

Abstract

Thermoresponsive materials are generating significant interest on account of the sharp and tunable temperature deswelling transition of the polymer chain. Such materials have shown promise in drug delivery devices, sensing systems, and self-assembly. Incorporation of nanoparticles (NPs), typically through covalent attachment of the polymer chains to the NP surface, can add additional functionality and tunability to such hybrid materials. The versatility of these thermoresponsive polymer/nanoparticle materials has been shown previously; however, significant and important differences exist in the published literature between virtually identical materials. Here we use poly(N-isopropylacrylamide) (PNIPAm)-AuNPs as a model system to understand the aggregation behavior of thermoresponsive polymer-coated nanoparticles in pure water, made by either grafting-to or grafting-from methods. We show that, contrary to popular belief, the aggregation of PNIPAm-coated AuNPs, and likely other such materials, relies on the size and concentration of unbound “free” PNIPAm in solution. It is this unbound polymer that also leads to an increase in solution turbidity, a characteristic that is typically used to prove nanoparticle aggregation. The size of PNIPAm used to coat the AuNPs, as well as the concentration of the resultant polymer–AuNP composites, is shown to have little effect on aggregation. Without free PNIPAm, contraction of the polymer corona in response to increasing temperature is observed, instead of nanoparticle aggregation, and is accompanied by no change in solution turbidity or color. We develop an alternative method for removing all traces of excess free polymer and develop an approach for analyzing the aggregation behavior of such materials, which truly allows for heat-triggered aggregation to be studied.
Keywords: gold, nanoparticle, aggregation, N-isopropylacrylamide, NIPAm, LCST, aggregate
Stimuli-responsive polymers exhibit changes in properties upon exposure to small variations in environmental conditions, e.g., light, ionic strength, pH, or temperature.1−4 This characteristic can be utilized in the formation of stimuli-responsive drug delivery materials, such as hydrogels,5−7 micelles,8−10 vesicles,11,12 and polymer–nanoparticle (NP) composites.13−15 Thermoresponsive polymers are commonly used in such systems on account of their biocompatibility and sharp yet tunable temperature-responsive phase transitions.16 A number of synthetic and natural polymers exhibit thermoresponsive behavior, such as poly[oligo(ethylene glycol) methacrylate] (POEGMA),17 poly(N-diethylacrylamide) (PDEAm),18 and poly(N-isopropylacrylamide) (PNIPAm),16 each with its own lower critical solution temperature (LCST) behavior and tunable temperature range.
PNIPAm, which is one of the most widely studied thermoresponsive polymers, undergoes a phase transition in pure water from hydrophilic, fully hydrated polymer chains to hydrophobic-collapsed chains at temperatures around 30–34 °C. Upon heating, PNIPAm becomes insoluble and precipitates from solution; the temperature at which this occurs is referred to as a lower critical solution temperature. This effect is independent of the concentration or molecular weight of the PNIPAm chains.19 The LCST of PNIPAm is also readily tunable by controlling the hydrophilic/hydrophobic balance of the polymer, often achieved by copolymerization with a second monomer such as N-hydroxyethylacrylamide (HEAm) or N,N′-dimethylacrylamide (DMAm).20 Such polymers can readily be attached to other materials such as gold surfaces and NPs, leading to thermoresponsive materials with enhanced properties over the individual components.
Figure 1.

Schematic representation of the two reported outcomes of heating PNIPAm-coated AuNPs (center) above their LCST. Left: Polymer layer collapse. Right: Aggregation of the PNIPAm-coated AuNPs.
Gold nanoparticles (AuNPs) are of particular interest to researchers due to their unique optical and electronic properties, and coating with PNIPAm produces a core–shell system whereby the AuNPs are encapsulated in a thermoresponsive polymer shell. Such composites have been shown to have interesting applications in drug delivery,21,22 catalysis,23 photodynamic therapy,24 triggered release mechanisms,25 and other biologically relevant applications.22,26 Much work has focused on the synthesis of PNIPAm-AuNP core–shell systems using techniques such as in situ reduction of gold salts in the presence of thiol-terminated PNIPAm,27 direct polymerization from the AuNP surface,28−30 referred to as “grafting-from”, and the “grafting-to” approach, where preformed polymers are used to coat AuNPs by ligand exchange.31,32 Methods utilizing the direct polymerization from the AuNP surface (grafting-from) can lead to poorly controlled and difficult to define polymer sizes. However, a grafting-from approach should not allow for any “free” polymers to form in solution, a fact that we show is important in the aggregation behavior of PNIPAm-AuNPs (vide infra). A grafting-to approach offers the most accurate and reproducible method for controlling both the size and dispersity of the AuNPs, as well as the size and PDI of the attached PNIPAm polymers, but leads to excess unbound polymer.33
Whether made by a grafting-to or grafting-from approach, the resultant PNIPAm-AuNPs are often very similar in size and structure and yet are regularly shown to have differing properties in response to temperature. This has led to a significant contradiction in the published literature of similar PNIPAm-AuNP core–shell structures, synthesized using different approaches. Choi etal. have shown that PNIPAm-AuNPs formed via a grafting-from approach exhibit a size decrease upon heating above the LCST,28 whereas Chakraborty etal. and Raula etal., who also made PNIPAm-AuNPs via a grafting-from approach, show aggregation in response to an increase in temperature.29,30 Typically, PNIPAm-AuNPs formed via a grafting-to approach are shown to aggregate upon heating, which is usually reported by observing a change in solution turbidity above the LCST.31 However, such grafting-to systems have also been reported that exhibit shell collapse.32 Studies have also investigated the effects of salt concentration on the aggregation behavior of grafting-to PNIPAm-AuNPs, with increased salt concentrations leading to greater degrees of aggregation.34,35 These apparent contradictions were highlighted in a recent review, with factors such as nanoparticle size and grafting density being suggested to play an important role in aggregation.36
Herein, we use PNIPAm-coated AuNPs to fully investigate the factors that control aggregation of thermoresponsive-nanoparticle materials. We report an alternative approach for the efficient removal of all unbound “free” PNIPAm present after synthesis, by manipulating the LCST of PNIPAm through solvent addition, which is confirmed by thermogravimetric analysis. We then proceed to show, for the first time, that PNIPAm-coated AuNPs can show both a decrease in size or aggregate upon heating, solely by controlling the presence of unbound “free” PNIPAm in solution. When no free PNIPAm remained in solution, no change in turbidity upon heating above the LCST was observed, highlighting that this method of characterization is less than optimal. On account of this, we have developed a new UV/vis-based method for characterizing thermoresponsive nanoparticle materials that allows for the degree of aggregation to be accurately monitored and absolute values calculated. UV/vis analysis was conducted alongside dynamic light scattering (DLS) analysis to confirm that shell collapse or aggregation can be readily controlled. It is shown that complete aggregation occurs only above a critical concentration of free PNIPAm and that this concentration is dependent on the molecular weight (MW) of the free PNIPAm in solution. Conversely, the concentration of PNIPAm-AuNPs and length of the PNIPAm coating of the AuNPs have little effect on the aggregation. This work explains the contradiction in the literature regarding PNIPAm-AuNP systems and offers procedures for effectively removing excess PNIPAm as well as for characterizing the aggregation behavior, which will lead to greater reproducibility in such thermally responsive materials.
