

Abstract
Nevoid basal cell carcinoma syndrome, or basal cell nevus syndrome (Gorlin syndrome), is a rare autosomal dominantly inherited disorder that is characterized by development of basal cell carcinomas from a young age. Other distinguishing clinical features are seen in a majority of patients, and include keratocystic odontogenic tumors (formerly odontogenic keratocysts) as well as dyskeratotic palmar and plantar pitting. A range of skeletal and other developmental abnormalities are also often seen. The disorder is caused by defects in hedgehog signaling which result in constitutive pathway activity and tumor cell proliferation. As sporadic basal cell carcinomas also commonly harbor hedgehog pathway aberrations, therapeutic agents targeting key signaling constituents have been developed and tested against advanced sporadically occurring tumors or syndromic disease, leading in 2013 to FDA approval of the first hedgehog pathway-targeted small molecule, vismodegib. The elucidation of the molecular pathogenesis of nevoid basal cell carcinoma syndrome has resulted in further understanding of the most common human malignancy.
Keywords: Nevoid basal cell carcinoma syndrome, Basal cell nevus syndrome, Gorlin syndrome, Keratocystic odontogenic tumor, PTCH1, SMO, SUFU, Vismodegib
Introduction
Nevoid basal cell carcinoma syndrome, or basal cell nevus syndrome (NBCCS; Gorlin syndrome) was purportedly first described in the literature in 1894. However, it was not until the mid-twentieth century when NBCCS was recognized as a distinct entity [1, 2]. The linking of patients with multiple basal cell carcinomas (BCCs), keratocystic odontogenic tumors (KOTs, formerly odontogenic keratocysts), palmar and plantar pitting, and a range of skeletal and developmental abnormalities laid the groundwork for establishing the genetic basis of NBCCS and targeted therapeutics exploiting knowledge of its pathogenesis.
Discussion
Clinical Features
NBCCS is a rare autosomal dominantly inherited entity that is characterized most strikingly by development of cutaneous BCCs from an early age—typically puberty—although in some cases occurring earlier in childhood [2]. The estimated prevalence is 1 in 57,000 to 1 in 164,000 [3] and no sex predilection has been observed [4]. BCCs encountered by an affected individual may number up to 500 in a lifetime and are often clinically aggressive, occurring more frequently in lighter pigmented individuals and in populations with higher ultraviolet (UV) light exposure [3, 5]. BCCs most commonly develop on the face, but also appear on the trunk and limbs [3].
Additional well-recognized clinical features are often present, occurring in 70–80 % of patients, and include KOTs (Figs. 1, 2), dyskeratotic palmar and plantar pitting (Fig. 3), rib and spine abnormalities, and early calcification of the falx cerebri. Characteristic facial features such as frontal bossing, hypertelorism, macrocephaly, and cleft lip and/or palate are also present in a significant fraction of patients [3]. Although occurring less commonly, patients are also susceptible to desmoplastic medulloblastoma during childhood, as well as numerous other neoplasms including ovarian and cardiac fibromas, mesenteric keratocysts, rhabdomyosarcomas, and meningiomas [3, 6]. Agenesis of the corpus callosum has also been reported [2] and additional less frequent clinical abnormalities are still being described. While many cases of NBCCS are inherited, sporadic occurrences are thought to account for up to 50 % of cases [7]. Although the disease is characterized by high penetrance, individuals display varying levels of expressivity even within families, sometimes making diagnosis challenging [5].
Fig. 1.
