

Abstract
Background
Transmission of the malaria parasite Plasmodium falciparum from humans to the mosquito vector requires differentiation of a sub-population of asexual forms replicating within red blood cells into non-dividing male and female gametocytes. The nature of the molecular mechanism underlying this key differentiation event required for malaria transmission is not fully understood.
Methods
Whole genome sequencing was used to examine the genomic diversity of the gametocyte non-producing 3D7-derived lines F12 and A4. These lines were used in the recent detection of the PF3D7_1222600 locus (encoding PfAP2-G), which acts as a genetic master switch that triggers gametocyte development.
Results
The evolutionary changes from the 3D7 parental strain through its derivatives F12 (culture-passage derived cloned line) and A4 (transgenic cloned line) were identified. The genetic differences including the formation of chimeric var genes are presented.
Conclusion
A genomics resource is provided for the further study of gametocytogenesis or other phenotypes using these parasite lines.
Electronic supplementary material
The online version of this article (doi:10.1186/s12936-016-1254-1) contains supplementary material, which is available to authorized users.
Keywords: Gametocytes, Plasmodium falciparum, A4, F12, ApiAP2 gene family, Whole genome sequencing
Background
The malaria parasite Plasmodium falciparum is a major threat to human health causing approximately 200 million clinical cases per year and an estimated 438,000 deaths [1]. Malaria control efforts focus mainly on the application of insecticides to kill the mosquito vector, and on the use of anti-malarial drugs that prevent the parasite from replicating/proliferating in the liver or red blood cell stages in the host. These tools reduce the prevalence of infection, but to reach elimination additional efforts will be required to interrupt transmission. For the malaria parasite to be transmitted from humans to the mosquito vector it requires differentiation of asexually replicating forms within red blood cells into non-dividing male and female gametocytes. Only one drug available in the clinic, primaquine, can kill mature gametocytes, but this is contra-indicated in patients with G6PD deficiency. Various pharmacological agents and even anti-malarial drugs, such as pyrimethamine and chloroquine have been shown to increase gametocyte production [2–4]. Also, high asexual parasite density and diffusible factors in cultures can promote gametocyte formation [5]. Gametocytaemia is a sensitive indicator of emerging drug resistance [6]. Increasing gametocyte prevalence has been shown to precede measurable changes in parasite clearance or a decrease in cure rates. Interventions that disrupt the switch to sexual development that occurs in the human host could represent a more effective tool for malaria control and eventually elimination.
Although gametocytes were first observed microscopically in blood films taken from patients in the tropics over a 100 years ago, the molecular mechanisms involved in commitment to gametocyte formation are still poorly understood. Certain pathways have been implicated in gametocytogenesis, including those producing phorbol ester and involved in cAMP signalling [7], but these findings have not been verified using genetic approaches. Evidence has been reported that a heterotrimeric G protein based pathway may be involved [8], but the absence of homologues in the parasite genome make this unlikely. A number of studies have sought to identify loci that might constitute the trigger for the switch from asexual replication to sexual commitment. Genes in the sub-telomeric region of the right arm of chromosome 9 have been implicated in the inability to produce gametocytes, particularly the gene PF3D7_0935400 (gametocyte development protein 1; Pfgdv1) [9–11]. Complementation with PF3D7_0935400 restores gametocyte production in a 3D7 gametocyte-deficient line, which has the subtelomeric region on chromosome 9 deleted [11]. A further 16 genes associated with gametocyte production were identified by random transposon mutagenesis [12]. It has been reported that the PF3D7_1222600 gene (a member of the ApiAP2 family encoding PfAP2-G) is an epigenetically regulated genetic master switch that triggers gametocyte development [13]. This finding was supported by several independent experimental approaches, including the identification of Pfap2-g mutations in two gametocyte non-producer (GNP) clonal lines, F12 and A4, both derived from the reference 3D7 clone. Using similar approaches the P. berghei AP2-G orthologue was shown to play the same role in this rodent malaria parasite [14].
The F12 strain is a well-studied GNP line [10], unable to produce either morphologically recognizable gametocytes or very early sexual stages [15]. A4 is a cloned transgenic line that was transfected with a plasmid designed to functionally delete the phosphodiesterase gene (PDEδ, PF3D7_1470500). A4 was generated with the intention of investigating whether the absence of PDEδ had any effect on exflagellation. However, the line lost the ability to produce gametocytes following drug selection, but this phenotype was unrelated to PDEδ function [16]. Both F12 and A4 clones do not have coding-sequence mutations or deletions within the sub-telomeric region of chromosome 9, nor on any other the 16 genes recently implicated in gametocyte development [13]. The only mutations found to be associated with the phenotype were those that disrupt the Pfap2-g gene [13].
Substantial genetic variation arises in clones that have been maintained in long-term culture, such as the F12 and A4 GNP lines. Genomic changes have been observed for several laboratory clones, including those derived from 3D7 [17, 18] and can be responsible for different phenotypes such as the non-production of gametocytes. Many of the mutations arising in vitro might not have an immediate selective advantage to the parasite. However in vivo, such apparently silent genetic changes might become important when a new environment (e.g., anti-malarial drugs) is presented and a selective advantage is suddenly introduced.
