

Abstract
Background
Tobacco-smoke is the major etiological factor related to lung cancer. However, other important factor is chronic wood smoke exposure (WSE). Approximately 30 % of lung cancer patients in Mexico have a history of WSE, and present different clinical, pathological and molecular characteristics compared to tobacco related lung cancer, including differences in mutational profiles. There are several molecular alterations identified in WSE associated lung cancer, however most studies have focused on the analysis of changes in several pathogenesis related proteins.
Methods
Our group evaluated gene expression profiles of primary lung adenocarcinoma, from patients with history of WSE or tobacco exposure. Differential expression between these two groups were studied through gene expression microarrays.
Results
Results of the gene expression profiling revealed 57 statistically significant genes (p < 0.01). The associated biological functional pathways included: lipid metabolism, biochemistry of small molecules, molecular transport, cell morphology, function and maintenance. A highlight of our analysis is that three of the main functional networks represent 37 differentially expressed genes out of the 57 found. These hubs are related with ubiquitin C, GABA(A) receptor-associated like protein; and the PI3K/AKT and MEK/ERK signaling pathways.
Conclusion
Our results reflect the intrinsic biology that sustains the development of adenocarcinoma related to WSE and show that there is a different gene expression profile of WSE associated lung adenocarcinoma compared to tobacco exposure, suggesting that they arise through different carcinogenic mechanisms, which may explain the clinical and mutation profile divergences between both lung adenocarcinomas.
Keywords: Non-small cell lung carcinoma , Wood smoke exposure, Gene expression profiles, Tobacco smoke, Microarray analysis
Background
Lung cancer is the first cause of death from cancer [1]. Approximately 85 % of diagnosed patients present Non-Small Cell Lung Cancer (NSCLC), and adenocarcinoma is the most frequent histological type. Despite efforts, innovations, and progress in diagnosis and treatment, 5-year overall survival is approximately 15 % with high mortality rates [2]. Tobacco smoking is the main risk of lung cancer. Other factors include pulmonary tuberculosis, genetic susceptibility, exposure to secondhand smoke, asbestos and radon [3]. In Mexico, the crude mortality rate of lung cancer is 6.68 per 105 individuals, representing nearly 9000 cases per year, most of them presenting metastatic stage at diagnosis [4, 5].
Nowadays about 15 % of lung cancer in men and 53 % in women is not associated to smoking [6]. Besides, due to the impact of tobacco control policies, a bigger percentage of non-smoking patients with lung cancer is expected in the following years. According to cancer statistics from the USA, lung cancer death rates declined 36 %, from 1990 to 2011, among males and 11 %, between 2002 and 2011, among females due to reduced tobacco use as a result of increased awareness of the health hazards of smoking and the implementation of comprehensive tobacco control [2]. There have been reports of a doubling in the annual incidence of lung cancer in never smokers, identifying as well that non-smoker NSCLC patients tend to be female and young [7, 8]. Regarding mortality, never smokers present lung cancer death rates greater in men than in women and a large fraction of cases have no identified risk factors [9]. Meanwhile former smokers present an increased risk of lung cancer but cumulative risk decreases with earlier smoking cessation compared to smokers who continue smoking [10].
Chronic wood smoke exposure (WSE) is related to obstructive pulmonary disease in developing, European and American countries [11, 12]. Wood dust has also been identified as a human carcinogen and a risk factor for lung cancer [3, 13]. Wood byproducts such as benzene, 1-butadiene, formaldehyde and acetaldehyde, are well-known carcinogens [14]. For more than 50 years, WSE has been associated with an increased risk of lung cancer as compared with pulmonary tuberculosis, interstitial lung disease and various pulmonary conditions (OR: 1.9; 95 % confidence interval (CI): 1.1–3.5) after adjusting for age, education, socioeconomic status and tobacco smoke exposure [13]. In Mexico, approximately 16 % of the population has long-term exposure to wood smoke for residential heating and/or cooking, and 30 % of lung cancers are associated with WSE [5, 15]. Molecular assays have shown up-regulation and phosphorylation of p53 in WSE related lung cancer [16]. Moreover, WSE is associated with macrophage dysfunction and an increase in the activity of metalloproteinases, like MMP-2 and MMP-9, which could be related to lung injury in chronic obstructive pulmonary disease and have a role in the physiopathology of lung cancer [17].
Ethnical origins and different risk factors for lung cancer might explain the distinct mutation profiles, as in the case of epidermal growth factor receptor (EGFR) and KRAS for Asians, Caucasians and Latins [18–20]. Our group previously reported a high rate of treatment response and a better outcome in patients with WSE related lung cancer treated with EGFR-Tyrosine Kinase Inhibitors (TKIs) [21]. We have further described that WSE related lung cancer is associated with an older age at diagnosis, adenocarcinoma histology, pleural effusion, high prevalence of EGFR mutations (55.4 %) and a low prevalence of KRAS mutation (6 %), compared to patients with smoking history [15]. These situations indicate clear differences in the molecular and clinical evolution of WSE related lung cancer compared with tobacco associated lung cancer.
In order to further analyze the molecular differences observed in WES-related lung cancer, the objective of our work was to compare the genetic expression profile of lung adenocarcinoma in patients with WSE or a smoking history.
Methods
Experimental design
This study used clinical, longitudinal, prospective, observational and analytical cohorts with the selection of a non–probabilistic sample type. The protocol was approved by the Scientific and Bioethical committees of the Instituto Nacional de Cancerología (INCan, 008102510M1, CB451).
Patients and tissue samples
Patients admitted to the INCan with a pulmonary lesion suggestive of primary lung cancer were prospectively biopsied from January 2008 to June 2011. After informed consent, tissue was obtained by computer tomography-guided tru-cut (Care fusion, San Diego, CA, USA) from the clinically suspected primary tumor. Data were excluded from the analysis if there was no histological diagnosis, a different type of primary cancer was present, or if the pathology report indicated a histology different from lung adenocarcinoma. The patients with histologically confirmed advanced lung adenocarcinoma (stages III B and IV) were eligible for inclusion in the study (Fig. 1).
Fig. 1.
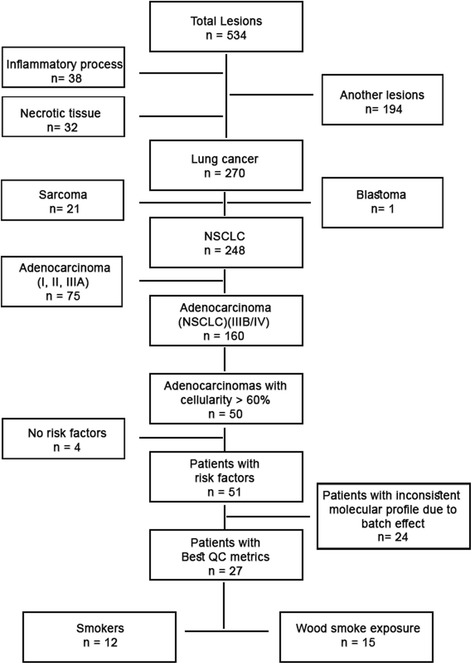
Consort
A complete medical history that included a detailed history of smoking, wood smoke exposure and a physical examination was obtained. Tumor specimens were collected at the time of diagnosis. WSE was defined as exposure to fumes resulting from burning wood in fireplaces and wood stoves for ≥ 4 h per day for ≥ 5 years. The WSE exposure index was calculated as the average number of hours spent on cooking daily per the total number of years spent cooking [22]. A smoker was defined as being someone having a lifetime exposure of more than 100 cigarettes [6]; the tobacco-smoking index was calculated by multiplying the number of cigarette packs consumed per day by the number of years spent smoking [15].
