

Abstract
Background
The c-Jun N-terminal kinase (JNK) signaling pathway plays an important role in neuronal pathophysiology. Using JNK inhibitors, we examined involvement of the JNK pathway in cultured rat retinal ganglion cell (RGC) death and in mouse retinal ischemia/reperfusion (I/R) injury of the visual axis. The in vitro effects of JNK inhibitors were evaluated in cultured adult rat retinal cells enriched in RGCs. Retinal I/R was induced in C57BL/6J mice through elevation of intraocular pressure to 120 mmHg for 60 min followed by reperfusion. SP600125 was administered intraperitoneally once daily for 28 days. Phosphorylation of JNK and c-Jun in the retina was examined by immunoblotting and immunohistochemistry. The thickness of retinal layers and cell numbers in the ganglion cell layer (GCL) were examined using H&E stained retinal cross sections and spectral domain optical coherence tomography (SD-OCT). Retinal function was measured by scotopic flash electroretinography (ERG). Volumetric measurement of the superior colliculus (SC) as well as VGLUT2 and PSD95 expression were studied.
Results
JNK inhibitors SP600125 and TAT-JNK-III, dose-dependently and significantly (p < 0.05) protected against glutamate excitotoxicity and trophic factor withdrawal induced RGC death in culture. In the I/R model, phosphorylation of JNK (pJNK) in the retina was significantly (p < 0.05) increased after injury. I/R injury significantly (p < 0.05) decreased the thickness of retinal layers, including the whole retina, inner plexiform layer, and inner nuclear layer and cell numbers in the GCL. Administration of SP600125 for 28 days protected against all these degenerative morphological changes (p < 0.05). In addition, SP600125 significantly (p < 0.05) protected against I/R-induced reduction in scotopic ERG b-wave amplitude at 3, 7, 14, 21 and 28 days after injury. SP600125 also protected against the I/R-induced losses in volume and levels of synaptic markers in the SC. Moreover, the protective effects of SP600125 in the retina and SC were also detected even with only 7 days (Days 1–7 after I/R) of SP600125 treatment.
Conclusions
Our results demonstrate the important role the JNK pathway plays in retinal degeneration in both in vitro and in vivo models and suggest that JNK inhibitors may be a useful therapeutic strategy for neuroprotection of RGCs in the retina.
Electronic supplementary material
The online version of this article (doi:10.1186/s13024-016-0093-4) contains supplementary material, which is available to authorized users.
Keywords: Retinal ischemia, c-Jun N-terminal kinase (JNK), Retinal ganglion cells, JNK inhibitors, Electroretinography
Background
Activation of c-Jun N-terminal kinase (JNK) is frequently observed in degenerative diseases in mammalian neuronal system such as Alzheimer’s disease, Parkinson’s disease and glaucoma [1–3]. As a member of the mitogen activated protein (MAP) kinase family, activation of JNK is mediated by phosphorylation of various biological and environmental stress factors [4, 5]. In particular, several major inflammatory cascades, including oxidative stress, inflammatory cytokines, pattern-recognition receptors, and neurotransmitter receptors, activate the JNK signaling pathway [6–9] followed by activation of downstream signaling molecules such as c-Jun, ATF-2, ELK-1, and Stat3 [4, 5, 10]. These molecular events result in gene transcription associated with various biological outcomes including neuronal cell migration, cytoskeletal reorganization, and cell death [11–15].
As part of the central nervous system, the visual system, including retinal ganglion cells (RGC) and other neurons, has been investigated to identify the influence of JNK activation under various pathologic conditions. Tezel et al. demonstrated that JNK signaling is associated with RGC degeneration induced by tumor necrosis factor (TNF) receptor after optic nerve (ON) injury [16]. Bessero et al. reported that inhibition of JNK1 protected the retina from NMDA-induced excitotoxicity [17]. In addition, combined knockdown of Jnk2 and Jnk3 induced long-term protection of RGCs against axonal injury in mice [18]. Balaiya et al. also observed increased phosphorylated JNK (pJNK) in cultured RGCs exposed to hypoxic conditions [19]. More recently, Welsbie et al. showed that knockdown of the dual leucine zipper kinase, which is an upstream activator of JNK, improved survival and function of RGCs [20]. Taken together, the JNK pathway appears to play a pivotal role in RGC death under various insults and disease conditions.
Ischemia and subsequent reperfusion elicits severe damage in the visual system, leading to irreversible vision loss in many ocular diseases including retinal vessel occlusion, glaucoma, and diabetic retinopathy [21–23]. In particular, ischemia/reperfusion (I/R) injury in the retina causes RGC death, resulting in functional failure of transmitting visual information to specific receptive fields in the brain [24–26]. We previously reported that I/R damage in the retina induced morphological and functional degeneration and RGC death that was associated with temporal regulation of retinal gene expression [27]. In particular, various gene clusters, especially those related to cell death and inflammatory responses, were upregulated post injury and directly associated with the JNK signaling pathway in pathological stages of various diseases [28].
In this study, we evaluated the role JNK signaling pathway plays in retinal degeneration and RGC death using pharmacological JNK inhibitors in retinal cell culture and mouse retinal I/R injury models. We first examined their protective effects against cell death in an adult rat retinal cell culture. We further examined the effect of JNK inhibition on I/R-induced changes in the retina and SC. We found that JNK inhibition provided total morphological and functional protection to RGCs.