Here, a comprehensive analysis of the genomes of these two long-term cultured GNP lines using whole genome sequencing data and advanced computational methods is presented. The mutations identified in the two GNP lines were compared with the 3D7 Ref. [19] and with the sequence data of the parental 3D7 clone (referred here as “3D7A”) used to generate the A4 line. The data were also compared with 930 P. falciparum field isolates from several malaria endemic regions [20–22]. Any additional genetic changes were investigated, including structural variants and rearrangements in the var genes that previously have been identified in clones subjected to long-term culture. The var genes are the most polymorphic gene family in P. falciparum, with 60 loci distributed across the 14 chromosomes, both in the sub-telomeres and in internal regions. They encode the hypervariable P. falciparum erythrocyte membrane protein 1 (PfEMP1) that is critical for host immune evasion. Finally, a genomic resource for the two GNP lines is established, thereby enabling the research community to further study these clonal lines that have lost the ability to undergo sexual differentiation.
Methods
Whole genome sequencing and statistical analysis
Genomic DNA was prepared for 3D7A, F12 and A4, and underwent whole genome sequencing using Illumina technology with 76-base paired end fragment sizes [13, 23]. The raw sequence data (accession numbers ERS011445, ERS011446 and ERS011447) was processed as previously described [22]. In brief, the raw sequence data was aligned onto the 3D7 reference genome (version 3.0) using the bwa-mem short read alignment algorithm [25]. Single nucleotide polymorphisms (SNPs) and small insertions and deletions (indels) were identified using samtools and GATK, at a quality threshold of one error per one thousand basepairs (bp) [24, 25]. SNP genotypes were called using an established coverage-based approach, and polymorphisms excluded if they had missing or mixed genotypes. Larger structural variants, including deletions and amplifications were identified using alignment, sequence coverage and de novo assembly and applying the Delly software [26]. Unless performing candidate region analysis, we only considered genomic variants in regions that were unique (calculated by sliding 54 bp of contiguous sequence across the reference genome), non-sub-telomeric, and not in highly variable gene families (vars, stevors, rifins). De novo assembly was performed using velvet software [27], and was applied to localize the A4 plasmid insertion site. Genetic variation in the PF3D7_1222600 gene was characterized using the tools described above for a set of 930 field isolates sequenced by the MalariaGEN P. falciparum community project (accession numbers ERP000190 and ERP000199) [20–22, 28]. This set included isolates from Bangladesh (n = 54), East Africa (n = 86; Kenya 17, Malawi 69), West Africa (n = 224; Burkina Faso 39, Gambia 55, Guinea-Bissau 95, Mali 35) and Southeast Asia (n = 566, Thailand 91; Cambodia 253, Lao 35; Vietnam 187). Pairwise population FST was used to assess allele frequency differences between regions, and computed using the function stamppFst in the R package.
Var gene characterization
A coverage-based framework was used to characterize the highly polymorphic var gene family [15]. This approach consisted of the quantification of the total coverage and the number of trans locus reads across the genome using a sliding window of 100 and 25 bp overlap. Trans locus reads are defined as pairs of mate reads that have been improperly paired, where one of the mate pairs is located inside a var gene and the other mate read is located outside the same gene. A peak on the coverage in the trans locus reads together with an alteration on the total coverage (increase or decrease) in a specific region can suggest the presence of a structural variant. The structural variants found in the var genes using this approach were compared with those found using the Delly software and were annotated accordingly.
Results and discussion
Polymorphisms identified in gametocyte non-producer lines
The sequencing technology yielded in excess of thirty-six million 76 base-pair reads for each of the 3D7A, F12 and A4 cloned lines; their alignment led to an excess of 100-fold median coverage and over 98 % of the genome with at least fivefold coverage (Table 1; Fig. 1). A set of 178 high quality SNPs were identified, of which 109 (61.2 %) positions were found in unique and non-sub-telomeric regions. The majority (98/109) of SNPs had the same genotype across the 3 clones but different from the reference allele (Table 1; Fig. 1). Others have also identified differences between 3D7 clones and the reference genome [17, 18]. These discrepancies could be due to mutations introduced during long-term culture. It is also possible that some are potential sequencing or assembly errors in the 3D7 reference. Six of these SNPs cause a non-synonymous change in the protein and one introduces a stop codon in comparison to the reference (Additional file 1: Table S1). In the mitochondria and apicoplast organelles we found 2 and 12 SNPs respectively, but again the three strains had the same alleles, different from the 3D7 reference genome. No SNPs were unique for the 3D7A clone. Only 11 SNPs were unique to either two GNP strains, with no overlap in genes (Fig. 1; Additional file 1: Table S1). It was assessed whether any of these SNPs were present in a set of P. falciparum field isolates from countries in Africa and Asia [20–22]. Only one SNP, found in the Pfap2-g gene (PF3D7_1222600) that we previously described to affect gametocytes production in the F12 strain (as it introduces a stop codon) was identified in one sample from Guinea Bissau, but with a different mutation (S1308L). It is possible that this polymorphism has an effect on a gametocyte phenotype, but still allows transmission to mosquitoes. Unfortunately, no phenotypic data are available for the field isolates to study the effect of this SNP.
Table 1.
Genome sequencing and genetic variation
| Clone | Number of read pairs (Millions)b | Median coveragec | % genome with at least 5× coverage | Genetic variationd | ||
|---|---|---|---|---|---|---|
| SNPs | Indels | Structural variants | ||||
| 3D7A | 24.6 | 124 | 99.9 | 98 | 143 | 66 |
| F12a | 24.1 | 118 | 99.9 | 103 | 157 | 51 |
| A4a | 18 | 132 | 99.9 | 104 | 168 | 44 |
aGametocyte non-producers
bFrom Illumina GA II 76 bp paired-end
cMapped to 3D7 version 3.0 and excluding multi-copy apicoplast and mitochondria
dTotal variation per sample, including shared genetic variation but only polymorphisms located in non-sub-telomeric and unique regions
Fig. 1.