RNA isolation and RNA preparation for microarrays
Primary tumor core-biopsy was performed prior to any treatment and snap-frozen in nitrogen for RNA extraction. A trained pathologist confirmed histological diagnosis and quantified tumor cell percentage.
The procedure for extraction and purification of total RNA from tissue (up to 5 mg tissue) was done using RNeasy Micro Kit (QIAGEN, Germany) (cat. 217084). RNA integrity was evaluated by capillary electrophoresis using the Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA). Samples with RNA integrity number (RIN) of six or higher were included for microarray analysis.
RNA amplification and expression microarray analysis
Gene expression analysis was done using the Affymetrix GeneChip® Human Gene 1.0 ST Array System, which evaluates the expression of 28,869 different genes. Sample processing was done following the manufacturer’s instructions.
Strategy for microarray gene-expression analysis
Statistical analysis for differential expression was conducted using R and Bioconductor. Background correction for non-specific hybridization was performed with Robust Multiarray Average (RMA) [23] which uses a fairly complex statistical model that supposes both additive and multiplicative noise components. After background correction to the individual probes, quantile normalization [24] was applied, both steps are implemented in the oligo package [25]. Normalized and corrected probes are summarized into probe sets using the median polish algorithm, which is a type of robust 2-way ANOVA, where one factor is the array and the other is the probe set. The algorithm is robust to outliers, making single probes with large values are down-weighted. Batch correction was also applied to all samples using combat in the sva package [26]. Differential expression was identified through linear models implemented in the limma package [27], genes were selected as significant according to two summary statistics: p-value (<10−2) and fold- changes larger than 1.2 in absolute values.
For the biological networks and functional analysis, we used QIAGEN’s Ingenuity Pathway Analysis (IPA®, QIAGEN Redwood City, http://www.ingenuity.com/). IPA was used to identify gene-signaling pathways that were involved in biological processes of WSE versus tobacco exposure. Networks of these genes were algorithmically generated based on their connectivity and assigned a score. The score ranks networks according to how relevant they are to the input genes not necessarily to the quality or significance of the network. The network indicates the molecular relationships between genes/gene products. Node color indicates up- or down-regulation and intensity is associated to degree of regulation. It is important to remark that uncolored genes were not identified as differentially expressed in our experiment, however, IPA integrate those into the computationally generated networks based on the collected evidence indicating a relevance to this network.
Identification of differentially expressed genes
Significant changes in gene expression were selected according to p-values <10−2 and fold changes in absolute values larger than 1.2. Differences in gene expression are shown in volcano plots representing fold changes in log2 base along the x-axis and the level of trust in the form of –log10(p-values) along the y-axis. A value of 2 on the y-axis represents our cut-off of 10−2 (top right and left corners). Heat maps show gene profiles clustered, result of an unsupervised hierarchical clustering of genes significantly different (p < 0.01) between patients with different risk factors.
Results
Overall 53 tumor samples were collected, and 29 samples were suitable for gene expression analysis. Two samples with suitable material were excluded due to patients’ history of asbestos exposure and thoracic radiotherapy. The 27 remaining samples included 12 patients with an exclusive history of tobacco exposure and 15 patients with exclusive WSE history (Fig. 1).
Clinical and molecular results
The clinical characteristics of the 27 patients included in the microarray analysis are: mean age 62.9 ± 11.7 years, 55.6 % (15/27) were females, WSE was present on 55.6 % of patients (15/27) and 44.4 % (12/27) had tobacco exposure (Table 1). EGFR mutational status was statistically significant (p = 0.003); 53 % (8/15) of patients presented positive EGFR mutation status in the WSE group (Table 2).
Table 1.
Baseline clinical pathological and molecular characteristics (N = 27)
| Variable | % (n/N) |
|---|---|
| Gender | |
| Female | 55.6 (15/27) |
| Male | 44.4 (12/27) |
| Age | |
| Mean (±SD) | 62.96 ± 11.7* |
| Wood smoke exposure | |
| Present | 55.6 (15/27) |
| Absent | 44.4 (12/27) |
| WES index | |
| Median (IQR) | 104 ± 10 |
| Tobacco-smoking index | |
| Median (IQR) | 14.1 ± 14.2 |
| Smoking exposure | |
| Present | 44.4 (12/27) |
| Absent | 55.6 (17/27) |
| Stage | 3.7 (1/27) |
| IIIB | |
| IV | 81.5 (22/27) |
| Recurrent | 14.8 (4/27) |
| ECOG | |
| 0–1 | 74.1 (20/27) |
| 2–4 | 25.9 (7/27) |
| EGFR mutation | |
| Positive | 33.3 (9/27) |
Abbreviations: NSCLC non–small-cell lung cancer, SD standard deviation, ECOG PS eastern cooperative oncology group performance status, EGFR epidermal growth factor receptor, WSE wood smoke exposure
*Kolmogorov-Smirnov test p value: 0.20
Table 2.
Clinical and molecular characteristics related to exposure history in all patients
| Variable | WSE (N = 15) | Smoking exposure (N = 12) | P* |
|---|---|---|---|
| Gender | |||
| Female | 12 | 5 | 0.49 |
| Male | 3 | 7 | |
| Age Mean (±SD) |
63 (11.88) |
62.9 (12.12) |
0.89 |
| aEGFR mutation (N = 9) | |||
| Positive | 8 | 1 | |
| Negative | 7 | 11 | 0.003 |
| Stage | |||
| IIIB | 0 | 1 | |
| IV | 13 | 9 | |
| Recurrent | 2 | 2 | 0.70 |
*Non-parametric test: Mann-Whitney U test
a9/27 patients had EGFR mutation
On differentially expressed genes in WSE compared with tobacco smoke exposure
Figures 2 and 3 show differences in gene expression, the gene profile shown in the clustered heat map displays significantly different genes (p < 0.01) between patients with WSE versus patients with a tobacco smoking history.
Fig. 2.