Results and Discussion
In order to fully investigate the aggregation behavior of PNIPAm-coated AuNPs, it was necessary to synthesize nanoparticles of different sizes, to investigate the effect of surface curvature, by using both the grafting-from and grafting-to approaches. Grafting-from PNIPAm-AuNPs were synthesized using a one-phase approach and bis[2-(2-bromoisobutyryloxy)undecyl] disulfide as the initiator on the AuNP surface, giving AuNPs of ∼4 nm in size. Atom-transfer radical polymerization (ATRP) was then used to grow polymer chains from the surface of the AuNPs in an approach similar to that used on flat gold surfaces. A grafting-from approach will typically lead to a system in which polymer chains do not exist in solution, on account of initiators being present only on the nanoparticle surface. Nevertheless, these grafting-from PNIPAm-AuNPs were washed to remove any PNIPAm chains that may be present in solution on account of insufficient removal of all free initiator from solution.
Grafting-to PNIPAm-AuNPs were prepared by first synthesizing thiol-terminated polymers and AuNPs separately, followed by mixing. PNIPAm was synthesized with three MWs, small (15 000), medium (30 000), and large (60 000), using RAFT polymerization (Figure S1a). Cleaving the chain transfer agent (CTA) of the PNIPAm in the presence of hydrazine yielded thiol-terminated polymers (HS-PNIPAm) with controllable MWs (Figure S1b). Cleavage of the CTA using azobis(isobutyronitrile) (AIBN) and 1-ethylpiperidine hypophosphite (EPHP) afforded a hydrogen-terminated polymer (H-PNIPAm) (Figure S1C) for control systems and for use as free polymer in solution. The LCST of these polymers was found to be ∼32 °C in all cases, both before and after cleavage.
The majority of AuNPs used in this synthesis were synthesized using the Turkevich method,37 to give 14 nm AuNPs. A seed-mediated approach38 that yields AuNPs sub-10 nm in size was also used as a comparison to those synthesized via the grafting-from approach. Smaller AuNPs have much higher degrees of curvature and greater surface-area-to-volume ratios and have different amounts of PNIPAm per AuNP compared to those produced via the Turkevich route, a factor that has been suggested to affect the aggregation behavior of the PNIPAm-AuNPs.36 Mixing and incubation of the as-synthesized AuNPs with the HS-PNIPAm for 48 h yields PNIPAm-AuNPs. In comparison to a grafting-from approach, a grafting-to approach allows for control over both the AuNP (core) size and PNIPAm (shell) length/size but can lead to excess polymer in solution.
Initially, the as-prepared grafting-to polymer-coated nanoparticles were washed using 3× centrifugation washes with water. It was noted that before purification complete precipitation of the PNIPAm-AuNPs from solution was observed upon heating. During the purification procedure heating led to an increase in turbidity but no aggregation, and after complete purification (approximately >20 washes) there was no visible change in solution color or turbidity. A cooled centrifuge aids in the purification process by reducing the number of washes required; however more than three washes were still required to reach a point where no visible change in solution color or turbidity is observed upon heating. This observation led us to investigate the washing procedure in more detail and develop an improved, more versatile method. It was noted that when performing the standard literature washes, in an uncooled centrifuge, excess PNIPAm would aggregate and precipitate on account of an increase in temperature of the solution. In order to overcome this issue, methanol (∼2 equiv) was introduced into the solution before washing. By introducing methanol the LCST behavior of PNIPAm is arrested, and so when the solution was heated, no aggregation was observed. By using four centrifugation–wash cycles at 12100g, with methanol, with >95% supernatant removal, pure PNIPAm-AuNPs were readily obtained and their purity was confirmed by thermogravimetric analysis (Figure S3).
When pure PNIPAm-AuNPs (i.e., no free PNIPAm) synthesized using either approach were analyzed by DLS, using a temperature sweep from 25 to 40 °C, a decrease in Rh was observed, due to deswelling and collapse of the PNIPAm chains. Figure 2a shows the (as expected) decrease in shell size of pure graft-from PNIPAm-AuNPs upon heating (blue circles) and also that aggregation can be induced upon the addition of 40 μmol of “free” H-PNIPAm (red squares). Figure 2b shows the effect of heating on graft-to AuNPs coated with a 60k PNIPAm, both before and after the addition of free H-PNIPAm chains to the solution. For the PNIPAm-AuNPs formed via a grafting-to approach, aggregation above the LCST of the PNIPAm was expected, but was not observed even if the increased temperature was maintained for more than 18 h. Additionally, there was no visible change in the solution color, which is characteristic of AuNP aggregation, on account of the coupling of the AuNPs’ surface plasmon resonance (SPR). We observed that these PNIPAm-AuNPs were capable of aggregation before washing, and thus, we investigated the effect of adding free PNIPAm back into the previously cleaned samples.
Figure 2.

Dynamic light scattering (DLS) data showing the change in hydrodynamic radius (Rh) as the temperature is increased for (a) graft-from PNIPAm AuNPs and (b) graft-to PNIPAm-AuNPs before (blue circles) and after (red squares) the addition of 40 μmol (2.4 mg) of free 60k H-PNIPAm.