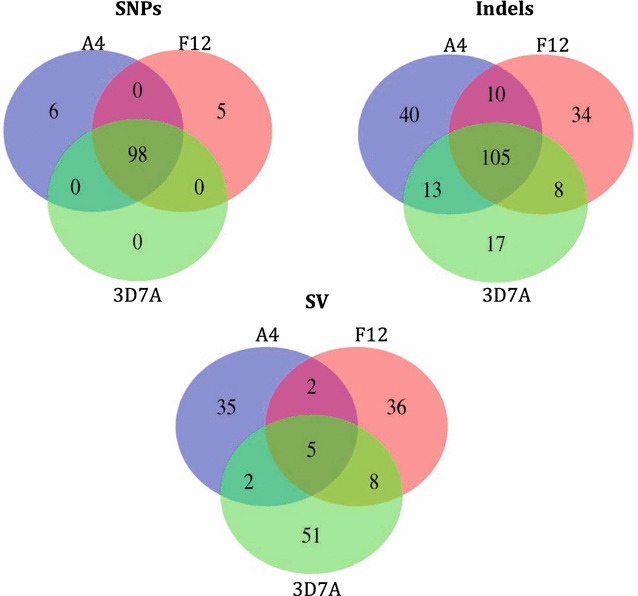
Venn diagram summarizing the polymorphisms found in A4, F12 and 3D7A clone lines. SNPs, insertions and deletions (indels) and structural variants (SVs) identified in unique and non-sub-telomeric regions of the core genome
The polymorphism list was extended by considering short indels. A set of high quality putative small indels (140 insertions/157 deletions, median length 2, range 1–20 bp) was identified (Additional file 2: Table S2; Fig. 1). The number of indels identified in unique and non-sub-telomeric genomic regions was 118 insertions/119 deletions. Eighty-three indels were only present in A4 or F12 (47 insertions, 36 deletions). Only two genes, PF3D7_0111200 (conserved Plasmodium protein, unknown function) and PF3D7_0726000 (a 28 rRNA encoding gene) have indels in both lines. In the PF3D7_0111200 gene there was an insertion in the intron. The PF3D7_0726000 gene has one deletion and one insertion present in the two lines. The same insertion was detected in 101 field isolates of the global set and the same deletion in 112. Only ten isolates have both. The indels detected in these genes most probably do not affect the production of gametocytes, as the insertion in PF3D7_0111200 is intronic, and the indels in the PF3D7_0726000 gene are frequent in the field isolates (~10 %).
In this set of indels, the insertion in the pfap2-g gene in the A4 strain that results in a reading frame shift and introduction of stop codons (Additional file 2: Table S2; Chromosome 12: 913765) was identified. This is the only protein-coding gene having polymorphisms in both strains that affect the coding sequence, introducing stop codons. These polymorphisms are responsible for the non-gametocyte production phenotype, as previously described [13].
Genetic diversity of Pfap2-g in Plasmodium falciparum field isolates collected across the globe
A polymorphism map using a set of P. falciparum genomes from Africa and Asia indicated that 91 SNPs (supported by at least two isolates) reside within the PF3D7_1222600 gene (Additional file 3: Table S3), and only 37 (41 %) are present at a minor allele frequency of more than 5 % (Fig. 2). Of these, 29 were non-synonymous mutations. Some of the mutations appear to be more common in malaria endemic regions, including the SNPs in positions 907264 (K21R, FST 0.41, East Africa vs Southeast Asia), 910153 (K984T, FST 0.35, West Africa vs Southeast Asia), 910741 (G1180E, FST 0.41, West Africa vs Southeast Asia), 913124 (K1974N, FST 0.31, West Africa vs Southeast Asia), 913157 (N1985 K, FST 0.53, East Africa vs Southeast Asia) and 914331 (Q2377K, FST > 0.67, Southeast Asia). Forty-six indels were identified but none change the protein frame shift nor introduce stop codons (Additional file 3: Table S3). These results emphasize the importance of this gene for parasite transmission, as no loss-of-function was detected in the field isolates.
Fig. 2.
