

Abstract
Background
Knowing the binding site of protein–protein complexes helps understand their function and shows possible regulation sites. The ultimate goal of protein–protein docking is the prediction of the three-dimensional structure of a protein–protein complex. Docking itself only produces plausible candidate structures, which must be ranked using scoring functions to identify the structures that are most likely to occur in nature.
Methods
In this work, we rescore rigid body protein–protein predictions using the optimized potential for efficient structure prediction (OPEP), which is a coarse-grained force field. Using a force field based on continuous functions rather than a grid-based scoring function allows the introduction of protein flexibility during the docking procedure. First, we produce protein–protein predictions using ZDOCK, and after energy minimization via OPEP we rank them using an OPEP-based soft rescoring function. We also train the rescoring function for different complex classes and demonstrate its improved performance for an independent dataset.
Results
The trained rescoring function produces a better ranking than ZDOCK for more than 50 % of targets, rising to over 70 % when considering only enzyme/inhibitor complexes.
Conclusions
This study demonstrates for the first time that energy functions derived from the coarse-grained OPEP force field can be employed to rescore predictions for protein–protein complexes.
Keywords: Protein–protein docking, Coarse graining, Rescoring, Flexible docking
Background
One of the main goals of proteomic research is to understand the biological function of proteins. Many proteins generate their function not as monomers but as part of complexes. Thus knowledge about protein–protein interactions is fundamental and allows regulation of protein structure and function. The Protein Data Bank (PDB) [1] contains more than one hundred thousand protein structures. However, structures of protein–protein complexes are often difficult to determine experimentally. These complexes are usually very big, which is a problem for elucidating structure via nuclear magnetic resonance (NMR), and the interactions are often too transient to be captured by X-ray crystallography.
Protein-protein docking is an in silico method for predicting the structures of protein–protein complexes. One can predict possible binding sites in a complex based on the protein structures in their unbound state. The binding partners can be single proteins or smaller protein–protein complexes. To increase computing efficiency, the proteins are usually modelled as rigid bodies at the first six-dimensional (6D) global search stage. Most of these global search methods are based on the convolution of grids, where the surface of the binding partners are parametrized such that an overlap between the surfaces of the two binding partners becomes possible. The aim of this surface description is to implicitly account for conformational changes upon binding. The convolution of the grids is accelerated by fast Fourier transformation (FFT) [2–5]. In the simplest approach, the convolution produces possible docking positions based solely on the shape of the proteins. However, more sophisticated grid maps exist which take chemical and knowledge-based properties into account. For refining the initial predictions, various methods are commonly applied, for instance Monte Carlo (MC) simulations [6, 7], clustering [8, 9], or side-chain optimization using rotamer libraries [10]. As computation time is usually the limiting factor, an MC simulation should start from a conformation close to the binding site. A complete global search with this method in a reasonable computing time would be impossible.
The global search, which is performed via ZDOCK in this study [11], usually finds many similar solutions [4]. Therefore, it is common practice to cluster and rerank the docking predictions. Reranking classifies and distinguishes native or near-native solutions from non-native or wrong predictions [12, 13]. The number of predictions in a cluster can also be used for reranking [14]. The aim of both approaches is to narrow down the list of possible interaction sites, significantly decreasing computational cost and effort for further analysis of the remaining docking predictions.
To investigate protein–protein complexes produced by ZDOCK, docking approaches that allow for more protein flexibility than ZDOCK with low time expenditure are needed. A coarse-grained force field should be a good choice here. Various coarse-grained force fields have already been developed for the treatment of protein–protein complexes, including the calculation of thermodynamic and structural properties of multi-protein complexes with relatively low binding affinities [15]. Coarse-grained models are also used for molecular dynamics (MD) simulations of protein–protein association [16, 17], where the proteins are modelled using the MARTINI force field [18, 19] or with a Go-model approach [20]. In the latter approach [17], the electrostatic and hydrophobic interactions between proteins are modelled via a Coulomb potential with a distance dependent dielectric constant and the Miyzawa-Jernigan potential [21].
In the current study, we apply the coarse-grained ‘Optimized Potential for Efficient structure Prediction’ (OPEP) [22] to the protein–protein docking problem. A coarse-grained force field is used because of the reduced number of degrees of freedom, making it computationally more efficient than an all atom potential. Moreover, it is believed that a coarse-grained model will smooth the underlying free energy landscape, facilitating exploration of the corresponding phase space [23]. OPEP has already been successfully employed with different techniques, including MD and MC simulations. It was applied to RNA/DNA/protein systems to investigate the effect of crowding, to amyloid formation, and for protein 3D structure prediction. A recent overview of OPEP and its applications can be found in [22]. This work investigates OPEP’s applicability to protein–protein complexes. To test its performance for protein–protein docking, the first step is to investigate the discriminating power of OPEP to distinguish between correctly and wrongly docked complexes. We use global docking predictions produced by ZDOCK which we coarse grain and energy minimize using OPEP, followed by rescoring with an OPEP-based soft potential. Moreover, we enhance the performance of the rescoring function via an iterative learning procedure and test the resulting scoring function on a subset of the Dockground benchmark [24].
Methods
We perform unbound docking, which starts from the binding partners in their native conformations. The methods applied for predicting and rescoring protein–protein complexes can be summarized via the following pipeline: For each of the 96 targets we produce 54,000 docking predictions with ZDOCK and retain the best 2000 of these complexes, as recommended by the ZDOCK developers. These predictions are energy minimized using the OPEP force field (step (1) in Fig. 1). For each prediction we perform 140 minimization steps in full Cartesian space with the limited-memory Broyden–Fletcher–Goldfarb–Shanno (LBFGS) minimizer [25], which leads to minimization times between 3.5 s for the target with PDB ID 1AY7 (185 amino acids) and 250 s for the target with PDB ID 2HMI (1413 amino acids) on a single CPU core. This amounts to an overall minimization time for the 2000 ZDOCK predictions per target of less than 24 h for 85 % of targets. Afterwards, the minimized predictions are reranked. For this, we replaced the side chain–side chain interaction potential of OPEP with a softer 8-6 Lennard-Jones-potential, while preserving the optimal distances and energies (step (2) in Fig. 1). At this stage, the OPEP potentials for salt bridges, interactions involving backbone atoms, and H-bonds are not changed. In a further step, we trained the parameters of side chain–side chain interactions, including salt bridge interactions with an iterative learning approach with the aim of further improving the performance of the OPEP-based rescoring function (steps (3)–(6) in Fig. 1). The resulting scoring function is tested on another dataset to independently prove its ability to distinguish between native and non-native complexes.
Fig. 1.
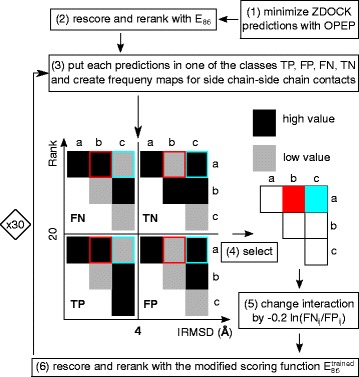
The training scheme for the side chain–side chain interactions. Every prediction is minimized (1) and rescored (2). Each prediction is classified as either TP, FP, FN, or TN (3). For each of these classes, an average contact map is created. Contact maps are shown for an artificial example containing only three residues. To train the potential, the side chain–side chain interaction a/b is selected because it is more frequent in TP and FN predictions than in FP and TN (4). The side chain–side chain interaction a/c, on the other hand, is selected because it is more frequent in FP and TN than in TP and FN predictions (4). The a/b interaction is strengthened by decreasing its energy, while the a/c interaction is disfavoured by increasing its energy (5). This leads to the new scoring function , with which the predictions are rescored. Steps (3) to (6) are iterated 30 times on the training dataset
The dataset
We use two different benchmarks to perform unbound docking. ZDOCK benchmark 4.0 is used as training dataset, while for further evaluation we use the Dockground benchmark 2.0. We used a subset of ZDOCK benchmark 4.0 [26]. We downloaded the docking predictions for 6° angular sampling from the ZDOCK website, which were obtained using ZDOCK 3.02 [27]. Ninety-six complexes were selected, including 39 enzyme/inhibitor, 19 antigen/antibody, and 38 other types of complexes. The latter will be called ‘other complexes’ for the remainder of this paper. One condition for selecting these complexes is that ZDOCK found at least one hit in the top 2000 predictions. A hit is defined as a prediction with an interface root mean square deviation (IRMSD) from the target of lower than 4 Å. Complexes that contain small molecules like ATP and GTP, for which OPEP is not parametrized, were not considered. The 1N2C complex could not be used, because it has more than 15,000 beads after coarse graining and the fixed file format for parametrization in OPEP currently only allows for up to 9999 beads.
The second dataset is a subset of the Dockground benchmark [24]. Here we follow the same selection criteria as for the ZDOCK benchmark. Furthermore, we remove complexes present in ZDOCK benchmark 4.0 in order to generate an independent and unbiased test set. The resulting test set contains 74 targets with 18 enzyme/inhibitor, 16 antigen/antibody, and 40 other complexes. As before, to generate complex predictions we applied ZDOCK with 6° sampling, using a local ZDOCK 3.02 installation and keeping the top 2000 predictions. As in the ZDOCK dataset, the docking for the antigen/antibody complexes was restricted to the complementarity determining regions (CDRs).