A portion of the 60k PNIPAm that had originally been reacted to yield a thiol terminus was cleaved to remove all trace of thiol and leave a H-terminus (H-PNIPAm). When the H-PNIPAm was added to pure PNIPAm-AuNPs, aggregation above the LCST began to occur. As the amount of free H-PNIPAm was increased, the degree of aggregation increased until complete aggregation from solution was observed. Figure 2 (red squares) shows the effect of heating on the same 14 nm AuNPs coated with 60k PNIPAm in the presence of 2.4 mg/mL (4 × 10–5 mol mL–1) free 60k H-PNIPAm. It can clearly be seen that above the LCST aggregation occurs and the Rh increases drastically, leading to complete precipitation of the AuNPs from solution, suggesting cooperativity between free polymer chains and polymer chains on the surface of the NPs leading to aggregation.16 This aggregation was accompanied by the solution becoming turbid upon heating. The same trend was observed for small AuNPs made via either a grafting-to or grafting-from approach (Figures 2 and S2), highlighting again that nanoparticle size has no effect on the aggregation behavior.
From analysis of the literature it is expected that the larger PNIPAm-AuNPs, synthesized using a grafting-to approach, would aggregate upon heating31 and PNIPAm-AuNPs synthesized using a grafting-from approach would show shell collapse.28 However, in both cases, PNIPAm-AuNPs that had been washed fully showed no signs of aggregation (Figure 2). The addition of free PNIPAm was necessary to cause aggregation in all of the synthesized (and completely purified) PNIPAm-AuNPs. As this observation was contrary to the published literature, a more detailed investigation was needed to fully understand and explain these results. For this, due to their more controlled synthesis and reproducibility, PNIPAm-AuNPs synthesized via a grafting-to approach were selected as our model system, a decision supported by the observation that, once pure, each type of PNIPAm-AuNP system behaves the same as the other.
Using larger (14 nm) PNIPAm-AuNPs coated with 60k PNIPAm formed by a grafting-to approach, it was possible to investigate how much free H-PNIPAm is required to completely aggregate the AuNPs. Larger particles also allow for the investigation into the reversibility of the system. Figure 3a shows the purified 14 nm PNIPAm-AuNPs (1 mg/mL), above and below the LCST of PNIPAm (red and blue, respectively), before the addition of free H-PNIPAm. It can clearly be seen that no aggregation occurs above the LCST. Figure 3b shows the same sample, above and below the LCST, after the addition of 1.6 mg of H-PNIPAm. When this sample is heated above the LCST it is clear that a small degree of aggregation takes place, on account of the slight change in solution color and turbidity and the formation of visible aggregates. Figure 3c shows the same sample after the addition of a further 1.8 mg of H-PNIPAm. It can clearly be seen that the degree of aggregation of the AuNPs is much greater as the amount of free H-PNIPAm is increased. If the aggregation is caused by free H-PNIPAm only, then its removal should yield a sample that exhibits the same polymer chain collapse and decrease in size as the initially purified PNIPAm-AuNPs.
Figure 3.

Images of 14 nm 60k-PNIPAm-coated AuNPs (1 mg/mL) taken above and below the LCST, red and blue, respectively. (a) Pure PNIPAm-AuNPs, (b) PNIPAm-AuNPs after the addition of 1.6 mg of H-PNIPAm, and (c) PNIPAm-AuNPs after the addition of a further 1.8 mg of H-PNIPAm.
The sample of PNIPAm-AuNPs that now contains 3.4 mg of free H-PNIPAm and exhibits a reversible aggregation of the AuNPs upon heating was subjected to an alternative wash procedure. After washing with methanol four times the AuNPs were dried and redispersed in H2O. This material now exhibited none of the aggregation behavior observed in the presence of free H-PNIPAm, instead exhibiting a decrease in Rh upon heating. This experiment demonstrates that the reversible aggregation of PNIPAm-AuNPs is caused by excess PNIPAm in solution and that the amount of free polymer can determine the degree of aggregation, especially considering that removal of the free PNIPAm halts the aggregation behavior.
In order to quantify the amount of free polymer required to fully aggregate the PNIPAm-AuNPs, a UV/vis study was developed. This method involved the addition of small portions of H-PNIPAm to 14 nm AuNPs coated with 60k PNIPAm. After addition, the samples were heated to 45 °C and centrifuged at 1200g for 6 min. Centrifugation removes all large aggregates but is not able to remove unaggregated PNIPAm-AuNPs. After centrifugation the supernatant is collected, cooled, and then analyzed using UV/vis spectroscopy. As the concentration of AuNPs in solution is closely linked to the absorbance of the solution, it was possible to determine if any AuNPs had been removed. The absorbance intensity at 400 nm was used to monitor AuNP aggregation, as this wavelength corresponds mainly to the absorbance for gold atoms, due to interband transitions, and is not red-shifted by any aggregation of the AuNPs, as would be the case for the plasmon absorbance at 525 nm.39,40 As the concentration of 60k H-PNIPAm was increased, a clear and sharp decrease in the absorbance of the solution was observed (Figure 4a). This indicates that the PNIPAm-AuNPs had aggregated and been removed from the solution upon reaching a critical concentration of H-PNIPAm. We believed that the concentration of H-PNIPAm required to cause aggregation would be linked to the MW of the free H-PNIPAm. In order to investigate this, 60k PNIPAm-coated AuNPs were mixed with H-PNIPAm with MWs of 60k, 30k, and 15k. It was observed that as the MW of the free H-PNIPAm decreased, the concentration required to cause aggregation increased (Figure 4b).
Figure 4.
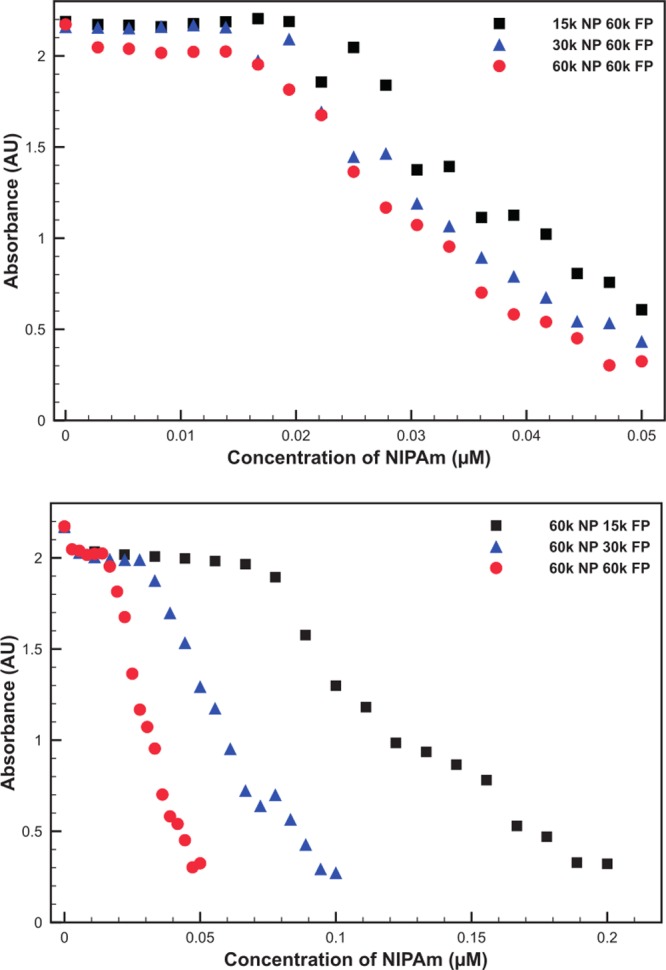
Absorbance of 14 nm AuNPs coated with 60k PNIPAm, (a) 14 nm AuNPs coated with 15k, 30k, and 60k PNIPAm, mixed with aliquots of 60k H-PNIPAm and (b) mixed with aliquots of 15k, 30k, and 60k H-PNIPAm, after heating above the LCST, and large aggregates have been removed via heated centrifugation at low g.