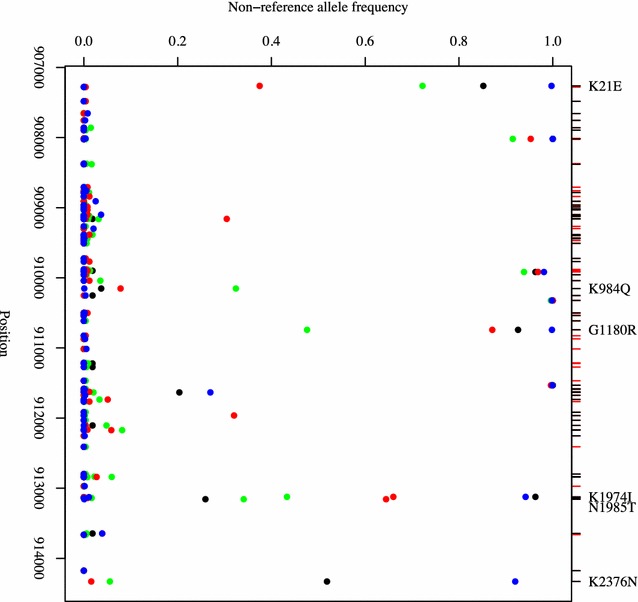
Allele frequencies of the SNPs in the PF3D7_1222600 gene across regions: West Africa (green), East Africa (red), Bangladesh (black), and Southeast Asia (blue). All mutations shown on upper axis (red tick marks = synonymous changes, black tick marks = non-synonymous; black text = with FST values > 0.3)
Structural variation in the core genome and sub-telomeric regions
Deletions, translocations and amplifications of large chromosomal stretches have been observed in P. falciparum lines following long-term in vitro culture as well as in field isolates [17, 18]. The sequence data were examined for structural variants in these parasite lines. Few structural variants were found within the core genome (Additional file 4: Table S4; Fig. 1), consisting mostly of deletions with an average length of 250 bp, and located predominantly in non-coding regions. No truncation on chromosome 9, which has been previously implicated in the inability to produce gametocytes in vitro, was observed in these parasite lines, as previously described (Additional file 5: Figure S1) [9–11]. In the A4 strain we detected a possible deletion in a non-coding region in the telomeric region of chromosome 9 (Additional file 5: Figure S1). Rearrangements in chromosomal sub-telomeric regions were found, particularly between the var family gene members. Previous studies have observed recombination between sub-telomeric var genes involving those located either in the immediate vicinity on the same chromosome, or on different chromosomes [17, 18]. These rearrangements retain the frame-shift of the var genes and therefore form functional loci. Four putative rearrangements all involving neighbouring var genes were detected. Two chimeric genes generated by recombination between two adjacent var genes were identified: (i) PF3D7_0421100 and PF3D7_0421300 (chromosome 4) found in F12, and (ii) PF3D7_1240400 and PF3D7_1240600 (chromosome 12) detected in all three lines (Fig. 3a, b). Chimeric var genes involving exactly these pairs of adjacent genes have been reported previously in 3D7 clones [17]. More complex rearrangements were detected, including duplication of var genes and formation of chimeras in the same region and chimeric genes formed by multiple cross-overs in between the same two adjacent genes (Fig. 3b–d). These were only detected in single isolates. The generation of chimeric var genes is a critical mechanism for immune invasion, and it has been proposed that these genes have a recombination rate of up to 0.2 % per life cycle (in vitro) [17]. This means that millions of new antigenic structures could possibly be produced in a single infected individual per day, giving the parasite one-upmanship in relation to the host immune system.
Fig. 3.
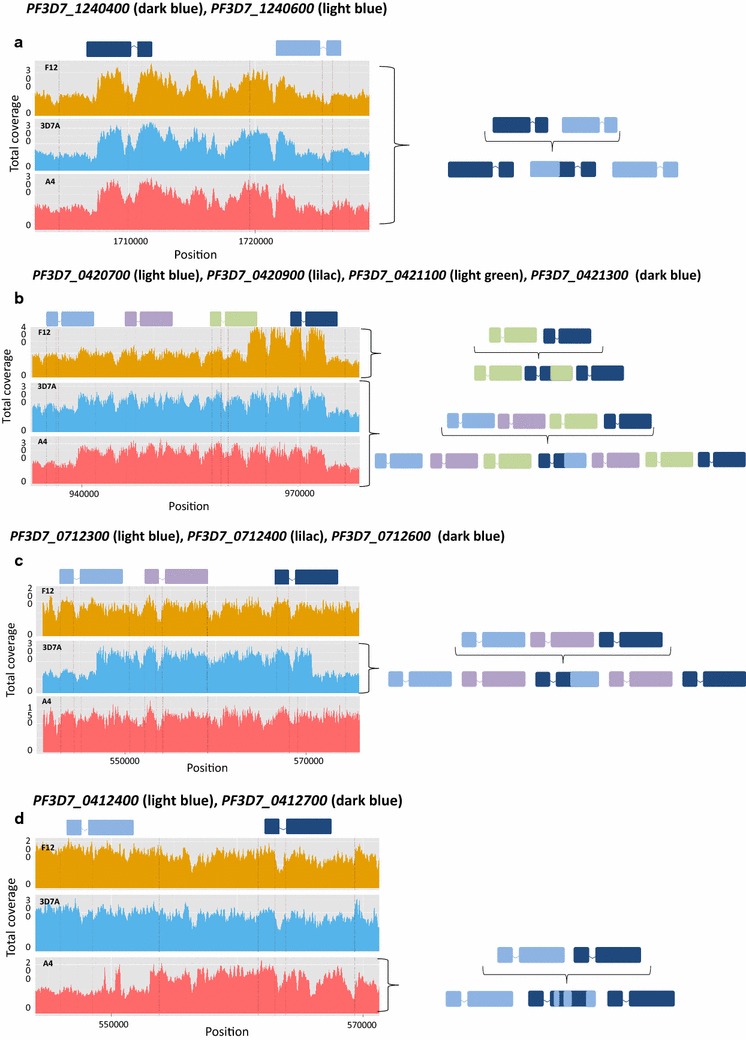
Chimeric var genes. a For all clone lines, there is twofold increase coverage for the second part of PF3D7_1240400 (dark blue) and the first part of the adjacent gene, PF3D7_1240600 (light blue). The duplication-chimera was formed by the fusion of the half of PF3D7_1240600 and half of PF3D7_1240400, the var gene immediately adjacent. b For F12, a twofold increase coverage was observed for the second part of PF3D7_0421100 (green), and the first part of the adjacent gene, PF3D7_0421300 (dark blue) indicating a duplication-chimera formed by duplicating half of PF3D7_0421100 and the other half of PF3D7_0421300. For the A4 and 3D7A strains an increased coverage of the second half of PF3D7_0420700, the full genes PF3D7_0420900 and PF3D7_0421100 and first half of gene PF3D7_0421300, was observed. This indicates a duplication of the genes PF3D7_0420900 and PF3D7_0421100 and a chimeric gene formed by duplicating half of PF3D7_0420700 and half of PF3D7_0421300. c For 3D7A, there is twofold increase coverage for the second part of PF3D7_0712300 (light blue), the whole of PF3D7_0712400 (lilac) and the first part of the adjacent gene PF3D7_0712600 (dark blue). It indicates a duplication of the full gene PF3D7_0712400 and a duplication-chimera formed by duplicating half of PF3D7_0712300 and half of PF3D7_0712600; d For A4, there is twofold increase in coverage of several regions of PF3D7_0412400 (light blue) and PF3D7_0412700 (dark-blue) only for A4. It indicates a chimeric gene formed by multiple cross-overs between the two adjacent genes
Typically, var genes contain two exons. Across the three strains, we detected a highly conserved area in the intronic region between the two exons. This intronic region seems to have very high coverage because the mate sequenced paired-reads map to almost all of the other var genes in all chromosomes. This region has been reported previously and experimental studies have shown that the conserved var introns are important regulatory elements [29, 30]. The regulation of var gene expression and silencing is closely linked to the regulation of sexual commitment, and both mechanisms are controlled by the same epigenetic machinery. The knockdown of the heterochromatin protein 1 (PF3D7_1220900) and a histone deacetylase (PF3D7_1008000) promote gametocytogenesis and leads to de-regulation of mutually exclusive var gene expression [30–32]. It is not known whether disruptions in var gene expression, due to e.g., chromatin modifications, also affect var gene recombination and, therefore, antigenic diversity. It has been suggested that introducing malaria control programs may increase parasite virulence in some circumstances [33]. The share of the core epigenetic machinery between these fundamental parasitic processes should be taken into account during control interventions (e.g., vaccine introduction) as blocking one process might unexpectedly affect the other process.