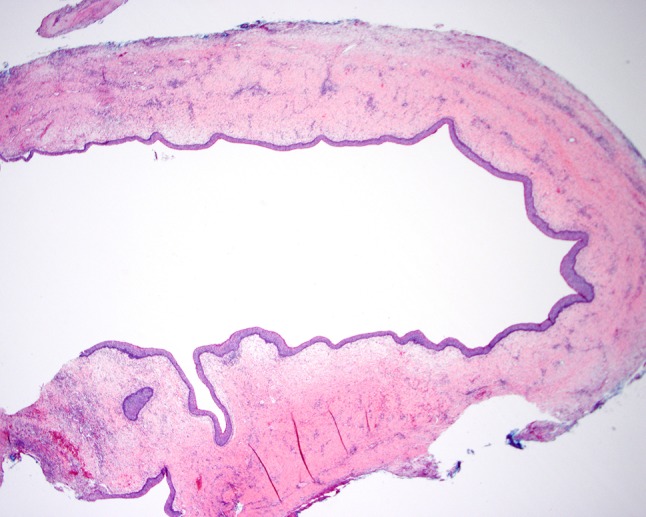
This unilocular keratocystic odontogenic tumor is lined by uniform layer of squamous cells with no rete ridges (H&E, original magnification ×20). Image courtesy of Sook-Bin Woo, D.M.D., M.M.Sc., Director of Clinical Affairs, Division of Oral Maxillofacial Surgery, Brigham and Women’s Hospital
Fig. 2.

At higher power, this keratocystic odontogenic tumor exhibits basal and parabasal hyperplasia with nuclear palisading and overlying parakeratosis (H&E, original magnification ×200). Basal and parabasal cell hyperplasia should not be mistaken for dysplasia. Image courtesy of Sook-Bin Woo, D.M.D., M.M.Sc., Director of Clinical Affairs, Division of Oral Maxillofacial Surgery, Brigham and Women’s Hospital
Fig. 3.
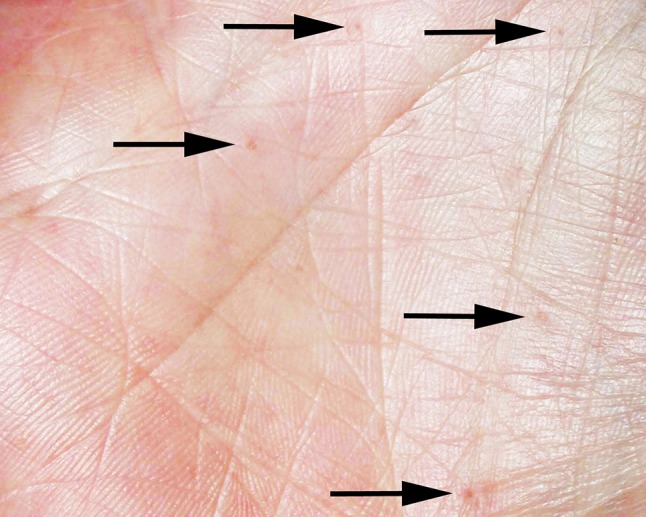
Dyskeratotic palmar pitting (arrows)
Molecular Pathogenesis
Molecular alterations in components of the highly conserved hedgehog signaling pathway have been implicated in the pathogenesis of NBCCS and result in constitutive signaling activity, with most mutations occurring as loss of function mutations in the patched (PTCH1) gene located on chromosome 9q22.3 [6, 8–10]. Associated developmental deficits and malignancies are thought to develop via a two-hit mechanism [11].