It was also possible to investigate the effect of PNIPAm MW when bound to the AuNPs. A range of PNIPAm-AuNPs coated with 15k, 30k, and 60k polymers were prepared and washed as before. Each PNIPAm-AuNP solution was mixed with aliquots of 60k H-PNIPAm and analyzed using the UV/vis method. The results show that the MW of the surface-bound PNIPAm has little, if any, effect on the concentration of free H-PNIPAm required to aggregate the AuNPs (Figure 4b).
In order to confirm that the aggregation of PNIPAm-AuNPs is indeed due to the presence of free polymer in solution, a second PNIPAm with a higher LCST was synthesized. By incorporating 10% of the hydrophilic monomer N-hydroxyethyl acrylamide into the PNIPAm chain (P[NIPAM-co-HEAm]) it is possible to increase the LCST to 38 °C. When P[NIPAM-co-HEAm] is added to AuNPs coated with 60k PNIPAm, the same trend is observed; that is, an excess (3.4 mg) of H-P[NIPAM-co-HEAm] must be added to cause complete aggregation of the PNIPAm-AuNPs (Figure S5). As the LCST of the PNIPAm bound to the AuNPs is 32 °C and that of the free polymer has now been increased to 38 °C, it is possible to confirm which polymer is responsible for the aggregation. By heating the solution of PNIPAm-AuNPs with free P[NIPAM-co-HEAm] to 35 °C, the free polymer in solution will not collapse, whereas the nanoparticle-bound polymer will. It can be seen in Figure 5 that when this sample is heated to 35 °C, no aggregation occurs, as the H-P[NIPAM-co-HEAm] is not above its LCST. This observation further confirms that the aggregation behavior is dependent on the free polymer chains in solution.
Figure 5.

60k PNIPAm-coated AuNPs (14 nm) with 3.4 mg of added H-PNIPAm (left vial) and P[NIPAM-co-HEAm] (right vial) taken above and below the LCST of the free polymer, right and left, respectively.
Conclusions
For the first time, we have studied in detail the factors that affect the aggregation behavior in water of PNIPAm-AuNPs as a model system for thermoresponsive polymer–nanoparticle materials. Contradictions in the published literature on virtually identical materials are common, with packing density due to the synthetic route being suggested to be the cause. We show that the aggregation is likely due to excess polymer chains still present after inadequate washing. By synthesizing PNIPAm-AuNPs of different sizes and via different grafting techniques we have fully investigated the factors that affect thermoresponsive polymer–nanoparticle composites. Clearly, the current literature procedure of washing using centrifugation for the purification of PNIPAm-AuNPs is inadequate. The alternative, more versatile, washing procedure allowing for the complete removal of excess polymer yields pure PNIPAm-AuNPs. Pure PNIPAm-AuNPs made via either a grafting-from or grafting-to approach exhibit the same property of shell collapse upon heating above the polymer LCST. The aggregation behavior has been shown to be dependent on the presence of free PNIPAm chains in solution, with the degree of aggregation being dependent on free PNIPAm concentration and MW. This was investigated using a UV/vis-based study that allows the amount of free PNIPAm required to cause aggregation to be easily determined. By synthesizing a P[NIPAM-co-HEAm] random copolymer, with a higher LCST, it was further confirmed that such aggregation is due purely to the presence and concentration of free polymer. This new-found understanding of PNIPAm-AuNP systems, in addition to the washing and aggregation determination procedures detailed herein, should assist in the further development of these and other similar dynamic materials, toward a variety of applications.
Methods
Materials and General Methods
All starting materials were purchased from Alfa Aesar and Sigma-Aldrich and used as received unless stated otherwise. 1H NMR (400 MHz) spectra were recorded using a Bruker Avance QNP 400. Chemical shifts are recorded in ppm (δ) in D2O with the internal reference set to δ 4.67 ppm. Spectroscopy studies were performed on a Varian Cary 100 Bio UV–vis spectrophotometer. The GPC system for determination of molecular weight distributions comprised a Jordi Gel DVB, a Shimadzu SPD-M20A Prominence diode array, and a Wyatt Optilab DSP interferometric refractometer. THF was used as the eluent at 35 °C and a flow rate of 1 mL/min–1. The GPC system was calibrated using narrow polystyrene standards. Samples were filtered through 0.45 μm PTFE filters before injection. DMF GPC was performed on two Jordi 5 μm DVB-Glucose columns connected in series with an SPD-M20A Prominence diode array detector and refractive index detector (both Shimadzu) calibrated in relation to poly(methyl methacrylate) standards. Samples were filtered through 0.45 μm nylon filters before injection using a 0.75 mL/min flow rate. Transmission electron microscopy (TEM) characterization was carried out by a JEOL 2000FX TEM under an accelerating voltage of 200 kV. Samples were prepared by applying one drop of the as-synthesized gold nanoparticles onto a holey carbon coated copper TEM grid (400 mesh). High-g centrifuge experiments were conducted using an Eppendorf Minispin centrifuge at 12100g for 30 min at room temperature. Low-g heated centrifugation was conducted using a Thermo Electron Corporation SPD131DDA SpeedVac concentrator at 60 °C. All aqueous solutions were made in deionized water treated with a Milli-Q reagent system ensuring a resistivity of >15 MΩ cm–1.