Location of the A4 plasmid insertion sites following transfection and drug selection
The A4 is a cloned transgenic line that was originally generated with the intention of investigating whether the absence of the phosphodiesterase gene (PDEδ, PF3D7_1470500) had any effect on exflagellation. The A4 clone harbours a whole plasmid (7 kbp; more than one copy) designed to delete the PDEδ gene [34]. Following drug selection, this transgenic line lost the ability to produce gametocytes, but this phenotype was unrelated to PDEδ function. Importantly, it was subsequently shown using another cloned line in which the PDEδ gene had been successfully disrupted that whilst the production of gametocytes was unaffected, there was a severe reduction in the ability to undergo gametogenesis [16]. The A4 sequence data was used to confirm the genomic insertion of the plasmid and the possibility that integration happened at more than one site and disrupted other genes. De novo assembly of the reads led to a contig of length ~10 kbp, containing the ~7 kbp intact plasmid (derived from pHTK [35]), and a flanking 3 kbp region. A blast search of the contig to the reference genome indicated that the P. falciparum-derived elements of the plasmid matched to their respective chromosome sites with at least 99 % identity. In particular, the 3D7 derived contig regions 1–845, 5510–6062, 6269–7134, 7360–10303 matched to chromosome 14 (2883802-2884646, PDEδ), 7 (381591-381037, HSP90), 14 (2882276-2883143, PDEδ), and 14 (1369973–1372928, PF3D7_1434300, HOP—Hsp70/Hsp90 organizing protein), with 99.8, 98.7, 99.1 and 99.5 % identity, respectively. Pulse field blot analysis of A4 indicated an insertion of the plasmid within chromosome 14 [34], leaving the PDEδ (located in 2882766–2886082) and HOP (PF3D7_1434300 located in 1372903–13745970) regions as potential candidates for insertion. Regions neighbouring the HOP and PDEδ (2882276–2883143) genes showed an excess of read coverage suggesting potential insertion sites for the plasmid (Additional file 6: Figure S2). To localize the exact plasmid integration site, the 3 kbp of the plasmid flanking sequence generated when performing de novo assembly of the plasmid was used. This flanking region contains pair-reads that mapped both the plasmid and the HOP region (1369961–1372928), confirming this location as a likely site of plasmid integration (Additional file 6: Figure S2). Excess coverage detected in two PDEδ regions corresponds to the two sequences that were inserted into the vector.
This sequence data analysis indicates that it is possible that integration occurred in a different genomic location to that intended, and that care must be taken when interpreting the observed phenotype. The application of whole genome sequencing and new reverse genetic methodology will ensure such issues will be less problematic in future.
Prediction of gametocyte/sexual development genes using DNA binding sequence motifs
A large proportion of P. falciparum genes lack a known function. Using a bioinformatic approach it is possible to identify genes that could function at specific P. falciparum developmental stages, and take into account similarities in gene expression patterns to predict function (‘guilt by association’). By applying this method, a study using stage-specific gametocyte mRNA preparations in conjunction with microarray analysis and using ‘guilt by association’ approach, generated a ‘sexual development’ cluster containing 246 genes [36]. A palindromic sequence (TGTANNTACA) was identified in the 5′ UTR of 65 genes in this cluster [36]. The A4 and F12 whole genome sequence data were scanned for this motif and 23 genes were found, seven overlapping with the 65 in the cluster. There was also a scan for the consensus DNA-binding preference of the transcription factor PfAP2-G that binds to specific motifs in the 5′ upstream region of early gametocyte genes to turn on transcription [37]. Fifty-one genes were identified, 11 also having the palindromic sequence and five of them overlapping with the 65 genes in the sexual development cluster (Additional file 7: Table S5). According to Illumina-based sequencing of P. falciparum 3D7 mRNA from gametocytes and erythrocytic stages [38], these 11 genes have higher expression during the sexual stages. Three of these genes were present in the guilt by association data set. This overlap could suggest that the two motifs, identified by independent means, may have a relationship in terms of determining timely expression of genes, particularly at the onset of gametocyte/sexual development, as the transcription factor PfAP2-G seems to regulate the expression of early gametocyte genes.