ZDOCK
ZDOCK is an FFT-based rigid-body protein–protein docking algorithm. During the search procedure one protein is kept fixed, while the other is moved around it. The fixed protein is usually the larger of the two and is called the receptor, while the other protein is the ligand. ZDOCK generates grid-based representations from the full atom chains of receptor and ligand and after each ligand rotation the grids can be fast convoluted via FFT. The three rotational angles of the ligand are sampled with a 6° spacing, and the 3 translational degrees of freedom are sampled with a 1.2 Å spacing. For each set of rotational angles, only the best (based on ZDOCK score) translationally sampled prediction is retained [28]. This leads to 54,000 ZDOCK predictions, of which we consider the top 2000 for further refinement. To account for some flexibility in ZDOCK, a soft docking approach is used where the receptor has a 3.4 Å thick surface layer [3]. This allows for some overlap between receptor and ligand and accounts for possible movements during docking. However, it may also lead to atom clashes between receptor and ligand. The ZDOCK scoring function contains a shape-complementary term [29], a knowledge-based contact term for atoms and residues [11], and an electrostatic term [30].
Missing residues and atoms
Some of the complex structures considered are missing certain residues in the receptor and/or ligand. Although this is no problem for a grid-based method like ZDOCK, it must be resolved for treatment with OPEP. Missing residues lead to gaps in the backbone chain and, if untreated, they would be considered overstretched bonds. In order to resolve this problem, polypeptides with missing residues are treated as separate chains. The distance between the terminal carbon and the terminal nitrogen of the gap is kept fixed via a harmonic potential with the equilibrium distance equal to the initial gap length and a force constant of 100 kcal/(mol·Å2).
OPEP
As rescoring function we use the coarse-grained potential OPEP or variations of it. OPEP uses a six bead representation for every amino acid except proline and glycine. The amino nitrogen N, the Cα, and the carbonyl carbon C’ atoms of the backbone are each modelled by one bead. In addition, the hydrogen H of the amino-group and carbonyl oxygen O are explicitly represented. Side chains are described by only one bead, except for proline where all heavy side chain atoms are modelled. The local energy terms in OPEP were developed based on the functional form of the Amber force field [31] and several rounds of minor adaptations to the side chain–side chain interactions have been conducted [22]. We use the latest version of OPEP, OPEPv5 [32], which for the first time includes an explicit potential for salt bridges that were parametrized with an iterative Boltzmann inversion method with parameters extracted from all atom MD simulations. A complete description of the OPEP potential can be found in the original OPEP publications [22, 31–33]. Here, we only present the nonbonded interactions, as they are used to rescore the protein–protein complexes. The nonbonded potential consists of four terms: (1) van der Waals interactions involving backbone atoms (EVDW), (2) hydrophobic and hydrophilic side chain–side chain interactions (ESS), (3) hydrogen bond (H-bond) interactions between backbone atoms (EHB), and (4) a potential for salt bridges (ESB). Interactions between side chains ESS are modelled differently for attractive and repulsive interactions [34]:
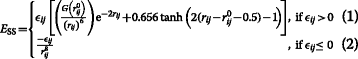
where rij is the distance between interacting beads i and j, the equilibrium distance σij is correlated with via
| (3) |
εij is the interaction strength, and
| (4) |
Figure 2a shows a matrix of the energies of the side chain–side chain interactions at the minimum distances σij. Equation (1) replaces the common 12-6 Lennard-Jones potential in order to limit ESS at longer distances. Figure 2b shows an example of the form of the potential for the Phe/Phe interaction. For proline and glycine the center of interaction is the Cα-atom, while for all other side chains the interaction center is a bead representing the center of mass of the side chain [33]. The potential ESS is not used for salt bridges between side chains. Instead, salt bridges are modelled with a potential, ESB, derived from all atom MD simulations [32], where the distance dependent contact probability is translated to free energy profiles. These free energy profiles have one minimum for Arg/Asp and Arg/Glu pairs and two minima for Lys/Asp and Lys/Glu interactions. To describe backbone–backbone and backbone–side chain interactions, OPEP contains a van der Waals term, EVDW, which is modelled via a 12-6 Lennard-Jones potential. H-Bond interactions, EHB, are modelled between the backbone N-H and the backbone C’-O atoms. In addition, OPEP has special terms for stabilizing α-helices and β-sheets. The two-body term for H-bonds between residues in the same chain has different equilibrium distances for H-bonds less than five residues apart and for H-bonds further than four residues apart. For stabilizing α-helices, the intra-chain potentials also contain a 4-body H-bond term. Furthermore, 11 side chain–side chain interactions were identified to be more frequently found in (i, i + 3) and (i, i + 4) contacts in α-helices. Therefore, these side chain–side chain interactions with this particular separation were made more attractive [34].
Fig. 2.
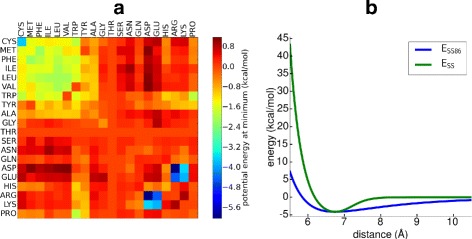
The OPEP force field. a The potential energy E SS+E SB for side chain–side chain interactions is shown at the energy minimum, which is at σ ij for E SS. For Arg/Asp and Arg/Glu, the average of the two minima for E SB is shown. Repulsive interactions, corresponding to energies higher than zero, are also plotted at σ ij. b The OPEP potential E SS is shown together with the soft function E SS86 for Phe/Phe
The scoring function
Before rescoring the predictions, we perform an energy minimization using OPEPv5 to relax the complexes after their transformation from the grid presentation to the coarse-grained model. We perform 140 minimization steps, as we found this to be the best compromise between computational efficiency and optimization result. We tested the effect of fewer and more minimization steps. Extending the minimization beyond 140 steps does not change the outcome of the rescoring result as for ∼90 % of the structures the energy only changes marginally at this point. Moreover, it happens especially for misdocked complexes that the energy minimum has not been reached within 140 minimization steps. However, there is no need to further optimize such misdocked decoys. Reducing the number of minimization steps below 140 bears the risk that also near-native structures have not been properly minimized yet, which would lead to a poor ranking for them. For the scoring function we found that it becomes more reliable if we introduce a softer potential, which allows for more overlap between the beads than the original OPEPv5 energy function. To obtain a softer scoring function we replace both the side chain–side chain interaction potential, ESS from Eq. (1), and the 12-6 Lennard-Jones potential EVDW with an 8-6 Lennard-Jones potential. This kind of soft potential is also used in the Attract force field that was developed for protein–protein docking [35]. We call the new potentials ESS86 and EVDW86, and the formula for ESS86 is given as:
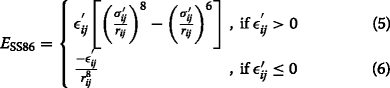
Here, σij′=0.866σij and εij′=9.481ESS(σij), with σij given in Eq. (3). The values σij′ and εij′ are chosen such that the minimum energies at the equilibrium distances are identical for ESS and ESS86. From Eq. (6), one can see that the repulsive-only potential is not modified. An example of the attractive ESS86 term is shown in Fig. 2b for the Phe/Phe side chain interaction. As the 8-6 potentials ESS86 and EVDW86 have broader wells than in OPEPv5, some overlap between beads is tolerated and, in addition, imperfectly fitted contacts are more strongly attractive at larger distances. The potentials for H-bonds and salt bridges were not modified, leading to our new scoring function, E86, with the modified potentials EVDW86 and ESS86:
| (7) |
which calculates the binding energy between receptor and ligand for scoring purposes. It should be noted that each binding partner can consist of several proteins (chains). We consider all chains from one binding partner as a single protein. Hence, we only consider non-bonded energies between the two binding partners, e.g., between receptor and ligand.
Interface RMSD
The interface RMSD (IRMSD) is defined as the RMSD between Cα interface atoms of the co-crystallized model and the prediction after superposition. Interface Cα atoms are all atoms within 10 Å distance of the binding partner in the co-crystallized complex [36]. For the superposition we use the corresponding function from Biopython [37].
Definition of a hit
As is standard [38, 39], we define a hit as a docked conformation with an IRMSD lower than 4 Å.
Performance evaluation
The performance is evaluated by ranking the predictions according to their (re)scoring energy in increasing order. From this list, the best ranked prediction with an IRMSD lower than 4 Å is reported. Furthermore, we calculate the success rate, which is a function of the number of predictions, Npred, that we consider from the sorted prediction list. This is averaged over the number of targets, Ntarget, and is calculated according to following equation:
| (8) |
where Si(Npred)=1 when the subset of Npred=1,2,…,2,000 predictions contains at least one hit, otherwise Si(Npred)=0. Thus, the success rate corresponds to the probability of finding the native complex among the Npred first models based on the (re)scoring energy.
Training the scoring function
After minimization, a residue-residue contact map between receptor and ligand is produced for each prediction. A contact is present if any of the beads of two residues are closer than 8 Å. Depending on the ranking with E86, one can classify the predictions for each complex into one of the four groups: true positive (TP), false negative (FN), false positive (FP), and true negative (TN). TPs have an IRMSD <4 Å and rank lower than or equal to 20, while the TN predictions have IRMSD ≥4 Å and a rank higher than 20. All other predictions are either FNs or FPs depending on whether their IRMSD is < or ≥ 4 Å and their ranking is > or ≤20. We only consider the first N=20 TPs or, if N<20 hits are found, we consider only those, because ideally one wants the correct predictions within the top hits. Twenty complexes is a small enough number for further processing by computationally more expensive approaches and visual inspection. We further limit the number of FNs and FPs to 20−N for training purposes. Thus, we do not consider FN and FP predictions if ≥20 hits are found for a target, as for such targets E86 already produces satisfying results. For each TP, FN, FP, and TN prediction considered, we calculate the frequency map for residue-residue contacts and average them over all targets for the enzyme/inhibitor, antigen/antibody, and other complexes. Next, we select residue-residue contacts where the frequency is higher in the maps for TP and FN than for the FP and TN maps. We assume these contacts need to be strengthened, so current FN predictions become TP without further favoring FP predictions. Therefore, we decrease the energy value ESS86 or ESB for this contact. The other contacts, for which we modify the potential, are those where the frequency of TPs and FNs is lower than FPs and TNs. It appears these contacts are not important for the complex class in question and should thus be disfavored, with the aim of transforming a current FP prediction into a TN prediction. Therefore, we increase ESS86 or ESB for such contacts. Figure 1 illustrates the training procedure.