Under inactive signaling conditions, PTCH1, a membrane-bound protein, maintains smoothened (SMO) in its inactive/unphosphorylated state, leaving it susceptible to endocytosis and degradation, incapable of activating GLI proteins, which are transcription factors necessary for activation (or repression) of pathway-dependent genes [12, 13]. Additionally, the protein suppressor of fused (SUFU, encoded by the SUFU gene), part of a corepressor complex, provides additional negative regulation of GLI factors through direct binding [14]. When a hedgehog ligand (members include the proteins Indian hedgehog, desert hedgehog, and the most common sonic hedgehog (SHH) [13]) binds to PTCH1, SMO becomes hyperphosphorylated and in turn allows GLI to relocalize to the nucleus. This results in transcriptional modulation and various downstream effects, including, under normal circumstances, proliferation, migration, and differentiation of progenitor cells during nearly every stage of vertebrate development [13]. Under pathogenic circumstances, constitutive activation of the hedgehog signaling pathway due to mutation of critical regulatory proteins leads to tumor cell proliferation. Loss of function mutations in PTCH1, which occur by a variety of mechanisms such as deletions, insertions, and nonsense and missense mutations [7], are thought to occur in ~70 % of patients who fulfill diagnostic criteria for NBCCS [15, 16]. In addition, loss of function SUFU germline mutations have also been recently associated with NBCCS in a subset of cases [16]. Interestingly, NBCCS patients with SUFU mutations, although they appear to have a lower incidence of major NBCCS criteria including KOTs, may have an increased risk for desmoplastic medulloblastoma [16].
Sporadic BCC is by far the most common cancer in fair-skinned individuals and many lesions require excision. Of the approximately 3,500,000 non-melanoma skin cancers (NMSCs) that occurred in the United States in 2006, BCC (commonly thought to represent ~80 % of NMSCs) was the most common, followed by squamous cell carcinoma, accounting for the bulk of the remainder of NMSCs [17]. As in cases of NBCCS, BCCs thought to arise due to UV exposure in non-NBCCS patients also frequently harbor aberrations in the hedgehog signaling pathway, most frequently with loss of function mutations in PTCH1 (~90 %). Consistent with these observations, nearly 50 % of sporadic KOTs harbor mutations in PTCH1 [18], as well as a subset of sporadic desmoplastic medulloblastomas [19]. The remaining sporadic BCC patients (~10 %) predominantly exhibit gain of function mutations in SMO [20], aberrations that to our knowledge have not yet been described in NBCCS. In addition, SUFU mutations are known to occur occasionally in sporadic BCCs [21].
Diagnostic Criteria and Differential Diagnosis
Diagnostic criteria for NBCCS were proposed nearly 20 years ago as the result of multiple large clinical case series and include several discrepancies, likely reflecting the variable expressivity of the syndrome [3, 22]. These criteria were further refined more recently, although a final consensus has not yet been reached [23]. The diagnosis of NBCCS requires the presence of either two major criteria or one major and two minor criteria (Table 1); a third satisfactory condition of one major criterion with molecular confirmation has also recently been proposed [23]. A patient suspected of having NBCCS should undergo a full dermatologic exam as well as an initial panel of radiologic tests including a baseline brain MRI, cardiac ultrasound, pelvic ultrasound (if female), a spine radiograph, and a panoramic dental radiograph [23].
Table 1.
Diagnostic criteria for nevoid basal cell carcinoma syndrome (adapted from [23])
| Major criteria |
| 1. Basal cell carcinoma before 20 years of age or excessive numbers of basal cell carcinomas out of proportion to prior sun exposure and skin type |
| 2. Keratocystic odontogenic tumor before 20 years of age |
| 3. Palmar or plantar pitting |
| 4. Lamellar calcification of the falx cerebri |
| 5. Medulloblastoma, typically desmoplastic |
| 6. First degree relative with nevoid basal cell carcinoma syndrome |
| Minor criteria |
| 1. Rib abnormalities |