The small overlap between the various datasets may indicate that other regulatory motifs besides the two analysed here, are important during the sexual stage. Overall, these types of approach, that integrate data from varying bioinformatics and gene expression studies, are important to identify target genes for further functional work, particularly when so many genes in the P. falciparum genome are hypothetical proteins. However, several genes may be missed, especially if they have low expressions levels, are regulated by different mechanisms or involved in negative regulation. Moreover, there is the need to support gene transcription data particularly with quantitative proteomics, to generate more robust hypotheses.
Conclusion
A comprehensive analysis is presented of the three P. falciparum genomes that were previously used as part of a study to determine that the transcriptional regulator AP2-G is a key determinant of sexual differentiation. These cloned lines have been in long-term culture, so as expected several SNPs, indels, structural variants and some new chimeric var genes were identified. The acquisition of genetic variants and the recombination of the var genes during mitosis provide the parasite with a mechanism that can generate an enormous amount of genetic diversity, which could lead to selective advantage. These mutations might be phenotypically undetectable or may cause a visible phenotype, as in the case of the gametocyte non-production in the F12 and A4 clones. Also, by performing whole genome sequencing of a transgenic line we have identified an unexpected integration site. This finding may motivate others to take this step to identify any additional changes that have occurred in transgenic lines that might potentially contribute to a phenotype. These lines were fundamental to identifying the Pfap2-g gene, and they provide an important tool to understand gametocytogenesis in vitro. There are still outstanding questions concerning how sexual commitment occurs in vivo, and whether this locus may be useful in predicting and monitoring transmission. Ultimately, fully understanding the molecular events underpinning gametocyte production could lead to a therapeutic which totally prevents sexual development and thereby blocks transmission. The A4 and F12 gametocyte non-producing lines and their sequence data will be a useful resource for future studies looking to translate biological insights into malaria control measures.
Availability of supporting data
The sequence data were generated by the Malaria Programme at the Wellcome Trust Sanger Institute and are publically available from the EBI short read archive (accession numbers: Clonal strains ERS011445, ERS011446 and ERS011447; field isolates ERP000190 and ERP000199). The data can also be accessed and browsed through the Pf3k web application (www.malariagen.net/apps//pf3k).
Authors’ contributions
SC, DAB, and TGC conceived and designed the study; SC, ET, LGD, CJT, ZG and PA performed laboratory experiments, contributed biological samples, microarray, sequencing or phenotypic data; SC, EDB, SAA and TGC performed the statistical analysis; SC and DPK led the sequencing effort. SC, TGC and DAB wrote/drafted and finalized the manuscript with contributions from all other authors. All authors read and approved the final manuscript.
Acknowledgements
DAB acknowledges support from the Wellcome Trust (Project Grant Ref WT094752, Senior Investigator Award Ref 106240). TGC is supported by the Medical Research Council UK (Grant numbers MR/K000551/1, MR/M01360X/1, MR/N010469/1), and SC by a MRC UK grant (MR/M01360X/1). This publication uses data from the MalariaGEN Plasmodium falciparum Community Project (www.malariagen.net/projects/parasite/pf) as described in refs 18 and 19. Genome sequencing was performed by the Wellcome Trust Sanger Institute and the Community Project is coordinated by the MalariaGEN Resource Centre with funding from the Wellcome Trust (098051, 090770).
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- SNPs
single nucleotide polymorphisms
- GNP
gametocyte non-producer
- indels
insertions and deletions
- bp
basepairs
- SVs
structural variants
Additional files
10.1186/s12936-016-1254-1 SNPs identified in the three lines 3D7A, A4 and F12.
10.1186/s12936-016-1254-1 Insertions (INS) and deletions (DEL) identified in the three lines 3D7A, A4 and F12.
10.1186/s12936-016-1254-1 SNPs and Indels identified in the PF3D7_1222600 gene among P. falciparum field isolates from Africa and Asia.
10.1186/s12936-016-1254-1 Structural variants identified in the three lines 3D7A, A4 and F12.
10.1186/s12936-016-1254-1 Coverage plots to evaluate evidence of truncation in the end of chromosome 9. (a) Coverage in the chromosome 9 candidate region (20 genes from PF3D7_0935400 to PF3D7_0937300, shaded) showing no evidence of deletion (also confirmed using CGH data (not presented)) (A4, blue; 3D7A, green; F12, red); (b) Evidence of a deletion in a sub-telomeric non-coding region of chromosome 9 in the A4 strain, but not 3D7A or F12.
10.1186/s12936-016-1254-1 Coverage plots showing potential insertion points of the A4 plasmid. Regions neighbouring the (a) HOP and (b) PDE-delta genes showed an excess of read coverage suggesting potential insertion sites for the plasmid. The flanking region of the de novo assembled plasmid-contig maps to the HOP region confirmed this location as a site of plasmid integration (Chromosome 14: 1369961–1372928). Excess coverage observed in two PDEδ regions corresponds to the two sequences that were inserted into the vector.