The amount of change for the selected interaction between residues i and j is determined by the ratio between the corresponding FN ij and FP ij frequencies. A value greater than one means this interaction energy has to be decreased, while the opposite indicates this interaction energy has to be increased. We do this by changing the interaction potentials ESS86(i,j) and ESB(i,j) according to
| (9) |
where EX=ESB or EX=ESS86 depending on the residue contact (i,j). For the parameter k, values between 0.1 and 0.6 were tested, and k=0.2 was found to be optimal. Equation (9) was iteratively applied. Thus, we had to determine when to stop the training for best parametrization and to avoid overfitting. To this end, we performed a 4-fold cross-validation on the enzyme/inhibitor training dataset, which gives us meaningful numbers for training and validation. This enzyme/inhibitor set contains 39 targets, of which 29 complexes were used for training, with the remaining 10 used for cross-validation. For these 10 targets, we measured the quality with , where rank() returns the rank of the best ranked hit. This function should decrease during training, while an increase is indicative of overfitting. We observe that overfitting becomes an issue after 30 iterations of Eq. (9). Therefore, we set the number of learning iterations to 30, yielding our new scoring function :
| (10) |
Results
Overall performance
The ranks of the first hit using ZDOCK and after rescoring are shown in Table 1. The ZDOCK column gives the results for ZDOCK 3.02. The column shows the rank after rescoring using Eq. (7) before energy minimization with the OPEP potential, while the E86 column reports the rank after minimization. Column five reports the rank of the first hit when using all intra- and interprotein contributions of the original OPEPv5 potential [32], while column six shows the rank of the first hit when the predictions are ranked by OPEPv5 energy when only the non-bonded energies between beads from the receptor and ligand are considered. These rescoring energies are denoted by EOPEP and in the following. Figure 3 represents the success rate as defined in Eq. (8) for the different complex classes. In general, ZDOCK and E86 perform better than EOPEP and and their performance is about equal if one considers the overall performance for all complex classes (Fig. 3a). However, there are differences between the three complex classes.
Table 1.
Best rank for (re)scoring with ZDOCK, , E 86, E OPEP, and for complexes from the ZDOCK benchmark 4.0. indicates the average rank for the complex class in question
| Complex | ZDOCK | E 86 | E OPEP | ||
|---|---|---|---|---|---|
| Enzyme/inhibitor | |||||
| 1ACB | 47 | 11 | 14 | 1 | 16 |
| 1AVX | 1 | 128 | 3 | 27 | 31 |
| 1AY7 | 330 | 205 | 358 | 730 | 858 |
| 1BVN | 1 | 39 | 1 | 98 | 1 |
| 1CGI | 28 | 52 | 9 | 2 | 70 |
| 1CLV | 1 | 2 | 1 | 1 | 1 |
| 1DFJ | 1 | 102 | 1 | 14 | 4 |
| 1EAW | 332 | 194 | 151 | 469 | 124 |
| 1EZU | 121 | 919 | 11 | 946 | 559 |
| 1F34 | 176 | 428 | 2 | 1664 | 79 |
| 1FLE | 1797 | 179 | 223 | 35 | 424 |
| 1GL1 | 49 | 116 | 8 | 107 | 34 |
| 1GXD | 229 | 84 | 2 | 4 | 47 |
| 1HIA | 389 | 88 | 901 | 1300 | 1392 |
| 1IJK | 1296 | 924 | 355 | 70 | 5 |
| 1JIW | 1350 | 504 | 553 | 989 | 851 |
| 1JTG | 1 | 5 | 1 | 23 | 9 |
| 1MAH | 1 | 80 | 87 | 9 | 27 |
| 1NW9 | 750 | 105 | 392 | 857 | 569 |
| 1N8O | 11 | 11 | 14 | 9 | 12 |
| 1OC0 | 225 | 82 | 240 | 70 | 83 |
| 1OPH | 28 | 150 | 422 | 903 | 822 |
| 2O8V | 34 | 20 | 502 | 112 | 308 |
| 1OYV | 3 | 34 | 1 | 2 | 381 |
| 1PPE | 1 | 7 | 1 | 1 | 2 |
| 1R0R | 533 | 896 | 40 | 151 | 856 |
| 1TMQ | 7 | 228 | 3 | 295 | 1 |
| 1UDI | 9 | 446 | 2 | 158 | 2 |
| 1YVB | 11 | 86 | 47 | 220 | 67 |
| 2ABZ | 689 | 670 | 772 | 619 | 1121 |
| 2B42 | 1 | 57 | 40 | 44 | 192 |
| 2J0T | 1730 | 677 | 179 | 579 | 178 |
| 2MTA | 1 | 122 | 104 | 13 | 43 |
| 2OUL | 1 | 30 | 1 | 23 | 1 |
| 2SIC | 1 | 145 | 1 | 3 | 5 |
| 3SGQ | 309 | 158 | 16 | 596 | 260 |
| 2UUY | 258 | 279 | 65 | 180 | 361 |
| 4CPA | 1 | 1 | 4 | 1 | 20 |
| 7CEI | 1 | 30 | 55 | 51 | 9 |
| 275.7 | 212.7 | 143.1 | 291.7 | 251.9 | |
| Antigen/antibody | |||||
| 1AHW | 1387 | 542 | 1087 | 1161 | 1563 |
| 1BJ1 | 1 | 20 | 132 | 89 | 230 |
| 1BVK | 10 | 143 | 356 | 6 | 283 |
| 1DQJ [a] | 1671 | 150 | – | – | – |
| 1E6J | 8 | 8 | 39 | 21 | 10 |
| 1FSK | 5 | 5 | 181 | 135 | 373 |
| 1I9R | 31 | 39 | 177 | 23 | 109 |
| 1IQD | 2 | 41 | 1 | 4 | 1 |
| 1JPS | 1261 | 221 | 14 | 1635 | 1930 |
| 1KXQ | 6 | 192 | 34 | 532 | 9 |
| 1K4C | 120 | 201 | 1077 | 14 | 166 |
| 1MLC | 188 | 1194 | 104 | 395 | 314 |
| 1NCA | 389 | 331 | 774 | 1091 | 1340 |
| 1VFB | 45 | 163 | 198 | 33 | 586 |
| 2FD6 | 2 | 82 | 20 | 16 | 268 |
| 2HMI | 62 | 178 | 516 | 4 | 766 |
| 2I25 | 12 | 2 | 7 | 45 | 39 |
| 2JEL | 22 | 85 | 18 | 49 | 139 |
| 2VIS | 725 | 557 | 1538 | 625 | 1095 |
| 313.0 | 246.1 | 435.4 | 414.6 | 485.3 | |
| Other complexes | |||||
| 1AKJ | 754 | 47 | 1823 | 787 | 1887 |
| 1B6C | 1 | 13 | 1 | 1 | 1 |
| 1BUH | 62 | 25 | 304 | 10 | 2 |
| 1DE4 | 4 | 472 | 38 | 39 | 52 |
| 1F51 | 9 | 22 | 32 | 166 | 176 |
| 1FC2 | 1286 | 1672 | 1654 | 896 | 224 |
| 1FFW | 41 | 47 | 47 | 111 | 433 |
| 1GLA | 210 | 326 | 1013 | 329 | 1296 |
| 1GPW | 3 | 2 | 1 | 111 | 997 |
| 1H9D | 203 | 53 | 386 | 297 | 345 |
| 1HE1 | 821 | 1837 | 871 | 1907 | 1702 |
| 1I2M | 354 | 828 | 3 | 482 | 262 |
| 1JK9 | 998 | 735 | 305 | 891 | 552 |
| 1JZD | 31 | 141 | 208 | 3 | 92 |
| 1K74 | 1 | 26 | 1 | 110 | 717 |
| 1ML0 | 1 | 12 | 2 | 2 | 3 |
| 1RV6 | 1 | 188 | 4 | 1 | 2 |
| 1S1Q | 1696 | 925 | 780 | 1635 | 932 |
| 1SYX | 149 | 345 | 123 | 24 | 781 |
| 1T6B | 439 | 224 | 14 | 130 | 30 |
| 1US7 | 150 | 107 | 772 | 283 | 341 |
| 1WDW | 1 | 3 | 23 | 12 | 326 |
| 1XD3 | 3 | 25 | 1 | 1 | 1 |
| 1XU1 | 37 | 69 | 5 | 6 | 1 |
| 1Z5Y | 31 | 1 | 1 | 1 | 1 |
| 1ZHI | 187 | 43 | 608 | 156 | 471 |
| 2AJF | 1115 | 318 | 1029 | 195 | 494 |
| 2A5T | 824 | 421 | 148 | 573 | 1149 |
| 2AYO | 6 | 112 | 3 | 700 | 902 |
| 2CFH | 1 | 18 | 1 | 3 | 3 |
| 2HLE | 54 | 29 | 20 | 52 | 74 |
| 2HRK | 2 | 76 | 64 | 30 | 5 |
| 2IDO | 171 | 314 | 73 | 577 | 11 |
| 2NZ8 | 1002 | 281 | 81 | 793 | 243 |
| 2VDB | 43 | 36 | 1 | 13 | 309 |
| 2Z0E | 123 | 181 | 187 | 684 | 1346 |
| 3BP8 | 998 | 375 | 131 | 538 | 242 |
| 3D5S | 524 | 29 | 1 | 34 | 246 |
| 324.6 | 273.1 | 283.1 | 331.1 | 438.2 | |
[a] The rank is set to 2000 for calculating the average
Fig. 3.