| 2. Other specific skeletal malformations and radiologic changes (i.e., vertebral anomalies, kyphoscoliosis, short fourth metacarpals, postaxial polydactyly) |
| 3. Macrocephaly |
| 4. Cleft lip or palate |
| 5. Ovarian or cardiac fibroma |
| 6. Lymphomesenteric cysts |
| 7. Ocular abnormalities (i.e., strabismus, hypertelorism, congenital cataracts, glaucoma, coloboma) |
The presence of multiple BCCs at an early age may also be seen in a variety of rare inherited disorders that require consideration at clinical presentation (Table 2). Bazex-Dupré-Christol syndrome (BDCS) is an X-linked dominantly inherited condition that results in multiple BCCs, typically with onset in the second decade of life. Dysgenesis of hair follicles resulting in hypotrichosis and follicular atrophoderma primarily occurring on the dorsum of the hands are also often seen in this disorder [24, 25]. These patients are thought to only exhibit cutaneous pathology with no skeletal abnormalities or predisposition to neoplasms of other organ systems, and approximately 20 cases have been reported in the literature [24]. The molecular pathogenesis of BDCS has not yet been elucidated, although the causative gene has been mapped to an 11.4 Mb region of chromosome Xq25-q27.1 [26]. Rombo syndrome is characterized by development of BCCs in the fourth decade of life, with follicular atrophoderma occurring on the elbows and cheeks as well as generalized cyanotic erythema, and is thought to occur in an autosomal dominant manner of inheritance, although the causative molecular aberration is yet to be determined [24]. Xeroderma pigmentosum (XP), an autosomal recessive disorder resulting from defects in DNA damage repair, predisposes to a range of cutaneous and ocular neoplasms including BCCs, squamous cell carcinomas (SCCs), and melanomas, as well as various malignancies of internal organs [24]. In addition to these rare genetic syndromes, multiple BCCs in a young person may develop largely due to environmental factors, such as excessive UV light exposure from the heavy use of indoor tanning [27].
Table 2.
Differential diagnosis for multiple basal cell carcinomas occurring at an early age
| Disorder | Additional clinical features | Causative gene(s) Manner of inheritance |
|---|---|---|
| Nevoid basal cell carcinoma syndrome | Keratocystic odontogenic tumors, palmar and plantar pitting, skeletal abnormalities, calcification of the falx cerebri, extracutaneous tumors (e.g. desmoplastic medulloblastoma, ovarian or cardiac fibroma) |
PTCH1, SUFU
Autosomal dominant |
| Bazex-Dupré-Christol syndrome | Hypotrichosis, follicular atrophoderma primarily occurring on the dorsum of the hands (no skeletal or extracutaneous developmental abnormalities) | Unknown, within Xq25-q27.1 X-linked dominant |
| Rombo syndrome | Follicular atrophoderma occurring on the elbows and cheeks, generalized cyanotic erythema | Unknown Autosomal dominant |
| Xeroderma pigmentosum | Freckle-like hyperpigmentation, squamous cell carcinoma, melanoma, malignancies of internal organs, neurologic abnormalities | Nuclear excision repair enzymes Autosomal recessive |
| Excess UV exposure | Skin neoplasia only without features of a heritable syndrome | UV signature mutations (pyrimidine dimers) N/A |
Histopathology
Clinically, BCCs typically appear as pearly papules, often with telangiectasias, occurring on sun exposed sites. Lesions can become ulcerated if left untreated, and can be locally invasive, with metastatic disease occurring rarely. BCCs developing in NBCCS-affected individuals are histologically indistinguishable from sporadically occurring lesions and are thought to begin as ‘buds’ of neoplastic basaloid cells protruding down from the epidermis. A histologic clue that is suggestive of, but probably not specific for, NBCCS is the presence of multiple incidental minute buds of early superficial BCC in otherwise unremarkable skin of an excision for BCC. BCCs in NBCCS exhibit a range of histologic patterns, including nodular, superficial, and infiltrative types. Lesions often exhibit more than one histologic pattern; however, the nodular subtype is the most common, characterized by well-circumscribed collections of basaloid epithelial cells with peripheral palisading, retraction artifact, and frequent mitoses.