Volcano Plot showing gene differential expression of patients with adenocarcinoma and history of wood smoke exposure vs. tobacco smoke exposure. Fold changes are represented in log2 base along the x-axis and the level of trust in the form of –log10 (p < 0.001) along the y-axis. The cut off value of 10−2 used is 2 on the y-axis (top right and left corners). Red and green dots represent up and down-regulated genes respectively: Fold change (≥1.2) and significance level (p < 0.001). Other colors: yellow indicates significant but low fold change, magenta shows not statistically robust changes and blue shows low fold change with low statistical significance
Fig. 3.
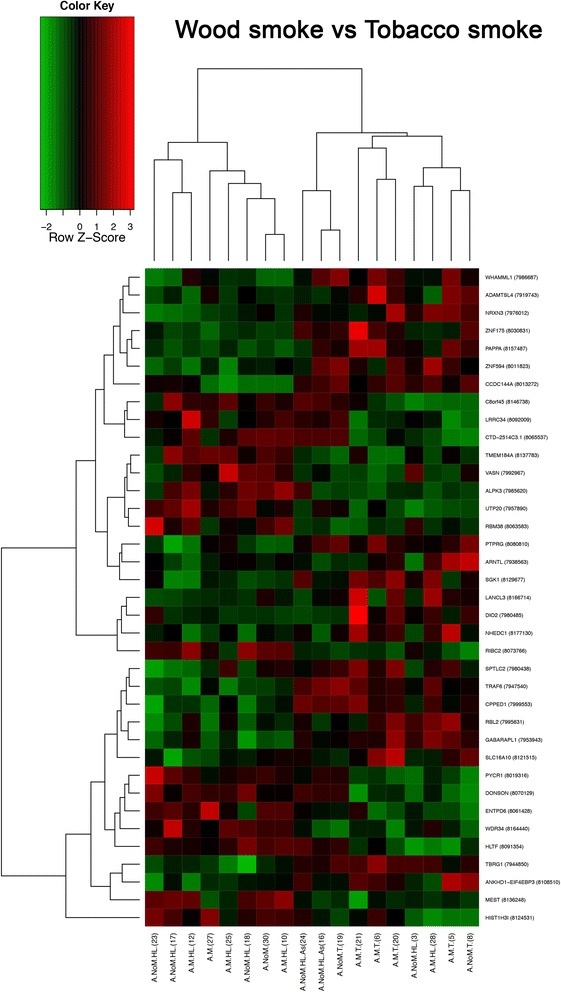
Heat map result of an unsupervised hierarchical clustering of genes significantly different (p < 0.01) of NSCLC adenocarcinoma exposed to wood smoke versus tobacco smoke. Each column represents a patient and each row a gene. The heat map indicates the level of gene expression. Red: high expression; Green: Low expression
The comparison of adenocarcinomas from patients exposed to tobacco smoke versus WSE revealed that both groups can be separated based on the differential expression of 57 genes (p < 0.01), 35 up-regulated and 22 down-regulated (Fig. 3).
Enrichment and functional analysis through biological networks was conducted using IPA software. The top functional networks were related to five different biological categories as follows: Lipid metabolism; Biochemistry of small molecules; Transport of molecules; Cell morphology; Function and cell maintenance. Table 3 lists the up- and down-regulated genes with significant changes in expression from patients with NSCLC exposed to tobacco smoke versus WSE (p < 0.01). The two main categories with greater differences were transport of molecules and cell function and maintenance.
Table 3.
List of genes with changed expression that are significantly (p < 0.01) over- and under-regulated in patients with NSCLC exposed to wood smoke versus tobacco (five IPA biological functions categories)
| Symbol | Log ratio | p-value | Lipid metabolism | Small mol. bioch. | Mol. transport | Cell morphology | Cell. func. and main. |
|---|---|---|---|---|---|---|---|
| Upregulated genes | |||||||
| HIST1H3I | 0.763 | 9.86E-03 | ✓ | ✓ | ✓ | ||
| DONSON | 0.554 | 3.82E-03 | ✓ | ✓ | ✓ | ||
| ENTPD6 | 0.472 | 2.98E-03 | ✓ | ✓ | ✓ | ||
| RIBC2 | 0.383 | 8.19E-04 | ✓ | ✓ | ✓ | ||
| VASN | 0.286 | 9.15E-03 | ✓ | ✓ | ✓ | ||
| ASL | 0.251 | 3.38E-03 | ✓ | ✓ | ✓ | ||
| DMC1 | 0.165 | 6.49E-03 | ✓ | ✓ | ✓ | ||
| ATP4B | 0.12 | 4.14E-03 | ✓ | ✓ | ✓ | ||
| MEST | 0.601 | 8.79E-03 | ✓ | ✓ | |||
| TMEM184A | 0.34 | 6.13E-03 | ✓ | ✓ | |||
| ATG9A | 0.262 | 6.82E-03 | ✓ | ✓ | |||
| PYCR1 | 0.499 | 8.28E-03 | ✓ | ✓ | |||
| RBM38 | 0.301 | 8.38E-03 | ✓ | ✓ | |||
| HNRNPD | 0.219 | 8.57E-03 | ✓ | ✓ | |||
| ZNF239 | 2.60E-01 | 9.77E-04 | ✓ | ✓ | |||
| Downregulated genes | |||||||
| SPTLC2 | −0.412 | 6.28E-03 | ✓ | ✓ | ✓ | ||
| TBRG1 | −0.374 | 3.42E-03 | ✓ | ✓ | ✓ | ||
| ZNF175 | −0.358 | 8.97E-03 | ✓ | ✓ | ✓ | ||
| ZNF594 | −0.351 | 7.40E-03 | ✓ | ✓ | ✓ | ||
| TEX12 | −0.125 | 7.24E-03 | ✓ | ✓ | ✓ | ||
| SLC16A10 | −0.777 | 9.14E-03 | ✓ | ✓ | |||
| GABARAPL1 | −0.61 | 9.62E-04 | ✓ | ✓ | |||
| PAPPA | −0.516 | 3.63E-04 | ✓ | ✓ | |||
| MED31 | −0.261 | 8.96E-03 | ✓ | ✓ | |||
| NDUFAF1 | −0.254 | 6.22E-03 | ✓ | ✓ | |||
| CCDC144NL | −0.224 | 9.26E-03 | ✓ | ✓ | |||
| CNTN4 | −0.189 | 9.02E-03 | ✓ | ✓ | |||
| NXPE2 | −0.145 | 9.07E-03 | ✓ | ✓ | |||
| RCOR3 | −0.143 | 8.15E-03 | ✓ | ✓ | |||
| SGK1 | −0.718 | 6.48E-03 | ✓ | ✓ | |||
| PTPRG | −0.686 | 2.63E-03 | ✓ | ✓ | |||
| DIO2 | −0.623 | 8.90E-03 | ✓ | ✓ | |||
| RBL2 | −0.369 | 6.19E-03 | ✓ | ✓ | |||
| ARNTL | −0.366 | 9.77E-03 | ✓ | ✓ | |||
| TRAF6 | −0.329 | 6.87E-03 | ✓ | ✓ | |||
| MBP | −0.246 | 4.43E-03 | ✓ | ✓ | |||
Three functional networks are shown, these networks involve the majority of the differentially expressed genes (37/57) and have: Ubiquitin C (UBC, score 28, Fig. 4), GABA(A) receptor-associated like protein (GABARAPL1, score 28, Fig. 5) and PI3K/AKT and MEK/ERK genes (score 26, Fig. 6) as main hubs of the network. Moreover, when the networks were overlapped, all up and down regulated genes appear around the PI3K/AKT and MEK/ERK signaling pathways (Fig. 7).