10.1186/s12936-016-1254-1 A scan for motifs in gene sequences (coding and UTR regions).
Footnotes
Susana Campino, Ernest Diez Benavente, David Baker and Taane Clark contributed equally.
Contributor Information
Susana Campino, Email: susana.campino@lshtm.ac.uk.
Ernest Diez Benavente, Email: Ernest.DiezBenavente@lshtm.ac.uk.
Samuel Assefa, Email: samuel.assefa@lshtm.ac.uk.
Eloise Thompson, Email: Eloise.Thompson@lshtm.ac.uk.
Laura G. Drought, Email: laura.drought@lshtm.ac.uk
Catherine J. Taylor, Email: Catherine.taylor@lshtm.ac.uk
Zaria Gorvett, Email: zaria.gorvett@lshtm.ac.uk.
Celine K. Carret, Email: Celine.carret@embo.org
Christian Flueck, Email: Christian.Flueck@lshtm.ac.uk.
Al C. Ivens, Email: Al.Ivens@ed.ac.uk
Dominic P. Kwiatkowski, Email: dominic@well.ox.ac.uk
Pietro Alano, Email: alano@iss.it.
David A. Baker, Email: david.baker@lshtm.ac.uk
Taane G. Clark, Email: taane.clark@lshtm.ac.uk
References
- 1.WHO . World Malaria Report 2015. Geneva: World Health Organization; 2015. [Google Scholar]
- 2.Baker DA. Malaria gametocytogenesis. Mol Biochem Parasitol. 2010;172:57–65. doi: 10.1016/j.molbiopara.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.A-Elbasit IE, Elbashir MI, Khalil IF, Alifrangis M, Giha HA. The efficacy of sulfadoxine-pyrimethamine alone and in combination with chloroquine for malaria treatment in rural Eastern Sudan: the interrelation between resistance, age and gametocytogenesis. Trop Med Int Health. 2006;11:604–612. doi: 10.1111/j.1365-3156.2006.01616.x. [DOI] [PubMed] [Google Scholar]
- 4.Puta C, Manyando C. Enhanced gametocyte production in Fansidar-treated Plasmodium falciparum malaria patients: implications for malaria transmission control programmes. Trop Med Int Health. 1997;2:227–229. doi: 10.1046/j.1365-3156.1997.d01-267.x. [DOI] [PubMed] [Google Scholar]
- 5.Williams JL. Stimulation of Plasmodium falciparum gametocytogenesis by conditioned medium from parasite cultures. Am J Trop Med Hyg. 1999;60:7–13. doi: 10.4269/ajtmh.1999.60.7. [DOI] [PubMed] [Google Scholar]
- 6.Barnes KI, Little F, Mabuza A, Mngomezulu N, Govere J, Durrheim D, et al. Increased gametocytemia after treatment: an early parasitological indicator of emerging sulfadoxine-pyrimethamine resistance in falciparum malaria. J Infect Dis. 2008;197:1605–1613. doi: 10.1086/587645. [DOI] [PubMed] [Google Scholar]
- 7.Trager W, Gill GS. Plasmodium falciparum gametocyte formation in vitro: its stimulation by phorbol diesters and by 8-bromo cyclic adenosine monophosphate. J Protozool. 1989;36:451–454. doi: 10.1111/j.1550-7408.1989.tb05822.x. [DOI] [PubMed] [Google Scholar]
- 8.Dyer M, Day K. Expression of Plasmodium falciparum trimeric G proteins and their involvement in switching to sexual development. Mol Biochem Parasitol. 2000;110:437–448. doi: 10.1016/S0166-6851(00)00288-7. [DOI] [PubMed] [Google Scholar]
- 9.Day KP, Karamalis F, Thompson J, Barnes DA, Peterson C, Brown H, et al. Genes necessary for expression of a virulence determinant and for transmission of Plasmodium falciparum are located on a 0.3-megabase region of chromosome 9. Proc Natl Acad Sci USA. 1993;90:8292–8296. doi: 10.1073/pnas.90.17.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alano P, Roca L, Smith D, Read D, Carter R, Day K. Plasmodium falciparum: parasites defective in early stages of gametocytogenesis. Exp Parasitol. 1995;81:227–235. doi: 10.1006/expr.1995.1112. [DOI] [PubMed] [Google Scholar]
- 11.Eksi S, Morahan BJ, Haile Y, Furuya T, Jiang H, Ali O, et al. Plasmodium falciparum gametocyte development 1 (Pfgdv1) and gametocytogenesis early gene identification and commitment to sexual development. PLoS Pathog. 2012;8:e1002964. doi: 10.1371/journal.ppat.1002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ikadai H, Shaw Saliba K, Kanzok SM, McLean KJ, Tanaka TQ, Cao J, et al. Transposon mutagenesis identifies genes essential for Plasmodium falciparum gametocytogenesis. Proc Natl Acad Sci U S A. 2013;110:E1676–1684. doi: 10.1073/pnas.1217712110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kafsack BFC, Rovira-Graells N, Clark TG, Bancells C, Crowley VM, Campino SG, et al. A transcriptional switch underlies commitment to sexual development in malaria parasites. Nature. 2014;507:248–252. doi: 10.1038/nature12920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sinha A, Hughes KR, Modrzynska KK, Otto TD, Pfander C, Dickens NJ, et al. A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature. 2014;507:253–257. doi: 10.1038/nature12970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silvestrini F, Bozdech Z, Lanfrancotti A, Di Giulio E, Bultrini E, Picci L, et al. Genome-wide identification of genes upregulated at the onset of gametocytogenesis in Plasmodium falciparum. Mol Biochem Parasitol. 2005;143:100–110. doi: 10.1016/j.molbiopara.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 16.Taylor CJ, McRobert L, Baker DA. Disruption of a Plasmodium falciparum cyclic nucleotide phosphodiesterase gene causes aberrant gametogenesis. Mol Microbiol. 