KOTs typically occur in the posterior mandible and are recognized radiographically by an area of lucency often associated with the crown of an unerupted tooth. Greater than 80 % are unilocular; however, they can also be multiloculated. Within the cystic space of the tumor is soft keratinaceous debris. Histologically, lesions exhibit a uniformly thick squamous epithelial lining (typically 5–10 cells in thickness; Figs. 1, 2) with parakeratosis and basal/parabasal cell palisading (Fig. 2), in contrast to other odontogenic cysts, which are orthokeratinized, do not display parakeratosis and basal/parabasal cell palisading, and have a low recurrence risk [28]. Inflammation is often present and can sometimes obscure diagnostic features. Satellite tumors are often seen and should be documented, since some authors have suggested that their presence may be associated with higher risk of recurrence, which for KOTs overall has been estimated to be 10–40 % [28]. Basal and parabasal cell hyperplasia is also often present (Fig. 2), and should not be mistaken for squamous dysplasia. Lesions often exhibit a high Ki67 proliferation index with p53 positivity of the basal and suprabasal cell layers by immunohistochemistry [29], and display loss of heterozygosity at several loci (including at TP53) [30]. Due to these reasons, in addition to a better understanding of the molecular pathogenesis, this entity has been re-classified as a neoplasm rather than a developmental phenomenon. As with BCCs, KOTs occurring in patients with NBCCS are histologically indistinguishable from those occurring sporadically.
Screening and Treatment
Frequent dermatologic surveillance is necessary for individuals with established NBCCS and sun protection is an important preventative care measure [24]. Radiologic screening for medulloblastomas (in patients younger than 8 years) and KOTs is typically performed annually [23, 31]. Therapeutic options for BCCs depend on the histologic subtype, size, and location of the tumor. Due to the large tumor burden often encountered, the number of excisions performed on an individual with NBCCS may be extensive, and significant disfigurement can occur. Conventional therapies other than surgical management for localized disease include topical application of various agents such as 5-fluorouracil (5-FU) and imiquimod. Radiotherapy is contraindicated in patients with NBCCS.
As detailed knowledge regarding the molecular pathogenesis of BCC has been elucidated, multiple targeted therapies have been developed to target aberrant HH signaling. Vismodegib, a small molecule agent that binds to and directly inhibits SMO was recently FDA approved for recurrent, locally advanced, or metastatic basal cell carcinoma [32]. Targeted therapy has also been attempted in NBCCS; for example, a phase II study examining the efficacy of vismodegib in NBCCS found a significant reduction in surgically eligible BCCs encountered in the treatment group (2 per patient) versus the control group (29 per patient). However, more than half of those randomized to vismodegib discontinued the drug due to adverse events including hair loss, weight loss, and muscle cramps [33]. In addition to issues with these effects, many vismodegib-treated sporadic BCCs (but not syndromic BCCs) go on to develop treatment resistance, with alterations in the inhibitor binding pocket of SMO occurring in the majority of cases with a functionally relevant point mutation [34]. As a result, second-generation inhibitors that are active against wild type as well as mutant SMO, or a multi-drug regimen that targets multiple proteins in the pathway, are needed to combat this emerging problem.
Conclusion
Although uncommon, NBCCS has provided critical genetic clues in our understanding of the most common human malignancy. Understanding of the underlying pathogenesis has led to greater knowledge of the HH signaling pathway and the development of targeted therapeutics that have considerable promise in the treatment of BCC. Additionally, the development of HH-targeted agents has served as a model for use of targeted therapy in the treatment of human malignancy in general.