Fig. 4.
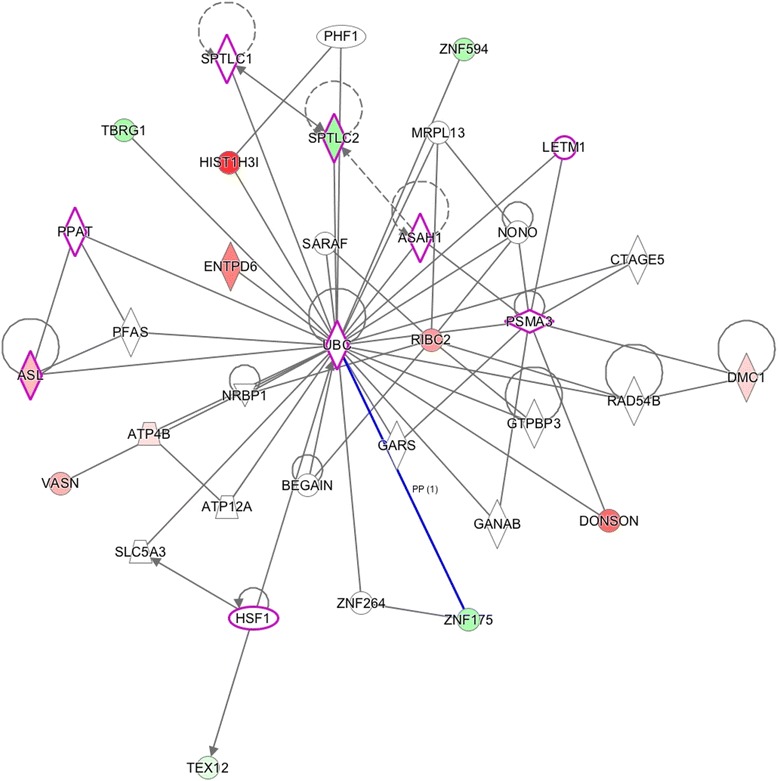
Network 1 (Score: 28/Ratio: 0.353/p value: 1.48E-24). Genes (p < 0.01) in wood smoke exposure in NSCLC around the UBC gene. Red mark: up-regulated genes. Green mark: down-regulated genes. The node shapes denote enzymes ( ), phosphatases (
), phosphatases ( ), kinases (
), kinases ( ), peptidases (
), peptidases ( ), G-protein coupled receptor (
), G-protein coupled receptor ( ), transmembrane receptor (
), transmembrane receptor ( ), cytokines (
), cytokines ( ), growth factor (
), growth factor ( ), ion channel (
), ion channel ( ), transporter (
), transporter ( ), translation factor (
), translation factor ( ), nuclear receptor (
), nuclear receptor ( ), transcription factor (
), transcription factor ( ) and other (
) and other ( )
)
Fig. 5.
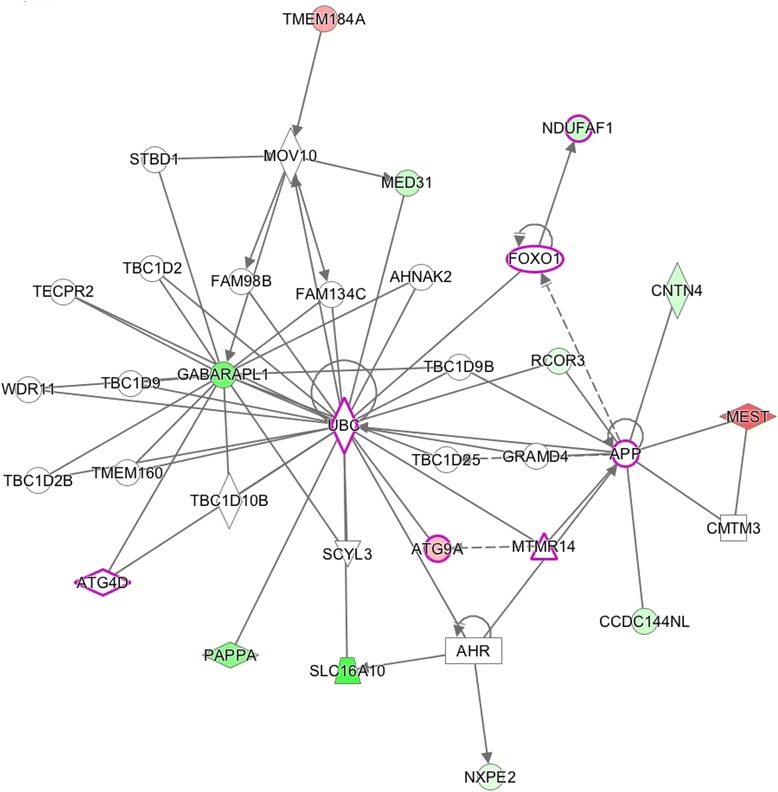
Network 2 (Score: 28/Ratio: 0.375/p value: 6.11E-25). Genes (p < 0.01) in wood smoke exposure in NSCLC around the GABARAPL1 gene. Red mark: up-regulated genes. Green mark: down-regulated genes. The node shapes denote enzymes ( ), phosphatases (
), phosphatases ( ), kinases (
), kinases ( ), peptidases (
), peptidases ( ), G-protein coupled receptor (
), G-protein coupled receptor ( ), transmembrane receptor (
), transmembrane receptor ( ), cytokines (
), cytokines ( ), growth factor (
), growth factor ( ), ion channel (
), ion channel ( ), transporter (
), transporter ( ), translation factor (
), translation factor ( ), nuclear receptor (
), nuclear receptor ( ), transcription factor (
), transcription factor ( ) and other (
) and other ( )
)
Fig. 6.

Network 3 (Score: 26/Ratio: 0.134/p value: 2.09E-17). Genes (p < 0.01) in wood smoke exposure in NSCLC around the PI3K/AKT and MEK/ERK signaling pathways. Red mark: up-regulated genes. Green mark: down-regulated genes. The node shapes denote enzymes ( ), phosphatases (
), phosphatases ( ), kinases (
), kinases ( ), peptidases (
), peptidases ( ), G-protein coupled receptor (
), G-protein coupled receptor ( ), transmembrane receptor (
), transmembrane receptor ( ), cytokines (
), cytokines ( ), growth factor (
), growth factor ( ), ion channel (
), ion channel ( ), transporter (
), transporter ( ), translation factor (
), translation factor ( ), nuclear receptor (
), nuclear receptor ( ), transcription factor (
), transcription factor ( ) and other (
) and other ( )
)
Fig. 7.