Synthesis of Alcohol-Terminated Chain Transfer Agent (CTA-OH)
CTA-OH was synthesized following a previously reported method.41 Briefly, ethanthiol (2 g, 32.2 mmol) was added to a stirred suspension of tribasic potassium phosphate (7.2 g, 33.9 mmol) in acetone (50 mL), and the mixture stirred at room temperature for 45 min. Carbon disulfide (7.35 g, 96.5 mmol) was added, and the mixture stirred for a further 45 min. 4-(Chloromethyl)benzyl alcohol (5.04 g, 32.2 mmol) was then added, and the reaction mixture left to stir overnight at room temperature. Once the reaction was complete, the mixture was filtered and the volatiles removed under reduced pressure. The solid residue was then dissolved in ethyl acetate, washed with water and brine, and dried over magnesium sulfate. The ethyl acetate was removed under reduced pressure, and the yellow oil product purified by column chromatography in ethyl acetate to give a yellow powder of CTA-OH (5.013 g, 19.4 mmol, 60%). 1H NMR (D2O, 400 MHz): 1.30 (3H, t, J = 7 Hz), 1.58 (1H, t, J = 6 Hz), 3.31 (2H, q, J = 7 Hz), 4.55 (2H, s), 4.60 (2H, d, J = 6 Hz), 7.26 (4H, m, phenyl).
General Synthesis of SSS-PNIPAm-OH
PNIPAm polymers were synthesized with a range of Mn’s (5k–60k g/mol). A general procedure for their synthesis was as follows. Quantities of each starting material used are given in Table S1, and yields and molecular weights in Table S2. N-Isopropylacrylamide, CTA-OH, and ACPA were dissolved in 1,4-dioxane in a Schlenk tube. The solution was degassed by bubbling nitrogen through it for 40 min. The tube was then sealed and heated in a 70 °C oil bath overnight. When completed, the polymerization mixture was quenched in liquid nitrogen and the product isolated as a yellow powder by precipitation from cold diethyl ether. 1H NMR (D2O, 400 MHz): 1.07 (6n H, br s), 1.50 (2n H, br t), 1.94 (1n H, br t), 3.82 (1n H, br s), 7.25 (4H, dd), where n is the number of repeat units in the polymer.
General Synthesis of HS-PNIPAm-OH
Cleavage of the trithiocarbonyl end group to a thiol was performed on all of the SSS-PNIPAm-OH polymers synthesized. A general method is as follows. SSS-PNIPAm-OH was dissolved in DMF, and hydrazine monohydrate added. The mixture was stirred for 2–3 h at room temperature. The resulting polymer was precipitated in cold, acidified diethyl ether to form a white powder. 1H NMR (D2O, 400 MHz): 1.07 (6n H, br s), 1.50 (2n H, br t), 1.94 (1n H, br t), 3.82 (1n H, br s), 7.25 (4H, dd), where n is the number of repeat units in the polymer.
General Synthesis of H-PNIPAm-OH
Cleavage of the trithiocarbonyl end group to a hydrogen was performed on SSS-PNIPAm-OH polymers. A general method is as follows. SSS-PNIPAm-OH and azobis(isobutyronitrile) were dissolved in DMF. 1-Ethylpiperidine hypophosphite was added to this solution, and the mixture degassed under nitrogen for 40 min. The solution was then heated at 100 °C for 3 h. Once the reaction was complete, the mixture was cooled in ice, precipitated in hexane, dried in an oven at 60 °C overnight, redissolved in water, and then freeze-dried to obtain a white powder. 1H NMR (D2O, 400 MHz): 1.07 (6n H, br s), 1.50 (2n H, br t), 1.94 (1n H, br t), 3.82 (1n H, br s), 7.25 (4H, dd), n being the number of repeat units in the polymer.
Synthesis of 14 nm Citrate Gold Nanoparticles
A HAuCl4 stock solution (30 mM, 1.9 mL, 0.057 mol) was diluted in 148 mL of Milli-Q water in a round-bottom flask (RBF), forming a pale yellow solution. The RBF was fitted with a condenser, and the solution heated to boiling under vigorous stirring. A stock solution of sodium citrate was made (MW = 294.1 g/mol, 0.3035 g, 1.03 mmol) in 6.78 mL of water (making a 152 mM solution). The required amount of sodium citrate stock solution was injected into the boiling gold salt solution quickly, with an injection time under one second. The diameter of nanoparticles synthesized depended on the ratio of sodium citrate to HAuCl4. For 14 nm AuNPs, 2 mL of sodium citrate was injected (1:5.33 Au:citrate). The solution changed to a red color within 10 min and was then left to cool. DLS, TEM, and UV–vis measurements were used to analyze the synthesized AuNPs.
Functionalization of 14 nm AuNPs with HS-PNIPAm-OH
A 20 mL amount of 14 nm AuNPs was concentrated to 1.75 mL and mixed with HS-PNIPAm (at least 100 mg). The solution was incubated for 48 h to yield PNIPAm-AuNPs.
Purification of 14 nm PNIPAm-AuNPs
The as-prepared PNIPAm-AuNPs (1.75 mL) were added to MeOH (∼3.5 mL). Centrifugation at 12100g for 30 min at room temperature allowed for more than 95% supernatant removal. PNIPAm-AuNPs were redispersed in MeOH, and the above process was repeated a further three times. The resultant pure PNIPAm-AuNPs were then dried and redissolved in 1.75 mL of H2O.
Synthesis of 5 nm AuNPs
AuNPs were synthesized via a seeded-growth method according to a modified literature procedure.38,42 A DDAB stock solution was first prepared by dissolving didecyldimethylammonium bromide (DDAB) (925 mg, 2.28 mmol) in toluene (20 mL). HAuCl4 (50 mg, 0.13 mmol) and dodecylamine (DDA) (450 mg, 2.42 mmol) were added to 12.5 mL of the stock solution and sonicated until dissolved. The gold salt was then reduced by dropwise addition of tetrabutylammonium borohydride (TBAB) (125 mg, 0.49 mmol) in 5 mL of DDAB stock solution under vigorous stirring, producing the (4 nm) seed solution, which was aged for 24 h. A growth solution was then prepared by adding 7 mL of the aged seed to a previously prepared solution containing toluene (50 mL) with HAuCl4 (200 mg, 0.51 mmol), DDAB (1 g, 2.46 mmol), and DDA (1.85 g, 7.19 mmol). Finally, hydrazine (131 μL, 4.22 mmol) in 20 mL of the DDAB stock solution was added dropwise under vigorous stirring, thus preparing a deep red solution of AuNPs (5 nm).