References
- 1.Howell B, Caro M. The basal-cell nevus. Arch Dermatol. 1959;79:67–80. doi: 10.1001/archderm.1959.01560130069008. [DOI] [PubMed] [Google Scholar]
- 2.Gorlin R, Goltz R. Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib. A syndrome. N Engl J Med. 1960;5:908–912. doi: 10.1056/NEJM196005052621803. [DOI] [PubMed] [Google Scholar]
- 3.Kimonis VE, Goldstein AM, Pastakia B, Yang ML, Kase R, Digiovanna JJ, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69:299–308. doi: 10.1002/(SICI)1096-8628(19970331)69:3<299::AID-AJMG16>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 4.Anderson DE, Taylor WB, Falls HF, Davidson RT. The nevoid basal cell carcinoma syndrome. Am J Hum Genet. 1967;19:12–22. [PMC free article] [PubMed] [Google Scholar]
- 5.Shanley S, Ratcliffe J, Hockey A, Haan E, Oley C, Ravine D, et al. Nevoid basal cell carcinoma syndrome: review of 118 affected individuals. Am J Med Genet. 1994;50:282–290. doi: 10.1002/ajmg.1320500312. [DOI] [PubMed] [Google Scholar]
- 6.Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 7.Klein RD, Dykas DJ, Bale AE. Clinical testing for the nevoid basal cell carcinoma syndrome in a DNA diagnostic laboratory. Genet Med. 2005;7:611–619. doi: 10.1097/01.gim.0000182879.57182.b4. [DOI] [PubMed] [Google Scholar]
- 8.Gailani MR, Ståhle-Bäckdahl M, Leffell DJ, Glynn M, Zaphiropoulos PG, Pressman C, et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet. 1996;14:78–81. doi: 10.1038/ng0996-78. [DOI] [PubMed] [Google Scholar]
- 9.Gailani MR, Bale SJ, Leffell DJ, DiGiovanna JJ, Peck GL, Poliak S, et al. Developmental defects in Gorlin syndrome related to a putative tumor suppressor gene on chromosome 9. Cell. 1992;69:111–117. doi: 10.1016/0092-8674(92)90122-S. [DOI] [PubMed] [Google Scholar]
- 10.Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–851. doi: 10.1016/S0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 11.Levanat S, Gorlin RJ, Fallet S, Johnson DR, Fantasia JE, Bale AE. A two hit model for developmental defects in Gorlin syndrome. Nat Genet. 1996;14:353–356. doi: 10.1038/ng1196-353. [DOI] [PubMed] [Google Scholar]
- 12.Atwood SX, Chang ALS, Oro AE. Hedgehog pathway inhibition and the race against tumor evolution. J Cell Biol. 2012;199:193–197. doi: 10.1083/jcb.201207140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22:2454–2472. doi: 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 14.Cherry AL, Finta C, Karlström M, Jin Q, Schwend T, Astorga-Wells J, et al. Structural basis of SUFU-GLI interaction in human Hedgehog signalling regulation. Acta Crystallogr Sect D Biol Crystallogr. 2013;69:2563–2579. doi: 10.1107/S0907444913028473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soufir N, Gerard B, Portela M, Brice A, Liboutet M, Saiag P, et al. PTCH mutations and deletions in patients with typical nevoid basal cell carcinoma syndrome and in patients with a suspected genetic predisposition to basal cell carcinoma: a French study. Br J Cancer. 2006;95:548–553. doi: 10.1038/sj.bjc.6603303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith MJ, Beetz C, Williams SG, Bhaskar SS, O’Sullivan J, Anderson B, et al. Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated With PTCH1 mutations. J Clin Oncol. 2014;32:4155–4161. doi: 10.1200/JCO.2014.58.2569. [DOI] [PubMed] [Google Scholar]
- 17.Rogers HW, Weinstock MA, Harris AR, Hinckley MR, Feldman SR, Fleischer AB, et al. Incidence estimate of nonmelanoma skin cancer in the United States. Arch Dermatol. 2006;2010(146):283–287. doi: 10.1001/archdermatol.2010.19. [DOI] [PubMed] [Google Scholar]