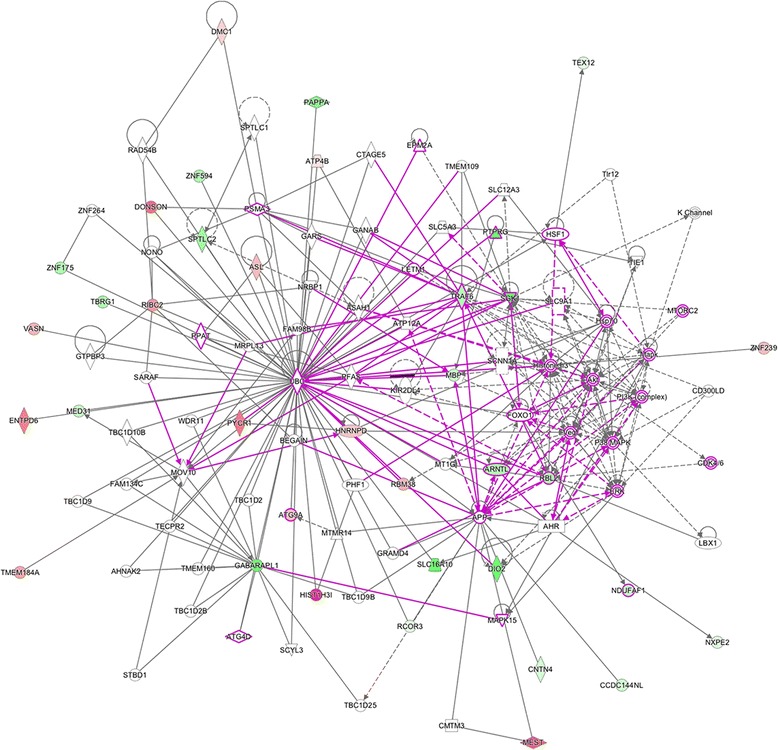
Overlapping networks and related genes (p < 0.01) in wood smoke exposure in NSCLC. Red mark: up-regulated genes. Green mark: down-regulated genes. Purple lines reflect the sites of intersections between our study genes (p < 0.01) and the main canonical networks (PI3K/AKT and MEK/ERK) associated with NSCLC. The node shapes denote enzymes (♦), phosphatases ( ), kinases (
), kinases ( ), peptidases (
), peptidases ( ), G-protein coupled receptor ( ), transmembrane receptor ( ), cytokines (
), G-protein coupled receptor ( ), transmembrane receptor ( ), cytokines ( ), growth factor (
), growth factor ( ), ion channel (
), ion channel ( ), transporter ( ), translation factor (
), transporter ( ), translation factor ( ), nuclear receptor (
), nuclear receptor ( ), transcription factor (
), transcription factor ( ) and other (
) and other ( )
)
Discussion
Although the majority of lung cancer occurs in smokers, 25 % of worldwide lung cancer occurs in life long never smokers [28], being the 7th largest cause of cancer-related mortality in this group [29], presenting a wide-ranging geographic incidence and risk factors such as asbestos, air pollution, radon, arsenic compounds, cadmium, chromium, ionizing radiation and WSE [30]. Additionally, molecular profiles observed in lung cancer are critically different among smokers and non-smokers particularly identified in genes such EGFR, KRAS, P53 and ALK [31]. In the case of WSE, there have been association with NSCLC and adenocarcinoma histology, EGFR mutations, a reverse association with KRAS mutations and higher response to EGFR-TKIs [15] making it a distinctive disease entity inside the group of never smokers which would be a good candidate for personalized diagnostic and therapeutic approaches. Therefore, lung cancer associated to WSE presents unique characteristics that make it a distinctive entity of disease within the group of never smokers; thus, it could be a good candidate for personalized diagnostic and therapeutic approaches.
There is evidence of differential expression profiles associated with the bronchial epithelium of tobacco-smokers that sustains carcinogenesis [32], as well as the determination of tobacco-smoke transcriptional changes in oncogenes and anti-oncogenes [33]. Our study shows that the gene expression profiling of samples from patients with WSE is different from patients using tobacco.
Our group has previously reported that lung cancer related to tobacco smoke and WSE exhibits different clinical and pathological characteristics that may be related to different mechanisms, and this is reflected in their response rate and overall survival in NSCLC patients [15]. However, in the present report we show a specific gene expression profile for WSE that involves 57 genes. Using biological or functional network analysis, 37 genes were identified around UBC, GABARAPL1 genes and PI3K/AKT and MEK/ERK signaling pathways.
The UBC hub in Network 1 (Fig. 4) is involved in cellular homeostasis and signaling. It was originally activated to degrade misfolded or disused proteins, but it has been recently associated with the cell cycle, DNA repair, endocytosis, antigen processing and apoptosis [34]. Recently, Tang et al. demonstrated that the inhibition of the ubiquitin system decreased the proliferation and radio-resistance in the H1299 cell line (NSCLC cells) [35]. In this regard, a clinically relevant observation is the approval of bortezomib as an inhibitor of the protein degradation system in human cancer [32].
The GABARAPL1 hub in Network 2 (Fig. 5) is a highly conserved protein throughout evolution. It is related to autophagy and vesicle intracellular transport [36]. Its participation in cancer is still not clear, but it has been reported that lower levels of this transcript correlates with decreased survival in patients with neuroblastoma [37] and increased metastasis in breast cancer [36]. On the other hand, the ectopic over-expression of GABARAPL1 inhibits cancer cell proliferation and tumor growth in mice [38]. There are other reports that relate low expression of this gene in several cancer cell lines [39].
Regarding the last network, there have been reports that show that PI3K/AKT and MEK/ERK signaling pathways are altered in NSCLC and their activation is associated with malignant transformation and drug resistance (Figs. 6 and 7). MEK and PI3K inhibitors can inhibit cell proliferation in NSCLC; however, for apoptosis activation, both signaling pathways must be simultaneously inhibited [40, 41], a situation that is directly related to the frequently observed EGFR-TKI resistance in this tumor. There are other reports showing that EGFR mutations function as inductors to sensitization to TKIs through PI3K/AKT and MEK/ERK signaling pathways [41–45]. It has also been demonstrated that cases with EGFR mutations have a major sensibility to the EGFR-TKIs, using inhibitors from PI3K/AKT and MEK/ERK [41–45]. On a clinical note, our group has previously reported the association between NSCLC adenocarcinoma and positive EGFR mutation status in patients with history of WSE compared to tobacco smoke exposure.