Functionalization of 5 nm Gold Nanoparticles with HS-PNIPAm-60k-OH
In order to successfully functionalize the synthesized AuNPs, it was necessary to remove excess capping ligands via a washing procedure. MeOH was added to the toluene AuNP solution in a 1:1 (v/v) ratio. After 2 h, precipitation of NPs allows for the removal of supernatant. The precipitate was redissolved in 1 volume of MeOH and allowed to stand until all NPs reprecipitated, the supernatant was removed, and the process was repeated once more. The resulting precipitate was easily dispersed in toluene/THF (1:1) to give a deep red solution of AuNPs. PNIPAm-60k-OH (100 mg, 1.7 μmol) was dissolved in toluene (5 mL) and THF (5 mL) and added to the cleaned 5 nm AuNPs. The solution was incubated for 48 h.
Purification of 5 nm PNIPAm-AuNPs
Diethyl ether (0.645 mL) was added to the functionalized AuNPs (1 mL) to precipitate the nanoparticles. The supernatant was removed and the AuNPs were redissolved in THF (0.5 mL). This process was repeated twice using fresh ether (0.8 mL), followed by drying. These were redissolved in Milli-Q water depending on the intended concentration. DLS measurements gave a diameter for these functionalized AuNPs of 156.0 nm.
Synthesis of Initiator-coated AuNPs
The initiator bis[2-(2-bromoisobutyryloxy)undecyl] disulfide was synthesized following a standard literature procedure.43 The as-synthesized initiator (30 mg, 0.04 mmol) was mixed with HAuCl4 (35.4 mg, 0.09 mmol) in ethanol (20 mL) and stirred vigorously at room temperature. NaBH4 (200 mg, 5.29 mmol) was dissolved in ethanol (20 mL), filtered, and then added dropwise to the gold salt solution. Once all of the NaBH4 solution had been added, the solution was stirred for a further hour at room temperature. The formed nanoparticles were precipitated with an excess of hexane and collected by centrifugation. These nanoparticles were washed extensively (over six times) by dissolving in the minimum amount of ethanol and precipitating with an excess of hexane.
Synthesis of Graft-from PNIPAm-AuNPs
A 5 mg amount of initiator–AuNPs was mixed with N-isopropylacrylamide (267 mg, 2.36 mmol) in 1 mL of ethanol/water (1:1) and degassed by bubbling with nitrogen for 30 min. Separately, CuCl (1.77 mg, 0.08 mmol) and tris[2-(dimethylamino)ethyl]amine (4.109 mg, 0.018 mmol) were dissolved in ethanol/water (1:1) and degassed by bubbling with nitrogen for 30 min. Once degassed, the copper solution was added to the nanoparticle/monomer solution. After 1 h the PNIPAm-AuNPs were washed several times using diethyl ether to precipitate the nanoparticles and centrifugation to collect them. Pure PNIPAm-AuNPs were then dried and dissolved in water.
UV–Vis Studies
A UV–vis spectroscopy method was developed for the analysis of PNIPAm-induced aggregation of AuNPs. In this method the PNIPAm-AuNPs are subjected to heating above the LCST (typically 45 °C) and are then centrifuged at 60 °C at low g using a Thermo Electron Corporation SPD131DDA SpeedVac concentrator. The supernant, after heated centrifugation, is then collected and analyzed. To study incremental increases in free PNIPAm concentrations, small amounts of PNIPAm were added to the supernant after analysis, and the solution was recombined with any precipitates generated via heated centrifugation. This mixture was then cooled on ice to fully dissolve all aggregates before another cycle of heated centrifugation and analysis was performed. As the method does not rely on the turbidity of the solution, as is typically used for such analysis, the actual degree of aggregation can be determined as opposed to the precipitation of free PNIPAm in solution and/or aggregation of AuNPs.
Acknowledgments
S.T.J. acknowledges support from an EPSRC NanoSci-E+CUBiHOLE grant (EP/H007024/1). S.J.B. thanks the European Commission for a Marie Curie Fellowship (NANOSPHERE, 658360). J.d.B. is grateful for a Marie Curie Intra-European Fellowship (PhotoTRAP 273807). We also acknowledge support from an ERC Starting Investigator Grant (ASPiRe, 240629) and a Next Generation Fellowship provided by the Walters-Kundert Foundation.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.5b04083.
1H NMR spectra, GPC measurements, and further information (PDF)
Additional data related to this publication is available at the University of Cambridge data repository (https://www.repository.cam.ac.uk/handle/1810/253658).
The authors declare no competing financial interest.
Supplementary Material
References
- Dai H.; Li X.; Long Y.; Wu J.; Liang S.; Zhang X.; Zhao N.; Xu J. Multi-Membrane Hydrogel Fabricated by Facile Dynamic Self-Assembly. Soft Matter 2009, 5, 1987–1989. 10.1039/b821363a. [DOI] [Google Scholar]
- Dai S.; Ravi P.; Tam K. C. pH-Responsive Polymers: Synthesis, Properties and Applications. Soft Matter 2008, 4, 435–449. 10.1039/b714741d. [DOI] [PubMed] [Google Scholar]
- Stuart M. A. C.; Huck W. T. S.; Genzer J.; Muller M.; Ober C.; Stamm M.; Sukhorukov G. B.; Szleifer I.; Tsukruk V. V.; Urban M.; Winnik F.; Zauscher S.; Luzinov I.; Minko S. Emerging Applications of Stimuli-Responsive Polymer Materials. Nat. Mater. 2010, 9, 101–113. 10.1038/nmat2614. [DOI] [PubMed] [Google Scholar]