- 18.Guo YY, Zhang JY, Li XF, Luo HY, Chen F, Li TJ. PTCH1 Gene mutations in keratocystic odontogenic tumors: a study of 43 Chinese patients and a systematic review. PLoS One. 2013;8:1–9. doi: 10.1371/journal.pone.0077305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pietsch T, Waha A, Koch A, Kraus J, Albrecht S, Tonn J, et al. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res. 1997;57:2085–2088. [PubMed] [Google Scholar]
- 20.Epstein EH. Basal cell carcinomas: attack of the hedgehog. Nat Rev Cancer. 2008;8:743–754. doi: 10.1038/nrc2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reifenberger J, Wolter M, Knobbe CB, Köhler B, Schönicke A, Scharwächter C, et al. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br J Dermatol. 2005;152:43–51. doi: 10.1111/j.1365-2133.2005.06353.x. [DOI] [PubMed] [Google Scholar]
- 22.Evans DG, Ladusans EJ, Rimmer S, Burnell LD, Thakker N, Farndon PA. Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J Med Genet. 1993;30:460–464. doi: 10.1136/jmg.30.6.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bree AF. Shah MR for the BCNS Colloquium Group. Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS) Am J Med Genet Part A. 2011;155:2091–7. doi: 10.1002/ajmg.a.34128. [DOI] [PubMed] [Google Scholar]
- 24.Parren LJMT, Frank J. Hereditary tumour syndromes featuring basal cell carcinomas. Br J Dermatol. 2011;165:30–34. doi: 10.1111/j.1365-2133.2011.10334.x. [DOI] [PubMed] [Google Scholar]
- 25.Abuzahra F, Parren LJ, Frank J. Multiple familial and pigmented basal cell carcinomas in early childhood - Bazex-Dupre-Christol syndrome. J Eur Acad Dermatol Venereol. 2012;26:117–121. doi: 10.1111/j.1468-3083.2011.04048.x. [DOI] [PubMed] [Google Scholar]
- 26.Parren LJMT, Abuzahra F, Wagenvoort T, Koene F, Van Steensel MAM, Steijlen PM, et al. Linkage refinement of Bazex-Dupré-Christol syndrome to an 11·4-Mb interval on chromosome Xq25-27.1. Br J Dermatol. 2011;165:201–203. doi: 10.1111/j.1365-2133.2011.10219.x. [DOI] [PubMed] [Google Scholar]
- 27.Karagas MR, Zens MS, Li Z, Stukel TA, Perry AE, Gilbert-Diamond D, et al. Early-onset basal cell carcinoma and indoor tanning: a population-based study. Pediatrics. 2014;134:e4–e12. doi: 10.1542/peds.2013-3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woo S-B. Oral pathology: a comprehensive atlas and text. Philadelphia: Elsevier Saunders; 2012. [Google Scholar]
- 29.Gurgel CAS, Ramos EAG, Azevedo RA, Sarmento VA, da Silva Carvalho AM, dos Santos JN. Expression of Ki-67, p53 and p63 proteins in keratocyst odontogenic tumours: an immunohistochemical study. J Mol Histol. 2008;39:311–316. doi: 10.1007/s10735-008-9167-0. [DOI] [PubMed] [Google Scholar]
- 30.Henley J, Summerlin D-J, Tomich C, Zhang S, Cheng L. Molecular evidence supporting the neoplastic nature of odontogenic keratocyst: a laser capture microdissection study of 15 cases. Histopathology. 2005;47:582–586. doi: 10.1111/j.1365-2559.2005.02267.x. [DOI] [PubMed] [Google Scholar]
- 31.John AM, Schwartz RA. Basal cell nevus syndrome: an update on Genetics and treatment. Br J Dermatol 2015; Sept. 26, Epub ahead of print. [DOI] [PubMed]
- 32.Axelson M, Liu K, Jiang X, He K, Wang J, Zhao H, et al. U.S. Food and Drug Administration approval: vismodegib for recurrent, locally advanced, or metastatic basal cell carcinoma. Clin Cancer Res. 2013;19:2289–2293. doi: 10.1158/1078-0432.CCR-12-1956. [DOI] [PubMed] [Google Scholar]
- 33.Tang JY, Mackay-Wiggan JM, Aszterbaum M, Yauch RL, Lindgren J, Chang K, et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N Engl J Med. 2012;366:2180–2188. doi: 10.1056/NEJMoa1113538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342–53. doi: 10.1016/j.ccell.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]