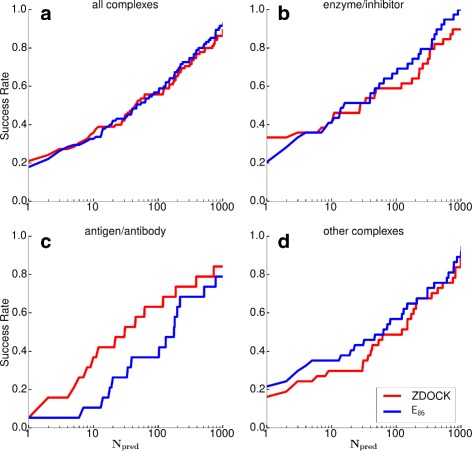
Results for the training dataset ZDOCK benchmark 4.0. The success rate is shown for (a) all complexes and separately for the three complex classes, (b) enzyme/inhibitor, (c) antigen/antibody and (d) other complexes using ZDOCK (red) and E 86 (blue)
Enzyme/inhibitor
For enzyme/inhibitor complexes, E86 finds equal or more hits if more than four predictions are considered, i.e., Npred≥5 (Fig. 3b). When considering more than 50 predictions, E86 becomes substantially better than ZDOCK. Table 1 shows that we can improve or maintain the rank using E86 for 25 out of 39 enzyme/inhibitor targets. For 1AVX, the rank is only slightly worse, increasing from 1 with ZDOCK to 3 with E86. Comparing the performance of E86 to , it becomes evident that the 140 minimization steps are not always sufficient to put every side chain in the minimum of the well, because the rank with is considerably higher than for E86. Thus rescoring with the softer potential is necessary. When using EOPEP for ranking, the ranks of only 16 targets are kept or improved. The average rank shows that E86 is generally better than ZDOCK, while produces a similar ranking to ZDOCK, and EOPEP performs worst.
Antigen/antibody
For antigen/antibody complexes, rescoring with E86 was least successful. For Npred≲500, the success rate of E86 is clearly smaller than for ZDOCK (Fig. 3c). Out of 19 antigen/antibody complexes, E86 improves the rank for only six targets and worsens it for the other 13. Using EOPEP only improves the ranking of six complexes, while the rank of only one complex can be improved using . The average rank shows that ZDOCK performs considerably better than any of the OPEP-based rescoring approaches. However, it should be noted that ZDOCK is not a perfect scoring function either for antigen/antibody complexes, as revealed by comparing the average ZDOCK ranks with enzyme/inhibitor complexes.
Other complexes
For other complexes, the success rate is always higher for rescoring with E86 than scoring with ZDOCK, independent of the number of predictions considered (Fig. 3d). The E86 score improves or maintains the rank of 21 complexes and worsens it for the other 17; however, for 1ML0 the rank only changes from 1 to 2 and 1RV6 from 1 to 4. While EOPEP improves the rank of 20 targets and worsens the rank of 18 targets, the improvements mostly occur for higher ranks, and only four predictions have rank 1, compared with eight for E86. can improve the rank of only 15 targets; it worsens the rank of the other 23. On average, for other complexes rescoring with E86 performs best, is least suited for this task, and EOPEP predicts a similar ranking as ZDOCK. From the strikingly different performance of E86 and it seems that optimal shape complementarity implying favourable residue-residue interactions are very important for protein binding in this complex category.
Structural changes upon energy minimization
We tested whether the structures of the complexes are affected as a result of energy minimization with the OPEP potential. To this end, the secondary structures of the complexes are determined before and after their energy minimization using STRIDE [40]. Since we use crystal structures of the unbound receptor and ligand as input, all 2000 ZDOCK predictions per target have the same secondary structures before minimization, while the secondary structures can change during minimization with the OPEP potential. However, we find that the changes in secondary structure are generally small (<5 %). Especially the near-native structures with IRMSD <3 Å are least affected by energy minimization, indicating that the correct binding helps stabilize the complex structure. However, the overall changes of secondary structure are small and do not follow a pattern, which prevents us from generalizing a dependency between IRMSD and secondary structure.
We further tested if the IRMSD is affected by minimization with OPEP and found it changes only slightly. A plot showing the average change of IRMSD as a function of the initial IRMSD as obtained from ZDOCK can be seen in Fig. 4. For most predictions, the IRMSD slightly increases due to minimization with the average IRMSD change fluctuating aroud 0.1 Å. For some of the complexes, the IRMSD also decreases: for 4.3 % of the predictions with IRMSD <4 Å before minimization, which increases to 8.7 % if one considers all predictions. The preferred IRMSD increase for near-native predictions is likely to be an effect of the tight packing at the binding site, which leads to more bead clashes after transformation from the grid to the coarse-grained representation, causing the atoms or beads to reorient during minimization. Nonetheless, the structures stay close to the conformations predicted by ZDOCK, as Fig. 4 testifies. Only for severely misdocked complexes (IRMSD Å) the IRMSD change increases to around 0.2 Å.
Fig. 4.
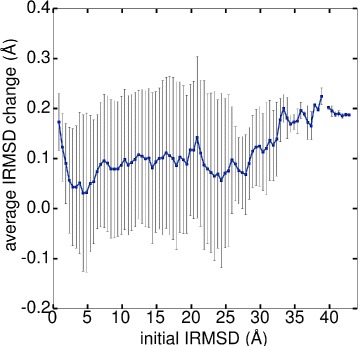
The average change in IRMSD as a result of energy minimization of the ZDOCK predictions using OPEP. Averages, calculated over all targets and complex classes, are shown in blue together with one standard deviation
Comparison of columns three and four of Table 1 reveals the effect of minimizing the energy before rescoring with E86. Column three reports the best rank without energy minimization, which we denote as . For the comparison we concentrate on the complexes for which either E86 or , or both, predict a best rank ≤10 as in the Critical Assessment of PRedicted Interactions (CAPRI) experiment [41] one can only upload 10 predictions per target. Thus, the aim is to score the decoys closest to the native structure in the top 10. For enzyme/inhibitor complexes, energy minimization is most successful as E86 identifies for more than 38 % a hit in the top 10 predictions (see success rate for Npred=10 in Fig. 3b). For only four of these 15 complexes (namely 1CLV, 1JTG, 1PPE and 4CPA) also predicts best ranks in the top 10, while it does not occur for enzyme/inhibitor complexes that finds a hit in the top 10, which is lost upon energy minimization. In two cases (1F34 and 1UDI) energy minimization improves the rank by more than 400 places, leading to second places in the rank list. A similar picture emerges for other complexes, for which for more than 34 % of the complexes a best rank in the top 10 is found with E86 (see success rate for Npred=10 in Fig. 3d). With , on the other hand, for only three complexes a top-10 rank is achieved. For one of these three (1WDW) the rank increases from 3 to 23 upon energy minimization, while the other two are also top-10 ranked with E86. Only for antigen/antibody complexes preceding energy minimization of the complexes offers no advantage over direct application of the rescoring function. E86 and find for 2 and 3, respectively, of the 19 complexes a hit in the top-10 rank list. For two complexes (1E6J and 1FSK) the top-10 rank is lost after energy minimization, while for 1IQD the best rank climbed 40 places and is ranked first with E86. However, it should be noted that the average rank for is considerably lower than for both ZDOCK and E86. Thus, energy minimization of antigen/antibody complexes is not absolutely necessary. Though apart from saving us computing time, omitting this step would also not (considerably) increase our chances of identifying the right prediction as the increase of the average rank for E86 originates mainly from further deterioration of the already high ranks obtained with (e.g., complexes 1AHW, 1K4C and 2VIS). More crucial would be a general improvement of the E86 scoring function for its application to antigen/antibody complexes.
Energy contributions to the protein-protein interactions
Figure 5 shows the different contributions to the E86 energy for predictions sorted by their IRMSD using a bin size of 1 Å. We show the averaged values of ESS86, ESB, and EHB for the three complex classes. For the enzyme/inhibitor complexes, a minimum in ESS86 is present for predictions up to 5 Å. However, for IRMSD values above 25 Å ESS86 becomes small again, in some cases even smaller than for the hits. This is more than counterbalanced by the H-bond energy, as only near-native hits have more and better oriented H-bonds, leading to EHB values more than 10 kcal/mol smaller than for all other predictions. Salt bridges seem to be of minor importance for the protein binding in enzyme/inhibitor complexes, as there is no correlation between the ESB values and the IRMSD, and the contribution of ESB to E86 is generally small, with all values fluctuating around −5 kcal/mol. Thus, the sum of ESS86 and EHB is mainly responsible for distinguishing between correct and incorrect complex predictions. This partly agrees with previous findings that protease-inhibitor complexes interact predominantly through main chain–main chain interactions [42], which are represented by H-bonds in the E86 function.
Fig. 5.
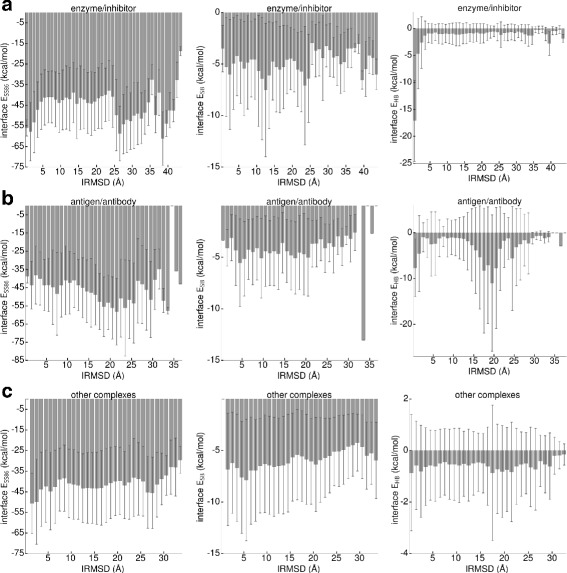
The different contributions of the rescoring function E 86 for the different complex classes, (a) enzyme/inhibitor, (b) antigen/antibody and (c) other complexes as a function of the IRMSD: (left) E SS86, (middle) E SB, (right) E HB. Averages, calculated over all targets and predictions for each target belonging to one of the three complex classes, are shown together with one standard deviation
For antigen/antibody complexes, none of the three energy contributions clearly decreases with decreasing IRMSD. Instead, both ESS86 and EHB adopt their smallest values for IRMSD ≈20 Å, which explains why E86 does not perform well for this complex class. Compared to enzyme/inhibitor complexes, backbone H-bonds are less important for the native complex. This agrees with the previous observation that antigen and antibody complexes predominantly bind through side chain–side chain or side chain–main chain interactions [42], which are represented by other contributions from E86 but not by EHB. For antigen/antibody complexes, the formation of salt bridges is also of minor importance. There is only one exception, at IRMSD ≈34 Å, where with ESB≈−13 kcal/mol the smallest salt bridge energy is observed, also taking the other two complex classes into account.