- Kost J.; Langer R. Responsive Polymeric Delivery Systems. Adv. Drug Delivery Rev. 2001, 46, 125–148. 10.1016/S0169-409X(00)00136-8. [DOI] [PubMed] [Google Scholar]
- Stile R. A.; Burghardt W. R.; Healy K. E. Synthesis and Characterization of Injectable Poly(N-isopropylacrylamide)-Based Hydrogels That Support Tissue Formation in vitro. Macromolecules 1999, 32, 7370–7379. 10.1021/ma990130w. [DOI] [Google Scholar]
- Yoshida R.; Uchida K.; Kaneko Y.; Sakai K.; Kikuchi A.; Sakurai Y.; Okano T. Comb-Type Grafted Hydrogels with Rapid Deswelling Response to Temperature Changes. Nature 1995, 374, 240–242. 10.1038/374240a0. [DOI] [Google Scholar]
- Li C.; Tang Y.; Armes S. P.; Morris C. J.; Rose S. F.; Lloyd A. W.; Lewis A. L. Synthesis and Characterization of Biocompatible Thermo-Responsive Gelators Based on ABA Triblock Copolymers. Biomacromolecules 2005, 6, 994–999. 10.1021/bm049331k. [DOI] [PubMed] [Google Scholar]
- Chung J. E.; Yokoyama M.; Yamato M.; Aoyagi T.; Sakurai Y.; Okano T. Thermo-Responsive Drug Delivery from Polymeric Micelles Constructed using Block Copolymers of Poly(N-isopropylacrylamide) and Poly(butylmethacrylate). J. Controlled Release 1999, 62, 115–127. 10.1016/S0168-3659(99)00029-2. [DOI] [PubMed] [Google Scholar]
- Luo S.; Hu X.; Zhang Y.; Ling C.; Liu X.; Chen S. Synthesis of Thermoresponsive Unimolecular Polymeric Micelles with a Hydrophilic Hyperbranched Poly(glycidol) Core. Polym. J. 2011, 43, 41–50. 10.1038/pj.2010.93. [DOI] [Google Scholar]
- Cheng Y.; Hao J.; Lee L. A.; Biewer M. C.; Wang Q.; Stefan M. C. Thermally Controlled Release of Anticancer Drug from Self-Assembled γ-Substituted Amphiphilic Poly(ϵ-caprolactone) Micellar Nanoparticles. Biomacromolecules 2012, 13, 2163–2173. 10.1021/bm300823y. [DOI] [PubMed] [Google Scholar]
- Wei K.; Su L.; Chen G.; Jiang M. Does PNIPAM Block Really Retard the Micelle-to-Vesicle Transition of its Copolymer?. Polymer 2011, 52, 3647–3654. 10.1016/j.polymer.2011.06.005. [DOI] [Google Scholar]
- Otsuka I.; Fuchise K.; Halila S.; Fort S.; Aissou K.; Pignot-Paintrand I.; Chen Y.; Narumi A.; Kakuchi T.; Borsali R. Thermoresponsive Vesicular Morphologies Obtained by Self-Assemblies of Hybrid Oligosaccharide-Block-Poly(N-isopropylacrylamide) Copolymer Systems. Langmuir 2010, 26, 2325–2332. 10.1021/la902743y. [DOI] [PubMed] [Google Scholar]
- Oishi M.; Tamura A.; Nakamura T.; Nagasaki Y. A Smart Nanoprobe Based On Fluorescence-Quenching PEGylated Nanogels Containing Gold Nanoparticles for Monitoring the Response to Cancer Therapy. Adv. Funct. Mater. 2009, 19, 827–834. 10.1002/adfm.200801164. [DOI] [Google Scholar]
- Tang H.; Guo J.; Sun Y.; Chang B.; Ren Q.; Yang W. Facile synthesis of pH Sensitive Solymer-Coated Mesoporous Silica Nanoparticles and Their Application in Drug Delivery. Int. J. Pharm. 2011, 421, 388–396. 10.1016/j.ijpharm.2011.10.013. [DOI] [PubMed] [Google Scholar]
- Sundaresan V.; Menon J. U.; Rahimi M.; Nguyen K. T.; Wadajkar A. S. Dual-responsive Polymer-Coated Iron Oxide Nanoparticles for Drug Delivery and Imaging Applications. Int. J. Pharm. 2014, 466, 1–7. 10.1016/j.ijpharm.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild H. G. Poly(N-isopropylacrylamide): Experiment, Theory and Application. Prog. Polym. Sci. 1992, 17, 163–249. 10.1016/0079-6700(92)90023-R. [DOI] [Google Scholar]
- Lutz J.-F.; Hoth A. Preparation of Ideal PEG Analogues with a Tunable Thermosensitivity by Controlled Radical Copolymerization of 2-(2-Methoxyethoxy)ethyl Methacrylate and Oligo(ethylene glycol) Methacrylate. Macromolecules 2006, 39, 893–896. 10.1021/ma0517042. [DOI] [Google Scholar]
- Idziak I.; Avoce D.; Lessard D.; Gravel D.; Zhu X. X. Thermosensitivity of Aqueous Solutions of Poly(N,N-diethylacrylamide). Macromolecules 1999, 32, 1260–1263. 10.1021/ma981171f. [DOI] [Google Scholar]
- Fujishige S.; Kubota K.; Ando I. Phase Transition of Aqueous Solutions of Poly(N-isopropylacrylamide) and Poly(N-isopropylmethacrylamide). J. Phys. Chem. 1989, 93, 3311–3313. 10.1021/j100345a085. [DOI] [Google Scholar]
- Appel E. A.; del Barrio J.; Loh X. J.; Dyson J.; Scherman O. A. High Molecular Weight Polyacrylamides by Atom Transfer Radical Polymerization: Enabling Advancements in Water-Based Applications. J. Polym. Sci., Part A: Polym. Chem. 2012, 50, 181–186. 10.1002/pola.25041. [DOI] [Google Scholar]
- Budhlall B. M.; Marquez M.; Velev O. D. Microwave, Photo- and Thermally Responsive PNIPAm-Gold Nanoparticle Microgels. Langmuir 2008, 24, 11959–11966. 10.1021/la8019556. [DOI] [PubMed] [Google Scholar]
- Salmaso S.; Caliceti P.; Amendola V.; Meneghetti M.; Magnusson J. P.; Pasparakis G.; Alexander C. Cell Up-Take Control of Gold Nanoparticles Functionalized with a Thermoresponsive Polymer. J. Mater. Chem. 2009, 19, 1608–1615. 10.1039/b816603j. [DOI] [Google Scholar]
- Carregal-Romero S.; Buurma N. J.; Pérez-Juste J.; Liz-Marzán L. M.; Hervés P. Catalysis by Au@pNIPAM Nanocomposites: Effect of the Cross-Linking Density. Chem. Mater. 2010, 22, 3051–3059. 10.1021/cm903261b. [DOI] [Google Scholar]