2008;69:110–118. doi: 10.1111/j.1365-2958.2008.06267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Claessens A, Hamilton WL, Kekre M, Otto TD, Faizullabhoy A, Rayner JC, et al. Generation of antigenic diversity in Plasmodium falciparum by structured rearrangement of Var genes during mitosis. PLoS Genet. 2014;10:e1004812. doi: 10.1371/journal.pgen.1004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bopp SER, Manary MJ, Bright AT, Johnston GL, Dharia NV, Luna FL, et al. Mitotic evolution of Plasmodium falciparum shows a stable core genome but recombination in antigen families. PLoS Genet. 2013;9:e1003293. doi: 10.1371/journal.pgen.1003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miotto O, Amato R, Ashley EA, MacInnis B, Almagro-Garcia J, Amaratunga C, et al. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat Genet. 2015;47:226–234. doi: 10.1038/ng.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manske M, Miotto O, Campino S, Auburn S, Almagro-Garcia J, Maslen G, et al. Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature. 2012;487:375–379. doi: 10.1038/nature11174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ocholla H, Preston MD, Mipando M, Jensen ATR, Campino S, MacInnis B, et al. Whole-genome scans provide evidence of adaptive evolution in Malawian Plasmodium falciparum isolates. J Infect Dis. 2014;210:1991–2000. doi: 10.1093/infdis/jiu349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Auburn S, Campino S, Clark TG, Djimde AA, Zongo I, Pinches R, et al. An effective method to purify Plasmodium falciparum DNA directly from clinical blood samples for whole genome high-throughput sequencing. PLoS One. 2011;6:4–11. doi: 10.1371/journal.pone.0022213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Preston MD, Assefa SA, Ocholla H, Sutherland CJ, Borrmann S, Nzila A, et al. PlasmoView: a web-based resource to visualise global Plasmodium falciparum genomic variation. J Infect Dis. 2014;209:1808–1815. doi: 10.1093/infdis/jit812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics. 2014;30:2843–2851. doi: 10.1093/bioinformatics/btu356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–339. doi: 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Samad H, Coll F, Preston MD, Ocholla H, Fairhurst RM, Clark TG. Imputation-based population genetics analysis of Plasmodium falciparum malaria parasites. PLoS Genet. 2015;11:e1005131. doi: 10.1371/journal.pgen.1005131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calderwood MS, Gannoun-Zaki L, Wellems TE, Deitsch KW. Plasmodium falciparum var genes are regulated by two regions with separate promoters, one upstream of the coding region and a second within the intron. J Biol Chem. 2003;278:34125–34132. doi: 10.1074/jbc.M213065200. [DOI] [PubMed] [Google Scholar]
- 30.Avraham I, Schreier J, Dzikowski R. Insulator-like pairing elements regulate silencing and mutually exclusive expression in the malaria parasite. Plasmodium falciparum. 2012;109:E3678–3686. doi: 10.1073/pnas.1214572109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brancucci NMB, Bertschi NL, Zhu L, Niederwieser I, Chin WH, Wampfler R, et al. Heterochromatin protein 1 secures survival and transmission of malaria parasites. Cell Host Microbe. 2014;16:165–176. doi: 10.1016/j.chom.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 32.Coleman BI, Skillman KM, Jiang RHY, Childs LM, Altenhofen LM, Ganter M, et al. A Plasmodium falciparum histone deacetylase regulates antigenic variation and gametocyte conversion. Cell Host Microbe. 2014;16:177–186. doi: 10.1016/j.chom.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mackinnon MJ, Gandon S, Read AF. Virulence evolution in response to vaccination: the case of malaria. Vaccine. 2008;26(Suppl 3):C42–52. doi: 10.1016/j.vaccine.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor CJ. The role of two cyclic nucleotide phosphodiesterases in the sexual development of Plasmodium falciparum and Plasmodium berghei. PhD Thesis. University of London; 2007.
- 35.Duraisingh MT, Triglia T, Cowman AF. Negative selection of Plasmodium falciparum reveals targeted gene deletion by double crossover recombination. Int J Parasitol. 2002;32:81–89. doi: 10.1016/S0020-7519(01)00345-9. [DOI] [PubMed] [Google Scholar]
- 36.Young JA, Fivelman QL, Blair PL, de la Vega P, Le Roch KG, Zhou Y, et al. The Plasmodium falciparum sexual development transcriptome: a microarray analysis using ontology-based pattern identification. Mol Biochem Parasitol. 2005;143:67–79. doi: 10.1016/j.molbiopara.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 37.Campbell TL, De Silva EK, Olszewski KL, Elemento O, Llinás M. Identification and genome-wide prediction of DNA binding specificities for the ApiAP2 family of regulators from the malaria parasite. PLoS Pathog. 2010;6:e1001165. doi: 10.1371/journal.ppat.1001165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.López-Barragán MJ, Lemieux J, Quiñones M, Williamson KC, Molina-Cruz A, Cui K, et al. Directional gene expression and antisense transcripts in sexual and asexual stages of Plasmodium falciparum. BMC Genomics. 2011;12:587. doi: 10.1186/1471-2164-12-587. [DOI] [PMC free article] [PubMed] [Google Scholar]