The hits for other complexes are stabilized by side chain–side chain interactions, as the lowest values for ESS86 are found for the complexes with IRMSD <4 Å. H-bonds seem to be of minor importance for binding receptor and ligand in this complex category, as all EHB values are >−1 kcal/mol, an order of magnitude higher than those in enzyme/inhibitor and antigen/antibody complexes. On the other hand, other complexes are the only ones where salt bridges contribute to stabilizing the complexes, as for IRMSD >5 Å, ESB increases. This trend only breaks for IRMSD ≤5 Å as ESB does not further decrease for the near-native predictions. This means that either ESS86 dominates these binding modes or the E86 potential can be further improved in this range.
Improving the rescoring function
Next we tested if the performance of E86 can be enhanced by training it according to Eq. (9), yielding the new rescoring function defined in Eq. (10). As the energy analysis revealed that complex formation in the three categories is driven by different interactions, we decided to optimize E86 separately for enzyme/inhibitor, antigen/antibody, and other complexes. The resulting leads to new energies at the optimal distances between the side chains at the binding sites, which can be presented as a matrix. Subtracting the new energy matrix from the original potential energy matrix shown in Fig. 2a gives a matrix for each complex category that represents the change in interaction energies. These matrices are shown in Fig. 6.
Fig. 6.
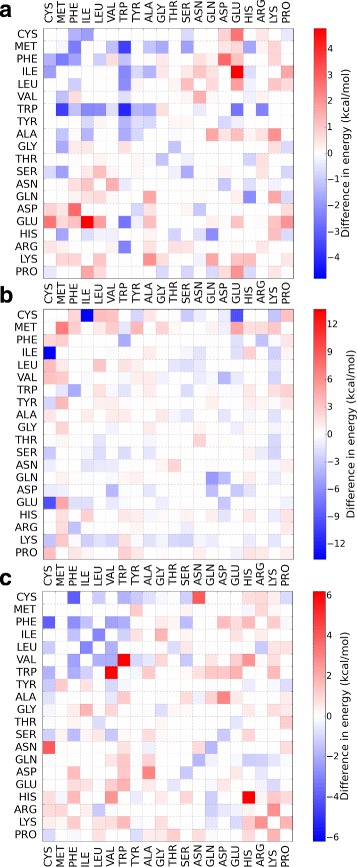
The change in minimum energy for side chain–side chain contacts as a result of training the scoring function E 86 for (a) enzyme/inhibitor, (b) antigen/antibody, and (c) other complexes. Blue values mean the interaction became more attractive, red means the interaction became more repulsive. Note the different energy scales for the protein classes
Enzyme/inhibitor
With few exceptions, the change in interaction energy follows the hydrophobicity of the amino acids. This confirms the findings from Fig. 5 that the enzyme/inhibitor complexes are stabilized by the interactions modeled by ESS86. The amino acids Phe to Ala are the hydrophobic amino acids, and interactions between them got stronger, except for Phe/Val and Phe/Ala. Most interactions involving polar amino acids do not change much, while some of the interactions involving charged residues become more repulsive. Previous studies also found that enzyme-inhibitor complexes contribute more hydrophobic interactions at the expense of polar contributions [42]. The most pronounced changes occur for Trp/Trp, Met/Trp, Glu/Ile, and Asp/Phe. The increased stability for Trp/Trp agrees with the Ravikant-Elber matrix [43], which was derived as the most likely interaction from a statistical analysis of protein–protein complexes. The Met/Trp interaction is also favoured by the Ravikant-Elber matrix. Both interactions were already attractive in the E86 rescoring function, but become even stronger as a result of the optimization procedure. Glu/Ile and Asp/Phe, on the other hand, were repulsive in E86 and become more so. Glu/Ile is also slightly repulsive in the Ravikant-Elber matrix, while Asp/Phe is slightly attractive. However, the Ravikant-Elber matrix includes protein–protein interactions independent of complex class, while our current finding only applies to enzyme/inhibitor complexes.
Antigen/antibody
Figure 6b shows the change in interaction energies for antigen/antibody complexes. Surprisingly, two interactions involving cysteine, namely Cys/Ile and Cys/Glu, considerably increase in strength. This probably results from the presence of a Cys residue just before the start of most of the CDRs [44], which is thus in contact with the antigen. The interaction between Met residues becomes the most repulsive. Before training it was slightly attractive. This change in energy is difficult to rationalize; many of the other changes are correlated to the frequency of residues at the antigen–antibody interface. At the paratope of the antibody, the residues that contribute most to the binding are Tyr, Trp, Asp, Glu, Asn, Ser, Thr, and Gly, while at the antigen epitope these are Arg, Lys, Asp, Tyr, Glu, Asn, Ser, Thr, and Gly [42]. Many of the interactions involving these residues become more attractive, while the remaining interactions do not change much in strength. This shows that our training scheme can strengthen interactions which have been previously shown to play an important role in antigen–antibody binding [42, 45].
Other complexes
The difference map for the other complexes can be seen in Fig. 6c. As with enzyme/inhibitor complexes, most hydrophobic interactions become more attractive. The exception is Trp/Val, for which the interaction became more repulsive. Previously, this interaction was only slightly repulsive. Almost all of the polar/hydrophobic interactions become more repulsive. Interestingly, the His/His interaction becomes considerably more repulsive, which corresponds well with repulsion of equal charges when His is positively charged. Before training, this interaction was attractive. The repulsion between the equally charged residues Glu/Asp and Arg/Lys also increased, but these were already repulsive before optimization. The salt bridges, with the exception of Lys/Asp, got stronger. Overall, this shows that electrostatics interactions play a more important role here than in the enzyme/inhibitor complexes. It also confirms the trend from Fig. 5, which revealed a general decrease (apart from a few exceptions) for ESS86 and ESB with decreasing IRMSD.
Test on a new dataset
To test the optimized rescoring function, we use the protein–protein complexes from the Dockground test set [24], removing all complexes which are also in the ZDOCK Benchmark 4.0 and were already used for training. The remaining 74 complexes are listed in Table 2. As before, we perform unbound docking with ZDOCK producing 2000 predictions for each target. However, ZDOCK is not able to produce a hit for all targets in the top 2000 predictions. In particular, ZDOCK is not very successful for other complexes, generating hits for only 19 out of 40 of these complexes. However, it is successful for 15 out of 18 enzyme/inhibitor complexes and 12 out of 16 antigen/antibody complexes. For complexes for which ZDOCK produced one or more hits, the 2000 predictions are rescored using E86 and .
Table 2.