WSE is also related to gene promoter methylation that synergistically increases the risk for reduced lung function in cigarette smokers [46]. A recent report describing the toxicological characteristics associated with WSE in A549 cell lines, including high levels of polycyclic aromatic hydrocarbons (PAH) and low level of water-soluble metals, showed an enhanced level of free radicals, DNA damage and the major expression of inflammatory/oxidative stress genes [47]. There is evidence that some potential molecular targets, such as EGFR and the ErbB family receptor, are usually altered in epithelial tumors [48]. EGFR mediates cell proliferation, differentiation, survival, angiogenesis and migration, and is overexpressed in approximately 40–80 % on NSCLC tumors [49–51].
Clinically, it is known that EGFR inhibitors in NSCLC extend survival after first-line or second-line therapy in patients with EGFR mutations [52]. These mutations are more frequent in specific populations, including women, Asian and Hispanic ethnicities, never-smokers and adenocarcinoma histology [18, 53, 54]. Activating mutations in EGFR leads to constitutive tyrosine kinase activation and oncogenic transformation of lung epithelial cells [12, 13]. In this sense, the presence of these common activating EGFR mutations is tightly associated with sensitivity to reversible EGFR- specific tyrosine kinase inhibitors (e.g.: erlotinib or gefitinib). Patients with these mutations display EGFR-TKIs response rates of approximately 70 % a median progression free survival (PFS) of approximately 9–12 months and overall survival rates that may exceed 20–32 months [55]. Most patients will experience disease progression and drug resistance attributed to the development of other second mutations or with the presence of other uncommon EGFR mutations [56]. Certain therapeutic relations in NSCLC include the main oncogenic protein KRAS-GTP with biological significance between EGFR and PI3K/AKT or MEK/ERK pathways [56]. The presence of KRAS mutations leads to an increased signal through the MEK/MAPK transduction pathway [56]. Rare cases of mutations of MEK have been reported in NSCLC [57]. Preclinical studies in both the KPC mouse model as well as patient-derived xenografts have shown that blocking the MAPK pathway at MEK results in a decrease of cell proliferation and a subsequent halt in tumor growth [58]. The activation of EGFR recruits PI3K to the cell membrane and phosphorylates phosphatidylinositol-2-phosphate (PIP2) to phosphatidylinositol-3-phosphate (PIP3), which in turn activates AKT and several downstream effectors [59]. Inhibitors of both PI3K and AKT have been developed [60], although inhibition of PI3K is complicated by the fact that there are multiple isoforms of the protein [61]. Another biological interaction takes place on KRAS is one that directly activates PI3KCA [62]. Unlike most oncogenic driver mutations on NSCLC, PI3K mutations may occur in association with EGFR or KRAS mutations [63]. Although rare, PI3K/AKT/mTOR pathway activation may occur through AKT mutations in NSCLC [64]. Clinical evidence has shown that reversible EGFR-TKIs are considered the frontline treatment for advanced NSCLC patients harboring EGFR mutations [65]. New emerging evidence suggests that the anti-tumor activity of EGFR-TKIs in resistant NSCLC cell lines can be enhanced by combined therapy with other regimens. Early efforts have shown that cetuximab, produced synergistic anti-proliferative effects when used in combination with gefitinib or erlotinib [66]. Our analyses provide biological networks relationships between 37 genes and PI3K/Akt and MEK signaling for understanding the biologic properties of WSE effects as a carcinogenic factor in NSCLC. It also shows useful common pathway maps for a future understanding of the disease and the development of new therapeutic targets.
Whilst the differences in gene expression patterns between WSE or tobacco-related lung cancer that we identified in this paper provide an important insight into the molecular basis of the clinical and biological differences between these two tumors, there is a limitation regarding the small sample size. However, this is countervailed by a thorough characterization of the samples, a detailed clinical history and close follow-up on all patients. It is imperative to continue further study to validate the potential biological and clinical implications of our findings.
Conclusion
In conclusion, our results suggest a differential gene expression profile for WSE or tobacco-related lung cancer, which suggests different carcinogenesis mechanisms between both risk factors and enlightens the clinical-pathological and mutational profiles between both groups with adenocarcinoma.
Acknowledgments
Institution at which the work was performed
Instituto Nacional de Cancerología (INCan) and Instituto Nacional de Medicina Genómica (INMEGEN) in Mexico City, Mexico.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
OA and CRE are the guarantors of the content of the manuscript, including the data and analysis. OA, EOMP, made substantial contributions to conception and design. AOG, CMR, AAS, and ALG acquired data. CRE, GAF, RRB, AHM and OA analyzed the data. AOG, CMR, GAF, EOMP, and OA drafted the manuscript. All authors revised the manuscript and approved the final version.
Contributor Information
Claudia Rangel-Escareño, Email: crangel@inmegen.gob.mx.
Oscar Arrieta, Phone: (+52) (55) 5628-0400, Email: ogar@unam.mx.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 3.Yu YH, Liao CC, Hsu WH, Chen HJ, Liao WC, Muo CH, et al. Increased lung cancer risk among patients with pulmonary tuberculosis: a population cohort study. J Thorac Oncol. 2011;6(1):32–7. doi: 10.1097/JTO.0b013e3181fb4fcc. [DOI] [PubMed] [Google Scholar]
- 4.IARC. Lung Cancer Estimated Incidence, Mortality and Prevalence. In: Cancer Fact Sheet. http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx. 2012. Accessed November 21 2015.
- 5.Arrieta O, Guzman-de Alba E, Alba-Lopez LF, Acosta-Espinoza A, Alatorre-Alexander J, Alexander-Meza JF, et al. National consensus of diagnosis and treatment of non-small cell lung cancer. Rev Invest Clin. 2013;65 Suppl 1:S5–84. [PubMed] [Google Scholar]
- 6.Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers--a different disease. Nat Rev Cancer. 2007;7(10):778–90. doi: 10.1038/nrc2190. [DOI] [PubMed] [Google Scholar]
- 7.Cufari ME, Proli C, Phull M, Raubenheimer H, Al Sahaf M, Asadi N, et al., editors. Increasing Incidence of Non-Smoking Lung Cancer: Presentation of Patients with Early Disease to a Tertiary Institution in the UK. 16TH WORLD CONFERENCE ON LUNG CANCER; 2015 September 6–9; Denver, CO: International Association of the Study of Lung Cancer (IASLC).
- 8.Pelosof L, Ahn C, Horn L, Madrigales A, Cox J, Roberts JN, et al., editors. Increasing Incidence of Never Smokers in Non Small Cell Lung Cancer (NSCLC) Patients. 16TH WORLD CONFERENCE ON LUNG CANCER; 2015 September 6–9; Denver, CO: International Association of the Study of Lung Cancer (IASLC).