- Huang X.; Jain P.; El-Sayed I.; El-Sayed M. Plasmonic Photothermal Therapy (PPTT) using Gold Nanoparticles. Lasers Med. Sci. 2008, 23, 217–228. 10.1007/s10103-007-0470-x. [DOI] [PubMed] [Google Scholar]
- Niikura K.; Iyo N.; Matsuo Y.; Mitomo H.; Ijiro K. Sub-100 nm Gold Nanoparticle Vesicles as a Drug Delivery Carrier enabling Rapid Drug Release upon Light Irradiation. ACS Appl. Mater. Interfaces 2013, 5, 3900–3907. 10.1021/am400590m. [DOI] [PubMed] [Google Scholar]
- Mastrotto F.; Caliceti P.; Amendola V.; Bersani S.; Magnusson J. P.; Meneghetti M.; Mantovani G.; Alexander C.; Salmaso S. Polymer Control of Ligand Display on Gold Nanoparticles for Multimodal Switchable Cell Targeting. Chem. Commun. 2011, 47, 9846–9848. 10.1039/c1cc12654g. [DOI] [PubMed] [Google Scholar]
- Shan J.; Nuopponen M.; Jiang H.; Kauppinen E.; Tenhu H. Preparation of Poly(N-isopropylacrylamide)-Monolayer-Protected Gold Clusters-Synthesis Methods, Core Size, and Thickness of Monolayer. Macromolecules 2003, 36, 4526–4533. 10.1021/ma034265k. [DOI] [Google Scholar]
- Kim D. J.; Kang S. M.; Kong B.; Kim W.-J.; Paik H.-j.; Choi H.; Choi I. S. Formation of Thermoresponsive Gold Nanoparticle/PNIPAAm Hybrids by Surface-Initiated, Atom Transfer Radical Polymerization in Aqueous Media. Macromol. Chem. Phys. 2005, 206, 1941–1946. 10.1002/macp.200500268. [DOI] [Google Scholar]
- Raula J.; Shan J.; Nuopponen M.; Niskanen A.; Jiang H.; Kauppinen E. I.; Tenhu H. Synthesis of Gold Nanoparticles Grafted with a Thermoresponsive Polymer by Surface-Induced Reversible-Addition-Fragmentation Chain-Transfer Polymerization. Langmuir 2003, 19, 3499–3504. 10.1021/la026872r. [DOI] [Google Scholar]
- Chakraborty S.; Bishnoi S. W.; Pérez-Luna V. H. Gold Nanoparticles with Poly(N-isopropylacrylamide) Formed via Surface Initiated Atom Transfer Free Radical Polymerization Exhibit Unusually Slow Aggregation Kinetics. J. Phys. Chem. C 2010, 114, 5947–5955. 10.1021/jp910417g. [DOI] [Google Scholar]
- Zhu M.-Q.; Wang L.-Q.; Exarhos G. J.; Li A. D. Q. Thermosensitive Gold Nanoparticles. J. Am. Chem. Soc. 2004, 126, 2656–2657. 10.1021/ja038544z. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Zhu X.; Jia F.; Auyeung E.; Mirkin C. A. Temperature-Activated Nucleic Acid Nanostructures. J. Am. Chem. Soc. 2013, 135, 14102–14105. 10.1021/ja408465t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B.; Brittain W. J. Polymer Brushes: Surface-Immobilized Macromolecules. Prog. Polym. Sci. 2000, 25, 677–710. 10.1016/S0079-6700(00)00012-5. [DOI] [Google Scholar]
- Yusa S.-i.; Fukuda K.; Yamamoto T.; Iwasaki Y.; Watanabe A.; Akiyoshi K.; Morishima Y. Salt Effect on the Heat-Induced Association Behavior of Gold Nanoparticles Coated with Poly(N-isopropylacrylamide) Prepared via Reversible Addition-Fragmentation Chain Transfer (RAFT) Radical Polymerization. Langmuir 2007, 23, 12842–12848. 10.1021/la702741q. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Maji S.; da Fonseca Antunes A. B.; Rycke R. D.; Zhang Q.; Hoogenboom R.; Geest B. G. D. Salt Plays a Pivotal Role in the Temperature-Responsive Aggregation and Layer-by-Layer Assembly of Polymer-Decorated Gold Nanoparticles. Chem. Mater. 2013, 25, 4297–4303. 10.1021/cm402414u. [DOI] [Google Scholar]
- Gibson M. I.; O’Reilly R. K. To Aggregate, or not to Aggregate? Considerations in the Design and Application of Polymeric Thermally-Responsive Nanoparticles. Chem. Soc. Rev. 2013, 42, 7204–7213. 10.1039/C3CS60035A. [DOI] [PubMed] [Google Scholar]
- Turkevich J.; Stevenson P. C.; Hillier J. A Study of the Nucleations and Growth Processes in the Synthesis of Colloidal Gold. Discuss. Faraday Soc. 1951, 11, 55–75. 10.1039/df9511100055. [DOI] [Google Scholar]
- Klajn R.; Olson M. A.; Wesson P. J.; Fang L.; Coskun A.; Trabolsi A.; Soh S.; Stoddart J. F.; Grzybowski B. A. Dynamic Hook-and-Eye Nanoparticle Sponges. Nat. Chem. 2009, 1, 733–738. 10.1038/nchem.432. [DOI] [PubMed] [Google Scholar]
- Kimling J.; Maier M.; Okenve B.; Kotaidis V.; Ballot H.; Plech A. Turkevich Method for Gold Nanoparticle Synthesis Revisited. J. Phys. Chem. B 2006, 110, 15700–15707. 10.1021/jp061667w. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Fernández J.; Pérez-Juste J.; de Abajo F. J.; Liz-Marzán L. M. Seeded Growth of Submicron Au Colloids with Quadrupole Plasmon Resonance Modes. Langmuir 2006, 22, 7007–7010. 10.1021/la060990n. [DOI] [PubMed] [Google Scholar]
- Biedermann F.; Appel E. A.; del Barrio J.; Gruendling T.; Barner-Kowollik C.; Scherman O. A. Postpolymerization Modification of Hydroxyl-Functionalized Polymers with Isocyanates. Macromolecules 2011, 44, 4828–4835. 10.1021/ma2008018. [DOI] [Google Scholar]
- Jana N. R.; Peng X. Single-Phase and Gram-Scale Routes toward Nearly Monodisperse Au and Other Noble Metal Nanocrystals. J. Am. Chem. Soc. 2003, 125, 14280–14281. 10.1021/ja038219b. [DOI] [PubMed] [Google Scholar]
- Shah R. R.; Merreceyes D.; Husemann M.; Rees I.; Abbott N. L.; Hawker C. J.; Hedrick J. L. Using Atom Transfer Radical Polymerization To Amplify Monolayers of Initiators Patterned by Microcontact Printing into Polymer Brushes for Pattern Transfer. Macromolecules 2000, 33, 597–605. 10.1021/ma991264c. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.