Best rank for (re)scoring with ZDOCK, E 86, and for complexes from the Dockground 2.0 benchmark
| Complex | ZDOCK | E 86 | |
|---|---|---|---|
| Enzyme/inhibitor | |||
| 1ARO | - | - | - |
| 1AVW | 2 (56/-) | 12 (76/-) | 46 (48/-) |
| 1BTH | 366 (-/-) | 218 (-/-) | 106 (-/-) |
| 1CHO | 3 (3/86) | 5 (5/59) | 1 (1/21) |
| 1GPQ | 1271 (-/-) | 438 (-/-) | 109 (-/-) |
| 1ID5 | 72 (-/-) | 11 (-/-) | 4 (-/-) |
| 1KU6 | 10 (62/-) | 19 (74/-)) | 14 (103/-) |
| 1OFH | - | - | - |
| 1PPF | 12 (12/36) | 12 (12/109) | 1 (1/13) |
| 1T6G | 2 (2/579) | 22 (22/311) | 5 (6/121) |
| 1TX6 | 539 (610/-) | 28 (28/-) | 87 (1090/-) |
| 1UGH | 1 (1/1098) | 1 (1/104) | 1 (1/44) |
| 1XX9 | 279 (-/-) | 17 (-/-) | 8 (-/-) |
| 2BKR | 4 (4/-) | 18 (24/-) | 33 (33/-) |
| 2D26 | - | - | - |
| 2FI4 | 335 (1287/1287) | 69 (69/182) | 48 (48/86) |
| 2KAI | 269 (737/-) | 287 (287/-) | 75 (75/-) |
| 3SIC | 1 (6/24) | 1 (1/1) | 1 (1/1) |
| 211.3 | 77.2 | 35.9 | |
| Antigen/antibody | |||
| 1A2Y | 5 (-/-) | 21 (-/-) | 3 (-/-) |
| 1G6V | 1344 (-/-) | 1485 (-/-) | 705 (-/-) |
| 1G9M | 1 (5/-) | 4 (38/-) | 3 (18/-) |
| 2BNQ | - | - | - |
| 1BZQ | 13 (13/13) | 22 (22/61) | 7 (43/274) |
| 1FBI | 609 (-/-) | 1113 (-/-) | 1174 (-/-) |
| 1FNS | 729 (-/-) | 1055 (-/-) | 1906 (-/-) |
| 1H0D | 159 (-/-) | 17 (-/-) | 1 (-/-) |
| 1JTP | 13 (-/-) | 1 (-/-) | 2 (-/-) |
| 1MQ8 | 16 (98/-) | 1479 (1548/-) | 565 (807/-) |
| 1NBY | - | - | - |
| 1NCB | - | - | - |
| 1NSN | 562 (-/-) | 695 (-/-) | 949 (-/-) |
| 1PKQ | - | - | - |
| 1SQ2 | 1 (16/-) | 1 (6/-) | 2 (8/-) |
| 1Z3G | 6 (6/-) | 1273 (1273/-) | 378 (378/-) |
| 288.2 | 597.2 | 474.6 | |
| Other complexes | |||
| 1BUI | 343 (-/-) | 1332 (-/-) | 573 (-/-) |
| 1F6A | - | - | - |
| 1FM9 | 1 (14/52) | 2 (3/26) | 2 (24/87) |
| 1G20 | 11 (132/-) | 15 (178/-) | 12 (34/-) |
| 1G4A | - | - | - |
| 1G4U | - | - | - |
| 1GHQ | - | - | - |
| 1GLB | 1021 (-/-) | 1356 (-/-) | 1352 (-/-) |
| 1HXY | - | - | - |
| 1JWM | - | - | - |
| 1K90 | - | - | - |
| 1K93 | - | - | - |
| 1L9B | - | - | - |
| 1MA9 | 1 (4/-) | 1 (3/-) | 1 (1/-) |
| 1NBF | 91 (-/-) | 105 (-/-) | 106 (-/-) |
| 1NVU | 1020 (-/-) | 555 (-/-) | 192 (-/-) |
| 1OMW | - | - | - |
| 1OOK | 171 (-/-) | 639 (-/-) | 237 (-/-) |
| 1P7Q | 3 (433/-) | 13 (139/-) | 21 (215/-) |
| 1R4M | 9 (-/-) | 201 (-/-) | 27 (-/-) |
| 1RQQ | - | - | - |
| 1S6V | 1 (-/-) | 9 (-/-) | 7 (-/-) |
| 1SQ0 | - | - | - |
| 1U0N | - | - | - |
| 1U7F | 149 (936/-) | 1508 (1875/-) | 969 (1740/-) |
| 1UEX | 25 (872/-) | 14 (578/-) | 11 (457/-) |
| 1V7P | 76 (-/-) | 52 (-/-) | 13 (-/-) |
| 1WLI | - | - | - |
| 1YI5 | - | - | - |
| 1ZY8 | 202 (325/435) | 143 (527/1097) | 54 (495/940) |
| 2A42 | - | - | - |
| 2ATQ | - | - | - |
| 2B4S | - | - | - |
| 2CKH | 1 (1/21) | 1 (1/178) | 1 (1/68) |
| 2G45 | 912 (-/-) | 682 (-/-) | 438 (-/-) |
| 2GD4 | - | - | - |
| 2GOO | - | - | - |
| 2GY7 | - | - | - |
| 3FAP | 143 (-/-) | 80 (-/-) | 95 (-/-) |
| 3PRO | 158 (350/-) | 41 (154/-) | 136 (412/-) |
| 228.3 | 355.2 | 223.5 | |
[a] The rank is set to 2,000 for calculating the average
indicates the average rank for the complex class in question. Targets without a hit in the top 2,000 are indicated by ‘–’. Values in brackets show the best rank for predictions with an IRMSD <2 Å and <1 Å, respectively. If such predictions are not found, no value is being reported
Enzyme/inhibitor complexes
Both OPEP-based rescoring functions can significantly improve the average ranking compared to ZDOCK, and performs better than E86. Compared to ZDOCK, can improve or maintain the rank for 11 targets and worsens the rank for 4 targets. However, for 1T6G the ranking decreased by only three places, from two to five. The standard soft rescoring function E86 can improve or maintain the ranking for 10 and worsens the ranking for 5 complexes. Figure 7a shows the success rate for the enzyme/inhibitor complexes. Both OPEP-based rescoring functions produce at least one hit in the top 1000 for all targets (i.e., the success rate is one for Npred=1000), which is not the case with ZDOCK. The performance of E86 is weak for Npred<10, but when considering more than 10 predictions the results improve, and E86 performs then better than ZDOCK and similar to . This means the selectivity of the E86 function near the native complex structure is not high enough; this is improved by training the scoring function, yielding . For Npred<10, the performance of is equal or better to ZDOCK. This finding shows that training the OPEP-based scoring function was successful for the enzyme/inhibitor complex class.
Fig. 7.
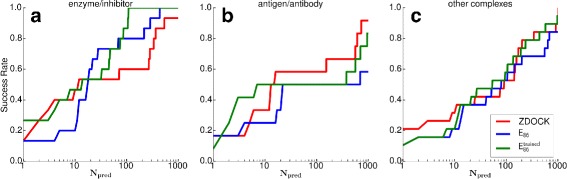
The success rate for the different complex complex classes, (a) enzyme/inhibitor, (b) antigen/antibody, and (c) other complexes for the Dockground test set when using ZDOCK (red), E 86 (blue), and (green) as (re)scoring function
Antigen/antibody complexes
ZDOCK finds hits in the top 2000 predictions for 12 out of 16 targets. can improve or maintain the rank for five complexes. For 1G9M and 1SQ2, the rank only decreases from 1 to 3 and from 1 to 2, respectively. E86 performs less well, only improving the ranks of three targets and worsening them for the other nine targets. Figure 7b shows E86 has as many top-1 hits as ZDOCK has, but its success rate dwindles when more predictions are taken into account. , on the other hand, performs best when between 2 and 12 predictions are considered, yet for Npred>12 ZDOCK is still most successful for antigen/antibody complexes. Nonetheless, training E86 was worthwhile, as for Npred>1 the trained potential always performs better than or equal to E86, improving the average rank by more than 120 places (see Table 2).
Other complexes
For the other complexes, can (considerably) improve the average ranking compared to ZDOCK and E86. Both and E86 improve the ranks of nine targets and worsen them for the other 10. However, with E86 the ranking of these 10 targets is considerably increased, leading to an average rank more than 120 places higher than the ZDOCK average. Figure 7c shows that performs slightly better than ZDOCK for Npred>20. However, the selectivity of should be further improved for near-native predictions, i.e., its performance should be increased for the top 20 predictions. However, this may prove difficult, as the other complexes are a collection of protein–protein complexes from different classes. Thus, the protein–protein binding may be driven by different interactions for the different complexes, making it difficult to fully accommodate all peculiarities within a scoring function.
Medium and high accuracy predictions
In the CAPRI evaluation [41], where the predictions are made blindly (i.e., without any knowledge of the correct answer), the predicted models are classified into four categories: incorrect, acceptable, medium, and high accuracy. To this end, the combination of three parameters is used, namely the fraction of native residue-residue contacts (fnat), the RMSD of the ligand molecules in the predicted versus the target complexes (LRMSD), and the IRMSD. A detailed description of these parameters and the corresponding thresholds used in classifying predictions can be found in previous CAPRI reports [41, 46]. In this work, only the IRMSD is used to assess the quality of the predictions. Application of fnat requires an atomisitic representation of the predicted complexes as it is defined based on contacts between any atoms of interacting residues. Therefore, a transformation from the coarse-grained OPEP to an atomistic representation would be first required for the calculation of fnat. This would probably entail an optimization of the side chain positions so that the correct residue-residue contacts can form. While desirable, this is, however, would be beyond the scope of current study, which focuses on testing of OPEP as rescoring function for protein-protein docking. Therefore, only the IRMSD is used to classify the accuracy of predictions as high if IRMSD≤1 Å, medium if IRMSD≤2 Å, and acceptable if IRMSD<4 Å [41, 46]. As we want to know whether finds more predictions of medium and high accuracy than E86, we determined the best ranks using these thresholds for the predictions obtained for the Dockground 2.0 test set. The results are listed in Table 2, together with the ones discussed above for threshold IRMSD<4 Å.
Table 2 reveals that one problem of our current approach is that ZDOCK does not produce many decoys of medium or high accuracy in the top 2000 predictions. This is particularly the case for antigen/antibody and other complexes. For only one antigen/antibody complex (1BZQ) decoys of high accuracy are predicted by ZDOCK, while for 5 of the 16 complexes predictions of medium accuracy are produced. For other complexes ZDOCK performs even worse as for only 9 (3) of the 40 complexes predictions of medium (high) accuracy are found. The ZDOCK results are somewhat better for the 18 enzyme/inhibitor complexes with decoys of medium accuracy being found for 10 complexes and of high accuracy for 6 complexes. As in the current study only energy minimization is used to optimize the geometry of the decoys, which has only minor effects on the docking pose (IRMSD changes of around 0.1 Å only, see Fig. 4), E86 and cannot find more decoys of high or medium quality as being produced by ZDOCK. More structural refinement of the ZDOCK predictions, for instance by using MC simulations as done by RosettaDock [6, 47], would be necessary for their further improvement. Comparison of the ZDOCK, E86 and scoring of decoys of medium accuracy and with top-10 ranks shows that performs best for enzyme/inhibitor complexes. In this category, ranks the docking models for four complexes first and for a fifth complex on sixth place. Also ZDOCK has for five complexes such models ranked in the top 10, however, for none on the first place. E86 predicts for only three complexes top-10 ranks, however, for two of them they are on the first place. For antigen/antibody complexes, ZDOCK finds for two complexes models in the top-10 rank list, while E86 and for only one complex. For other complexes, E86 is slightly better than ZDOCK and as it has for three complexes decoys in the top-10 list, while the other two scoring functions achieve this for only two complexes.
