cozzolino A. F., Britten J. F. & Vargas-Baca I. The effect of steric hindrance on the association of telluradiazoles through Te−N secondary bonding interactions. Crystal Growth Des. 6, 181–186 (2005). [Google Scholar]
- Cozzolino A. F., Whitfield P. S. & Vargas-Baca I. Supramolecular chromotropism of the crystalline phases of 4,5,6,7-tetrafluorobenzo-2,1,3-telluradiazole. J. Am. Chem. Soc. 132, 17265–17270 (2010). [DOI] [PubMed] [Google Scholar]
- Zhou A.-J., Zheng S.-L., Fang Y. & Tong M.-L. Molecular tectonics: self-complementary supramolecular Se…N synthons directing assembly of 1D silver chains into 3D porous molecular architectures. Inorg. Chem. 44, 4457–4459 (2013). [DOI] [PubMed] [Google Scholar]
- Lee L. M. et al. The size of the metal ion controls the structures of the coordination polymers of benzo-2,1,3-selenadiazole. CrystEngComm. 15, 7434–7437 (2013). [Google Scholar]
- Berionni G., Pégot B., Marrot J. & Goumont R. Supramolecular association of 1,2,5-chalcogenadiazoles: an unexpected self-assembled dissymetric [Se⋯N]2 four-membered ring. CrystEngComm. 11, 986–988 (2009). [Google Scholar]
- Aakeröy C. B. et al. The quest for a molecular capsule assembled via halogen bonds. CrystEngComm. 14, 6366–6368 (2012). [Google Scholar]
- Dumele O., Trapp N. & Diederich F. Halogen bonding molecular capsules. Angew. Chem. Int. Ed. 54, 12339–12344 (2015). [DOI] [PubMed] [Google Scholar]
- Beyeh N. K., Pan F. & Rissanen K. A halogen-bonded dimeric resorcinarene capsule. Angew. Chem. Int. Ed. 54, 7303–7307 (2015). [DOI] [PubMed] [Google Scholar]
- Kübel J., Elder P. J. W., Jenkins H. A. & Vargas-Baca I. Structure and formation of the first (–O–Te–N–)4 ring. Dalton Trans. 39, 11126–11128 (2010). [DOI] [PubMed] [Google Scholar]
- Lee S., Chen C.-H. & Flood A. H. A pentagonal cyanostar macrocycle with cyanostilbene CH donors binds anions and forms dialkylphosphate [3]rotaxanes. Nat. Chem. 5, 704–710 (2013). [DOI] [PubMed] [Google Scholar]
- Shimada K. et al. Synthesis of isochalcogenazole rings by treating β-(N,N-dimethylcarbamoylchalcogenenyl)alkenyl ketones with hydroxylamine-O-sulfonic acid. Bull Chem. Soc. Jpn 80, 567–577 (2007). [Google Scholar]
- Thomas P. A. The crystal structure and absolute optical chirality of paratellurite, α-TeO2. J. Phys. C: Solid State Phys. 21, 4611–4627 (1988). [Google Scholar]
- Robertson S. D., Ritch J. S. & Chivers T. Palladium and platinum complexes of tellurium-containing imidodiphosphinate ligands: nucleophilic attack of Li[(PiPr2)(TePiPr2)N] on coordinated 1,5-cyclooctadiene. Dalton Trans. 8582–8592 (2009). [DOI] [PubMed] [Google Scholar]
- Reed C. A. & Bolskar R. D. Discrete fulleride anions and fullerenium cations. Chem. Rev. 100, 1075–1120 (2000). [DOI] [PubMed] [Google Scholar]
- Krätschmer W., Lamb L. D., Fostiropoulos K. & Huffman D. R. Solid C60: a new form of carbon. Pure Appl. Chem. 347, 354–358 (1990). [Google Scholar]
- Cozzolino A. F., Dimopoulos-Italiano G., Lee L. M. & Vargas-Baca I. Chalcogen–nitrogen secondary bonding interactions in the gas phase—spectrometric detection of ionized benzo-2,1,3-telluradiazole dimers. Eur. J. Inorg. Chem. 2013, 2751–2756 (2013). [Google Scholar]
- Beale T. M., Chudzinski M. G., Sarwar M. G. & Taylor M. S. Halogen bonding in solution: thermodynamics and applications. Chem. Soc. Rev. 42, 1667–1680 (2013). [DOI] [PubMed] [Google Scholar]
- Sarwar M. G., Dragisic B., Salsberg L. J., Gouliaras C. & Taylor M. S. Thermodynamics of halogen bonding in solution: substituent, structural and solvent effects. J. Am. Chem. Soc. 132, 1646–1653 (2010). [DOI] [PubMed] [Google Scholar]
- El-Sheshtawy H. S., Bassil B. S., Assaf K. I., Kortz U. & Nau W. M. Halogen bonding inside a molecular container. J. Am. Chem. Soc. 134, 19935–19941 (2012). [DOI] [PubMed] [Google Scholar]
- Sarwar M. G., Ajami D., Theodorakopoulos G., Petsalakis I. D. & Rebek J. Jr Amplified halogen bonding in a small space. J. Am. Chem. Soc. 135, 13672–13675 (2013). [DOI] [PubMed] [Google Scholar]
- Garrett G. E., Gibson G. L., Straus R. N., Seferos D. S. & Taylor M. S. Chalcogen bonding in solution: interactions of benzotelluradiazoles with anionic and uncharged Lewis bases. J. Am. Chem. Soc. 137, 4126–4133 (2015). [DOI] [PubMed] [Google Scholar]
- Cho S. H., Hwang S. J. & Chang S. Palladium-catalyzed C−H functionalization of pyridine N-oxides: highly selective alkenylation and direct arylation with unactivated arenes. J. Am. Chem. Soc. 130, 9254–9256 (2008). [DOI] [PubMed] [Google Scholar]
- Levason W., Reid G. & Zhang W. The chemistry of the p-block elements with thioether, selenoether and telluroether ligands. Dalton Trans. 40, 8491–8506 (2011). [DOI] [PubMed] [Google Scholar]
- Zingaro R. A., Herrera C. & Meyers E. A. Isolation and crystal structure of an unusual ditelluride: bis(N,N-dimethylaminoformyl) ditelluride. J. Organomet. Chem. 306, C36–C40 (1986). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures 1-5, Supplementary Tables 1-8, Supplementary Methods and Supplementary References