- 9.Alberg AJ, Brock MV, Ford JG, Samet JM, Spivack SD. Epidemiology of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013;143(5 Suppl):e1S–29S. doi: 10.1378/chest.12-2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pua BB, Dou E, O'Connor K, Crawford CB. Integrating smoking cessation into lung cancer screening programs. Clin Imaging. 2015 doi: 10.1016/j.clinimag.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 11.Salvi SS, Barnes PJ. Chronic obstructive pulmonary disease in non-smokers. Lancet. 2009;374(9691):733–43. doi: 10.1016/S0140-6736(09)61303-9. [DOI] [PubMed] [Google Scholar]
- 12.Naeher LP, Brauer M, Lipsett M, Zelikoff JT, Simpson CD, Koenig JQ, et al. Woodsmoke health effects: a review. Inhal Toxicol. 2007;19(1):67–106. doi: 10.1080/08958370600985875. [DOI] [PubMed] [Google Scholar]
- 13.Hernandez-Garduno E, Brauer M, Perez-Neria J, Vedal S. Wood smoke exposure and lung adenocarcinoma in non-smoking Mexican women. Int J Tuberc Lung Dis. 2004;8(3):377–83. [PubMed] [Google Scholar]
- 14.Zelikoff JT, Chen LC, Cohen MD, Schlesinger RB. The toxicology of inhaled woodsmoke. J Toxicol Environ Health B Crit Rev. 2002;5(3):269–82. doi: 10.1080/10937400290070062. [DOI] [PubMed] [Google Scholar]
- 15.Arrieta O, Campos-Parra AD, Zuloaga C, Aviles A, Sanchez-Reyes R, Manriquez ME, et al. Clinical and pathological characteristics, outcome and mutational profiles regarding non-small-cell lung cancer related to wood-smoke exposure. J Thorac Oncol. 2012;7(8):1228–34. doi: 10.1097/JTO.0b013e3182582a93. [DOI] [PubMed] [Google Scholar]
- 16.Delgado J, Martinez LM, Sanchez TT, Ramirez A, Iturria C, Gonzalez-Avila G. Lung cancer pathogenesis associated with wood smoke exposure. Chest. 2005;128(1):124–31. doi: 10.1378/chest.128.1.124. [DOI] [PubMed] [Google Scholar]
- 17.Montano M, Beccerril C, Ruiz V, Ramos C, Sansores RH, Gonzalez-Avila G. Matrix metalloproteinases activity in COPD associated with wood smoke. Chest. 2004;125(2):466–72. doi: 10.1378/chest.125.2.466. [DOI] [PubMed] [Google Scholar]
- 18.Arrieta O, Cardona AF, Martin C, Mas-Lopez L, Corrales-Rodriguez L, Bramuglia G, et al. Updated frequency of EGFR and KRAS mutations in nonsmall-cell lung cancer in Latin America: the Latin-American Consortium for the Investigation of Lung Cancer (CLICaP) J Thorac Oncol. 2015;10(5):838–43. doi: 10.1097/JTO.0000000000000481. [DOI] [PubMed] [Google Scholar]
- 19.Arrieta O, Ramirez-Tirado LA, Baez-Saldana R, Pena-Curiel O, Soca-Chafre G, Macedo-Perez EO. Different mutation profiles and clinical characteristics among Hispanic patients with non-small cell lung cancer could explain the "Hispanic paradox". Lung Cancer. 2015;90(2):161–6. doi: 10.1016/j.lungcan.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 20.Luo YH, Wu CH, Wu WS, Huang CY, Su WJ, Tsai CM, et al. Association between tumor epidermal growth factor receptor mutation and pulmonary tuberculosis in patients with adenocarcinoma of the lungs. J Thorac Oncol. 2012;7(2):299–305. doi: 10.1097/JTO.0b013e31823c588d. [DOI] [PubMed] [Google Scholar]
- 21.Arrieta O, Martinez-Barrera L, Trevino S, Guzman E, Castillo-Gonzalez P, Rios-Trejo MA, et al. Wood-smoke exposure as a response and survival predictor in erlotinib-treated non-small cell lung cancer patients: an open label phase II study. J Thorac Oncol. 2008;3(8):887–93. doi: 10.1097/JTO.0b013e31818026f6. [DOI] [PubMed] [Google Scholar]
- 22.Behera D, Jindal SK. Respiratory symptoms in Indian women using domestic cooking fuels. Chest. 1991;100(2):385–8. doi: 10.1378/chest.100.2.385. [DOI] [PubMed] [Google Scholar]
- 23.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 24.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185–93. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 25.Carvalho BS, Irizarry RA. A framework for oligonucleotide microarray preprocessing. Bioinformatics. 2010;26(19):2363–7. doi: 10.1093/bioinformatics/btq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–3. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 28.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 29.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55(2):74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 30.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359(13):1367–80. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee YJ, Kim JH, Kim SK, Ha SJ, Mok TS, Mitsudomi T, et al. Lung cancer in never smokers: change of a mindset in the molecular era. Lung Cancer. 2011;72(1):9–15. doi: 10.1016/j.lungcan.2010.12.013. [DOI] [PubMed] [Google Scholar]
- 32.Word B, Lyn-Cook LE, Jr, Mwamba B, Wang H, Lyn-Cook B, Hammons G. Cigarette smoke condensate induces differential expression and promoter methylation profiles of critical genes involved in lung cancer in NL-20 lung cells in vitro: short-term and chronic exposure. Int J Toxicol. 2013;32(1):23–31. doi: 10.1177/1091581812465902. [DOI] [PubMed] [Google Scholar]
- 33.Lowe FJ, Luettich K, Gregg EO. Lung cancer biomarkers for the assessment of modified risk tobacco products: an oxidative stress perspective. Biomarkers. 2013;18(3):183–95. doi: 10.3109/1354750X.2013.777116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoeller D, Hecker CM, Dikic I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat Rev Cancer. 2006;6(10):776–88. doi: 10.1038/nrc1994. [DOI] [PubMed] [Google Scholar]
- 35.Tang Y, Geng Y, Luo J, Shen W, Zhu W, Meng C, et al. Downregulation of ubiquitin inhibits the proliferation and radioresistance of non-small cell lung cancer cells in vitro and in vivo. Sci Rep. 2015;5:9476. doi: 10.1038/srep09476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berthier A, Seguin S, Sasco AJ, Bobin JY, De Laroche G, Datchary J, et al. High expression of gabarapl1 is associated with a better outcome for patients with lymph node-positive breast cancer. Br J Cancer. 2010;102(6):1024–31. doi: 10.1038/sj.bjc.6605568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roberts SS, Mori M, Pattee P, Lapidus J, Mathews R, O'Malley JP, et al. GABAergic system gene expression predicts clinical outcome in patients with neuroblastoma. J Clin Oncol. 2004;22(20):4127–34. doi: 10.1200/JCO.2004.02.032. [DOI] [PubMed] [Google Scholar]
- 38.Klebig C, Seitz S, Arnold W, Deutschmann N, Pacyna-Gengelbach M, Scherneck S, et al. Characterization of {gamma}-aminobutyric acid type A receptor-associated protein, a novel tumor suppressor, showing reduced expression in breast cancer. Cancer Res. 2005;65(2):394–400. [PubMed] [Google Scholar]