In summary, E86 and rank docking models of medium accuracy on average better than ZDOCK (apart from antigen/antibody complexes). For complex 3SIC both OPEP-based rescoring functions even rank a high-accuracy decoy first, which ZDOCK fails to achieve for any complex; it does not even place any decoy of high accuracy in the top 10. However, mainly due to limited refinement of the docking models obtained from ZDOCK, both E86 and do not find quantitatively more docking models of medium and high quality. This is further supported by the fact that for seven complexes (1BTH, 1XX9, 2BKR, 1G20, 1NBF, 1OOK, 3PRO), for which no decoy of high (sometimes not even of medium) accuracy and also no top-10 hit are found after rescoring, the native (i.e., target) complex is ranked first or second by E86 and/or (data not shown). In these cases rescoring with E86 and would have worked if the correct decoys had been generated. It should be noted, however, that in many other cases the native complex has a much higher rank than the other decoys, also for complexes for which top-10 predictions of medium or even high accuracy have been found. A similar observation was made by Baker and co-workers [6] when the performance of RosettaDock was for the first time tested. There, the problem was solved by performing 50 rounds of side-chain repacking and minimization. We assume that also after the transformation of the PDB structures to the coarse grained representation, energy minimization is often not sufficient for an optimal positioning of the side-chain beads. In our future work we will test wether side-chain refinement will improve the scoring of the native complexes.
Discussion and conclusion
In this work we examined the applicability of the coarse-grained OPEP force field [22] for refining and rescoring rigid body protein–protein docking predictions. We use ZDOCK [11] to produce protein complex predictions, which also serves as quality control. The predictions from the ZDOCK benchmark 4.0 are transformed to the coarse-grained model and their energy minimized using the original OPEP potential, which is followed by rescoring with a softer energy function, denoted E86, based on the interprotein OPEP interactions. This approach produces a better rank for the best prediction than ZDOCK for 54 % of targets. However, the results differ significantly across the three complex classes: There is an improvement for 65 % of the enzyme/inhibitor complexes, for 55 % of other complexes, but for only 32 % of the antigen/antibody complexes. Furthermore, the average rank with E86 is for antigen/antibody complexes considerably higher than that obtained with ZDOCK. To improve these results, we developed a training scheme for the OPEP-based rescoring function based on false positive and false negative predictions. The resulting trained rescoring function callesd , which was applied to the targets from the test dataset taken from the Dockground benchmark [24], produces a lower best rank compared to the ZDOCK results for 54 % of the targets, while the untrained OPEP-based scoring function can only improve the rank of 48 % of targets. The trained scoring function performs particularly well for enzyme/inhibitor complexes, where the best rank of 73 % of targets can be improved. These figures are 47 % for other complexes, and 42 % for antigen/antibody complexes.
Performance analysis for different complex classes Training the OPEP-based rescoring function revealed that the complexes from different classes are stabilized by different protein–protein interactions. For enzyme/inhibitor and other complexes, interactions between hydrophobic residues are of general importance, and for enzyme/inhibitor complexes backbone–backbone hydrogen bonds are also important. For antigen/antibody complexes we found that training strengthens the interactions between residues, which have been previoulsy shown to be prevalent at the paratope of the antibody and the epitope of the antigen [42, 45]. The different performance and training potentials reflect the different protein–protein binding in enzyme/inhibitor and antigen/antibody complexes. Antibodies can recognize a wide spectrum of antigens, including proteins, polysaccharides, nucleic acids, and even lipids, while enzyme–ligand binding has developed in an evolutionary sense to enable specific binding of a ligand to its target enzyme. This diverse binding by antibodies is accommodated by the complementarity determining regions composed of six loops that are modified in shape and chemical nature to match the corresponding features of the antigen epitope. Furthermore, the paratopes are mainly discontinuous, and binding is usually mediated by only 4–13 residues. In contrast, the enzyme inhibitors are typically small proteins that form tight, substrate-like interactions with the enzyme, which is reflected in a much stronger binding energetics. The binding constants for enzyme/inhibitor complexes are in the femtomolar range, which is about six orders of magnitude smaller than the nanomolar binding constants between antigen and antibody [42]. Thus, it is not surprising that the more static and strong enzyme–inhibitor binding is more easily predicted than the protein–protein interface in antigen/antibody complexes, where already one missing or one wrong interresidue contact in a decoy can have a profound impact on the performance of the scoring result. Our results suggest that the collective complex class called ‘other complexes’ lies between the two ends of the spectrum bounded by enzyme/inhibitor and antigen/antibody complexes.
Comparison to other rescoring approaches In summary, this study demonstrates for the first time that energy functions derived from the coarse-grained OPEP force field can be employed to rescore predictions for protein–protein complexes. This expands the applicability of OPEP to new problems. While the performance of OPEP is already very good for enzyme/inhibitor complexes and better than ZDOCK, for the other complexes and especially for antigen/antibody complexes, ZDOCK is still better suited. The comparison to RDOCK results [13] shows that rescoring with an all-atom force field works somewhat better than rescoring with E86 and . In RDOCK, the ZDOCK predictions are subjected to a three-stage energy minimization scheme using the CHARMM force field [48] and amounting to 130 minimization steps, followed by the rescoring based on the CHARMM electrostatic and desolvation energies. This elaborate approach improves the success rate for Npred=10 (i.e., the success rate for finding a near-native hit within the first 10 predictions, as expected in the CAPRI experiment [41]) from 38 to 45 % for decoys obtained from ZDOCK(PDE), which is similar to the ZDOCK 3.02 version used in this work. In our study, the success rate for Npred=10 decreases by 1–2 % after rescoring with E86 and (see Figs. 3 and 7). In case of E86 it is due to the poor performance of this scoring function for antigen/antibody complexes, while does not perform well for Npred<12 for other complexes. It should be noted that also RDOCK is considerably less successful for antigen/antibody complexes compared to enzyme/inhibitor complexes, supporting our conclusion that the rescoring of the former is more challenging than that of the latter.
Comparison to other coarse-grained force fields shows that OPEP is better suited as scoring function for protein-protein docking than these. In addition to OPEP we also tested the coarse-grained force field developed by Bereau and Deserno (BD) [49] on a decoy set produced by ZDOCK consisting of 23 enzyme/inhibitor and 23 other complexes. The BD force field increased the rank of 31 complexes and decreased it for only four complexes, which is considerably worse than what we obtain with OPEP. Reasons for the failure of the BD force field when applied to protein-protein docking are that the side-chain beads have all the same size and that electrostatic interactions between charged residues are not modelled, features that are present in OPEP. Moreover, in a study performed similarly to ours, the UNRES coarse-grained force field was tested as rescoring function [50]. The number of hits that were retained in the top-10 predictions generated by FTDock [51] decreased by more than 50 %, while with our approach the success rate decreases by only 1–2 % at Npred=10. This shows that while OPEP is still not a perfect scoring function for protein-protein docking, it is clearly better suited than other coarse-grained force fields.
Outlook In our future work we strive to further improve the performance of the OPEP rescoring functions. Here, special attention will be devoted to antigen/antibody complexes, where improvement is most needed. In addition, we will not only rescore the decoys generated by ZDOCK but also refine them by performing Monte Carlo simulations with OPEP. One advantage of OPEP is that it is a physics-motivated force field defined based on continuous functions and is therefore ideally suited for flexible docking. Our aim is to produce a reliable refinement and rescoring protocol based on OPEP that only needs docking decoys generated by ZDOCK or another global search algorithm as additional input. For the participation in the CAPRI experiment, however, a final transformation from the coarse-grained to the atomistic level for the top-10 decoys will become necessary as only atomistic decoys can be submitted.
Availability of data and materials
All decoy structures as obtained from ZDOCK and after their transformation to the OPEP coarse-grained level are available upon request from the authors.
Acknowledgements
We thank Holger Gohlke for fruitful discussions. P.K. gratefully acknowledges financial support from the International NRW Research School BioStruct, granted by the Ministry of Innovation, Science and Research of the State North Rhine-Westphalia, the Heinrich Heine University of Düsseldorf, and the Entrepreneur Foundation at the Heinrich Heine University of Düsseldorf.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PK, PD, and BS designed the study. PK assembled input data, wrote code, ran the simulations, and analysed output data. PK and BS wrote the manuscript. All authors have read and approved the final version of the manuscript.
Contributor Information
Philipp Kynast, Email: p.kynast@fz-juelich.de.
Philippe Derreumaux, Email: philippe.derreumaux@ibpc.fr.
Birgit Strodel, Email: b.strodel@fz-juelich.de.
References
- 1.Bernstein FC, Koetzle TF, Williams GJ, Meyer EF, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. The protein data bank. Eur J Biochem. 1977;80(2):319–24. doi: 10.1111/j.1432-1033.1977.tb11885.x. [DOI] [PubMed] [Google Scholar]
- 2.Katchalski-Katzir E, Shariv I, Eisenstein M, Friesem AA, Aflalo C, Vakser IA. Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc Natl Acad Sci. 1992;89(6):2195–199. doi: 10.1073/pnas.89.6.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen R, Weng Z. Docking unbound proteins using shape complementarity, desolvation, and electrostatics. Proteins Struct Funct Bioinforma. 2002;47(3):281–94. doi: 10.1002/prot.10092. [DOI] [PubMed] [Google Scholar]
- 4.Garzon JI, Lopéz-Blanco JR, Pons C, Kovacs J, Abagyan R, Fernandez-Recio J, Chacon P. Frodock: a new approach for fast rotational protein–protein docking. Bioinformatics. 2009;25(19):2544–551. doi: 10.1093/bioinformatics/btp447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kozakov D, Brenke R, Comeau SR, Vajda S. Piper: An fft-based protein docking program with pairwise potentials. Proteins Struct Funct Bioinforma. 2006;65(2):392–406. doi: 10.1002/prot.21117. [DOI] [PubMed] [Google Scholar]
- 6.Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D, et al. Protein–protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J Mol Biol. 2003;331(1):281–300. doi: 10.1016/S0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 7.Fernández-Recio J, Totrov M, Abagyan R. ICM-DISCO docking by global energy optimization with fully flexible side-chains. Proteins Struct Funct Bioinforma. 2003;52(1):113–7. doi: 10.1002/prot.10383. [DOI] [PubMed] [Google Scholar]
- 8.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20(1):45–50. doi: 10.1093/bioinformatics/btg371. [DOI] [PubMed] [Google Scholar]
- 9.Tong W, Weng Z. Clustering protein-protein docking predictions. Conf Proc IEEE Eng Med Biol Soc. 2004;4:2999–3002. doi: 10.1109/IEMBS.2004.1403850. [DOI] [PubMed] [Google Scholar]
- 10.Jackson RM, Gabb HA, Sternberg MJE. Rapid refinement of protein interfaces incorporating solvation: application to the docking problem1. J Mol Biol. 1998;276(1):265–85. doi: 10.1006/jmbi.1997.1519. [DOI] [PubMed] [Google Scholar]
- 11.Vreven T, Hwang H, Weng Z. Integrating atom-based and residue-based scoring functions for protein–protein docking. Protein Sci. 2011;20(9):1576–1586. doi: 10.1002/pro.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pierce B, Weng Z. Zrank: reranking protein docking predictions with an optimized energy function. Proteins Struct Funct Bioinforma. 2007;67(4):1078–1086. doi: 10.1002/prot.21373. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Chen R, Weng Z. Rdock: Refinement of rigid-body protein docking predictions. Proteins Struct Funct Bioinforma. 2003;53(3):693–707. doi: 10.1002/prot.10460. [DOI] [PubMed] [Google Scholar]
- 14.Kozakov D, Clodfelter KH, Vajda S, Camacho CJ. Optimal clustering for detecting near-native conformations in protein docking. Biophys J. 2005;89(2):867–75. doi: 10.1529/biophysj.104.058768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim YC, Hummer G. Coarse-grained models for simulations of multi-protein complexes: Application to ubiquitin binding. J Mol Biol. 2008;375(5):1416. doi: 10.1016/j.jmb.2007.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.May A, Pool R, van Dijk E, Bijlard J, Abeln S, Heringa J, Feenstra KA. Coarse-grained versus atomistic simulations: realistic interaction free energies for real proteins. Bioinformatics. 2014;30:326–34. doi: 10.1093/bioinformatics/btt675. [DOI] [PubMed] [Google Scholar]
- 17.Ravikumar KM, Huang W, Yang S. Coarse-grained simulations of protein-protein association: An energy landscape perspective. Biophys J. 2012;103(4):837–45. doi: 10.1016/j.bpj.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH. The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B. 2007;111(27):7812–824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- 19.Monticelli L, Kandasamy SK, Periole X, Larson RG, Tieleman DP, Marrink SJ. The MARTINI coarse-grained force field: Extension to proteins. J Chem Theory Comput. 2008;4(5):819–34. doi: 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- 20.Taketomi H, Ueda Y, Gō N. Studies on protein folding, unfolding and fluctuations by computer simulation. Int J Pept Protein Res. 1975;7(6):445–59. doi: 10.1111/j.1399-3011.1975.tb02465.x. [DOI] [PubMed] [Google Scholar]
- 21.Miyazawa S, Jernigan RL, et al. Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J Mol Biol. 1996;256(3):623–44. doi: 10.1006/jmbi.1996.0114. [DOI] [PubMed] [Google Scholar]
- 22.Sterpone F, Melchionna S, Tuffery P, Pasquali S, Mousseau N, Cragnolini T, Chebaro Y, St-Pierre JF, Kalimeri M, Barducci A, et al. The opep protein model: from single molecules, amyloid formation, crowding and hydrodynamics to dna/rna systems. Chem Soc Rev. 2014;43(13):4871–93. doi: 10.1039/c4cs00048j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou J, Thorpe IF, Izvekov S, Voth GA. Coarse-grained peptide modeling using a systematic multiscale approach. Biophys J. 2007;92(12):4289–303. doi: 10.1529/biophysj.106.094425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu S, Gao Y, Vakser IA. Dockground protein–protein docking decoy set. Bioinformatics. 2008;24(22):2634–635. doi: 10.1093/bioinformatics/btn497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nocedal J. Updating quasi-newton matrices with limited storage. Math Comput. 1980;35(151):773–82. doi: 10.1090/S0025-5718-1980-0572855-7. [DOI] [Google Scholar]
- 26.Hwang H, Vreven T, Janin J, Weng Z. Protein–protein docking benchmark version 4.0. Proteins Struct Funct Bioinforma. 2010;78(15):3111–114. doi: 10.1002/prot.22830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pierce BG, Hourai Y, Weng Z. Accelerating protein docking in zdock using an advanced 3d convolution library. PloS ONE. 2011;6(9):24657. doi: 10.1371/journal.pone.0024657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hwang H, Vreven T, Pierce BG, Hung JH, Weng Z. Performance of zdock and zrank in capri rounds 13–19. Proteins Struct Funct Bioinforma. 2010;78(15):3104–110. doi: 10.1002/prot.22764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen R, Weng Z. A novel shape complementarity scoring function for protein-protein docking. Proteins Struct Funct Bioinforma. 2003;51(3):397–408. doi: 10.1002/prot.10334. [DOI] [PubMed] [Google Scholar]
- 30.Chen R, Li L, Weng Z. Zdock: an initial-stage protein-docking algorithm. Proteins Struct Funct Bioinforma. 2003;52(1):80–7. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- 31.Derreumaux P. From polypeptide sequences to structures using monte carlo simulations and an optimized potential. J Chem Phys. 1999;111(5):2301–310. doi: 10.1063/1.479501. [DOI] [Google Scholar]
- 32.Sterpone F, Nguyen PH, Kalimeri M, Derreumaux P. Importance of the ion-pair interactions in the opep coarse-grained force field: parametrization and validation. J Chem Theory Comput. 2013;9(10):4574–584. doi: 10.1021/ct4003493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maupetit J, Tuffery P, Derreumaux P. A coarse-grained protein force field for folding and structure prediction. Proteins Struct Funct Bioinforma. 2007;69(2):394–408. doi: 10.1002/prot.21505. [DOI] [PubMed] [Google Scholar]
- 34.Chebaro Y, Pasquali S, Derreumaux P. The coarse-grained opep force field for non-amyloid and amyloid proteins. J Phys Chem B. 2012;116(30):8741–752. doi: 10.1021/jp301665f. [DOI] [PubMed] [Google Scholar]
- 35.Zacharias M. Protein–protein docking with a reduced protein model accounting for side-chain flexibility. Protein Sci. 2003;12(6):1271–1282. doi: 10.1110/ps.0239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Méndez R, Leplae R, De Maria L, Wodak SJ. Assessment of blind predictions of protein–protein interactions: current status of docking methods. Proteins Struct Funct Bioinforma. 2003;52(1):51–67. doi: 10.1002/prot.10393. [DOI] [PubMed] [Google Scholar]
- 37.Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, et al. Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25(11):1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Viswanath S, Ravikant D, Elber R. Improving ranking of models for protein complexes with side chain modeling and atomic potentials. Proteins Struct Funct Bioinforma. 2013;81(4):592–606. doi: 10.1002/prot.24214. [DOI] [PubMed] [Google Scholar]
- 39.Tobi D. Designing coarse grained-and atom based-potentials for protein-protein docking. BMC Struct Biol. 2010;10(1):40. doi: 10.1186/1472-6807-10-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frishman D, Argos P. Knowledge-based protein secondary structure assignment. Proteins Struct Funct Bioinforma. 1995;23(4):566–79. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 41.Janin J. Assessing predictions of protein–protein interaction: The capri experiment. Protein Sci. 2005;14(2):278–83. doi: 10.1110/ps.041081905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jackson RM. Comparison of protein–protein interactions in serine protease-inhibitor and antibody-antigen complexes: Implications for the protein docking problem. Protein Sci. 1999;8(03):603–13. doi: 10.1110/ps.8.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ravikant D, Elber R. Energy design for protein-protein interactions. J Chem Phys. 2011;135:065102. doi: 10.1063/1.3615722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin AC. Antibody engineering. Berlin Heidelberg: Springer; 2001. Protein sequence and structure analysis of antibody variable domains. [Google Scholar]
- 45.Robin G, Sato Y, Desplancq D, Rochel N, Weiss E, Martineau P. Restricted diversity of antigen binding residues of antibodies revealed by computational alanine scanning of 227 antibody–antigen complexes. J Mol Biol. 2014;426(22):3729–743. doi: 10.1016/j.jmb.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 46.Méndez R, Leplae R, De Maria L, Wodak SJ. Assessment of blind predictions of protein–protein interactions: Current status of docking methods. Proteins Struct Funct Bioinforma. 2003;52(1):51–67. doi: 10.1002/prot.10393. [DOI] [PubMed] [Google Scholar]
- 47.Pierce B, Weng Z. A combination of rescoring and refinement significantly improves protein docking performance. Proteins Struct Funct Bioinforma. 2008;72(1):270–9. doi: 10.1002/prot.21920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4(2):187–217. doi: 10.1002/jcc.540040211. [DOI] [Google Scholar]
- 49.Bereau T, Deserno M. Generic coarse-grained model for protein folding and aggregation. J Chem Phys. 2009;130(23):235106. doi: 10.1063/1.3152842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Solernou A, Fernández-Recio J. Refinement of rigid-body protein–protein docking using backbone and side-chain minimization with a coarse-grained model. Open Access Bioinforma. 2010;2:19–27. [Google Scholar]
- 51.Gabb HA, Jackson RM, Sternberg MJE. Modelling protein docking using shape complementarity, electrostatics and biochemical information1. J Mol Biol. 1997;272(1):106–20. doi: 10.1006/jmbi.1997.1203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All decoy structures as obtained from ZDOCK and after their transformation to the OPEP coarse-grained level are available upon request from the authors.