- 39.Nemos C, Mansuy V, Vernier-Magnin S, Fraichard A, Jouvenot M, Delage-Mourroux R. Expression of gec1/GABARAPL1 versus GABARAP mRNAs in human: predominance of gec1/GABARAPL1 in the central nervous system. Brain Res Mol Brain Res. 2003;119(2):216–9. doi: 10.1016/j.molbrainres.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 40.Li H, Schmid-Bindert G, Wang D, Zhao Y, Yang X, Su B, et al. Blocking the PI3K/AKT and MEK/ERK signaling pathways can overcome gefitinib-resistance in non-small cell lung cancer cell lines. Adv Med Sci. 2011;56(2):275–84. doi: 10.2478/v10039-011-0043-x. [DOI] [PubMed] [Google Scholar]
- 41.Janmaat ML, Kruyt FA, Rodriguez JA, Giaccone G. Response to epidermal growth factor receptor inhibitors in non-small cell lung cancer cells: limited antiproliferative effects and absence of apoptosis associated with persistent activity of extracellular signal-regulated kinase or Akt kinase pathways. Clin Cancer Res. 2003;9(6):2316–26. [PubMed] [Google Scholar]
- 42.Cragg MS, Kuroda J, Puthalakath H, Huang DC, Strasser A. Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS Med. 2007;4(10):1681–89. doi: 10.1371/journal.pmed.0040316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng J, Shimamura T, Perera S, Carlson NE, Cai D, Shapiro GI, et al. Proapoptotic BH3-only BCL-2 family protein BIM connects death signaling from epidermal growth factor receptor inhibition to the mitochondrion. Cancer Res. 2007;67(24):11867–75. doi: 10.1158/0008-5472.CAN-07-1961. [DOI] [PubMed] [Google Scholar]
- 44.Engelman JA, Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr Opin Genet Dev. 2008;18(1):73–9. doi: 10.1016/j.gde.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 45.Gong Y, Somwar R, Politi K, Balak M, Chmielecki J, Jiang X, et al. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 2007;4(10):e294. doi: 10.1371/journal.pmed.0040294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sood A, Petersen H, Blanchette CM, Meek P, Picchi MA, Belinsky SA, et al. Wood smoke exposure and gene promoter methylation are associated with increased risk for COPD in smokers. Am J Respir Crit Care Med. 2010;182(9):1098–104. doi: 10.1164/rccm.201002-0222OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Danielsen PH, Moller P, Jensen KA, Sharma AK, Wallin H, Bossi R, et al. Oxidative stress, DNA damage, and inflammation induced by ambient air and wood smoke particulate matter in human A549 and THP-1 cell lines. Chem Res Toxicol. 2011;24(2):168–84. doi: 10.1021/tx100407m. [DOI] [PubMed] [Google Scholar]
- 48.Mendelsohn J, Baselga J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J Clin Oncol. 2003;21(14):2787–99. doi: 10.1200/JCO.2003.01.504. [DOI] [PubMed] [Google Scholar]
- 49.Janmaat ML, Giaccone G. The epidermal growth factor receptor pathway and its inhibition as anticancer therapy. Drugs Today. 2003;39 Suppl C:61–80. [PubMed] [Google Scholar]
- 50.Herbst RS, Shin DM. Monoclonal antibodies to target epidermal growth factor receptor-positive tumors: a new paradigm for cancer therapy. Cancer. 2002;94(5):1593–611. doi: 10.1002/cncr.10372. [DOI] [PubMed] [Google Scholar]
- 51.Sridhar SS, Seymour L, Shepherd FA. Inhibitors of epidermal-growth-factor receptors: a review of clinical research with a focus on non-small-cell lung cancer. Lancet Oncol. 2003;4(7):397–406. doi: 10.1016/S1470-2045(03)01137-9. [DOI] [PubMed] [Google Scholar]
- 52.Campos-Parra AD, Zuloaga C, Manriquez ME, Aviles A, Borbolla-Escoboza J, Cardona A, et al. KRAS mutation as the biomarker of response to chemotherapy and EGFR-TKIs in patients with advanced non-small cell lung cancer: clues for its potential use in second-line therapy decision making. Am J Clin Oncol. 2015;38(1):33–40. doi: 10.1097/COC.0b013e318287bb23. [DOI] [PubMed] [Google Scholar]
- 53.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123–32. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 54.Arrieta O, Cardona AF, Federico Bramuglia G, Gallo A, Campos-Parra AD, Serrano S, et al. Genotyping non-small cell lung cancer (NSCLC) in Latin America. J Thorac Oncol. 2011;6(11):1955–9. doi: 10.1097/JTO.0b013e31822f655f. [DOI] [PubMed] [Google Scholar]
- 55.Arrieta O, Cardona AF, Corrales L, Campos-Parra AD, Sanchez-Reyes R, Amieva-Rivera E, et al. The impact of common and rare EGFR mutations in response to EGFR tyrosine kinase inhibitors and platinum-based chemotherapy in patients with non-small cell lung cancer. Lung Cancer. 2015;87(2):169–75. doi: 10.1016/j.lungcan.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 56.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9(7):517–31. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sadiq AA, Salgia R. MET as a possible target for non-small-cell lung cancer. J Clin Oncol. 2013;31(8):1089–96. doi: 10.1200/JCO.2012.43.9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–90. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 59.Freudlsperger C, Burnett JR, Friedman JA, Kannabiran VR, Chen Z, Van Waes C. EGFR-PI3K-AKT-mTOR signaling in head and neck squamous cell carcinomas: attractive targets for molecular-oriented therapy. Expert Opin Ther Targets. 2011;15(1):63–74. doi: 10.1517/14728222.2011.541440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–62. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 61.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11(5):329–41. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 62.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 63.Yamamoto H, Shigematsu H, Nomura M, Lockwood WW, Sato M, Okumura N, et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res. 2008;68(17):6913–21. doi: 10.1158/0008-5472.CAN-07-5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Malanga D, Scrima M, De Marco C, Fabiani F, De Rosa N, De Gisi S, et al. Activating E17K mutation in the gene encoding the protein kinase AKT1 in a subset of squamous cell carcinoma of the lung. Cell Cycle. 2008;7(5):665–9. doi: 10.4161/cc.7.5.5485. [DOI] [PubMed] [Google Scholar]
- 65.Joshi M, Rizvi SM, Belani CP. Afatinib for the treatment of metastatic non-small cell lung cancer. Cancer Manage Res. 2015;7:75–82. doi: 10.2147/CMAR.S51808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang S, Armstrong EA, Benavente S, Chinnaiyan P, Harari PM. Dual-agent molecular targeting of the epidermal growth factor receptor (EGFR): combining anti-EGFR antibody with tyrosine kinase inhibitor. Cancer Res. 2004;64(15):5355–62. doi: 10.1158/0008-5472.CAN-04-0562. [DOI] [PubMed] [Google Scholar]