Results
Protection of RGC death by JNK inhibitors
Several insults are known to induce cell death of purified RGCs in vitro. Otori et al. showed that glutamate (5 to 500 μM) induced cell death of cultured rat RGCs in a dose-dependent manner [29]. Withdrawal of trophic factors also induced cultured RGC death [30]. In addition, TNFα from glia under ischemic conditions also induced RGC death in a co-culture system [31]. Based on previous findings, we further investigated whether these RGC death mechanisms are associated with JNK signaling. Death of cultured RGCs was induced by treating cells for 3 days with glutamate (100 μM), TNFα (10 ng/mL), or TFW (trophic factor withdrawal) in the presence or absence of various concentrations of the JNK inhibitors SP600125 or TAT-JNKi-III. Cells were then fixed and labeled with anti-Thy-1 antibody for RGC counting. SP600125 treatment significantly (p < 0.05) enhanced RGC survival in a dose-dependent manner against glutamate and TFW-induced cell death (Fig. 1a and c), but did not protect against TNFα-induced RGC death (Fig. 1e). Similarly, TAT-JNKi-III also dose-dependently protected against RGC death induced by glutamate and TFW, but not by TNFα (Fig. 1b, d and f).
Fig. 1.
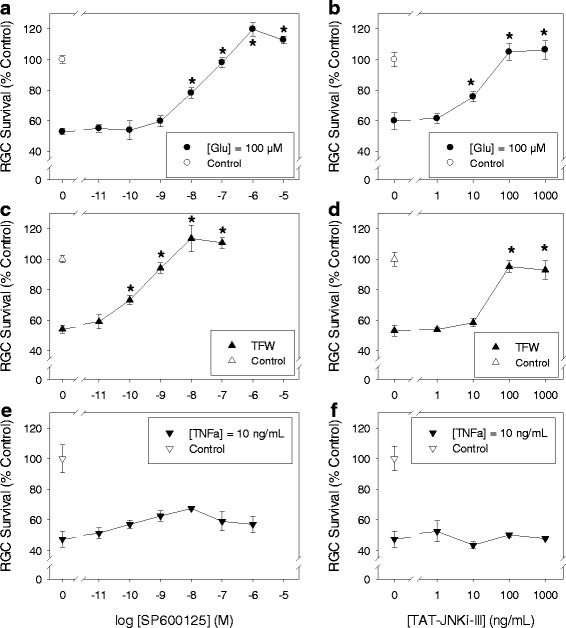
JNK inhibitors protected against RGC death induced by glutamate (Glu) excitotoxicity and trophic factor withdrawal (TFW), but not by tumor necrosis factor (TNF)-α. Cultured adult rat retinal cells were treated with the indicated concentration of JNK inhibitors SP600125 (a, c, e) or TAT-JNKi-III (b, d, f) in the presence of the cytotoxic insults: 100 μM of Glu (a, b), TFW (c, d), or 10 ng/mL TNFα (e, f) for 3 days. Surviving cells were fixed and labeled with anti-Thy-1 antibody and manually counted. Vehicle-treated (no insult) control group in each study defines 100 %. Symbols represent mean ± SEM (n = 6–10). Asterisk indicates statistical difference (p < 0.05) between the JNK inhibitor-treated groups versus the respective insult-only group by One-way ANOVA then Dunnett’s test
JNK activation induced by retinal I/R
JNK is activated via phosphorylation of threonine and tyrosine residues located in the activation loop in the carboxyl-terminus. Activated JNK subsequently phosphorylates c-Jun [32, 33]. Therefore, we examined I/R-induced phosphorylation of JNK and c-Jun in the whole retina at various time points after injury using immunoblotting analysis (Fig. 2). Retinal JNK phosphorylation was detected at 0, 1, 6, 12, 24, and 72 h after I/R injury. As previously reported, we also observed a basal level of phosphorylated JNK at 0 h [34, 35]. JNK phosphorylation appeared to show a bi-phasic increase with an initial peak at 1 h (p < 0.05 vs. 0 h control) and a delayed increase at 72 h after I/R injury (Fig. 2a). Interestingly, c-Jun phosphorylation also closely correlated with the JNK phosphorylation pattern (Fig. 2b).
Fig. 2.
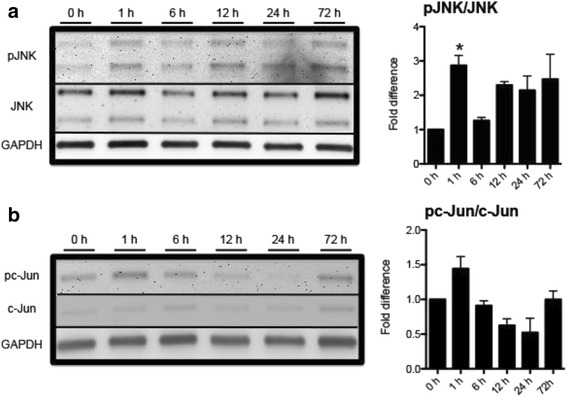
Retinal I/R induced phosphorylation of JNK and c-Jun. Mouse retinas were collected at 1, 6, 12, 24, and 72 h post I/R injury. The 0 h control represents the non-injured group. Western blotting analyses were conducted using total retinal proteins. a Representative images of phosphorylated JNK (pJNK), total JNK, and loading control GAPDH as well as ratio of pJNK versus total JNK, analyzed by ImageJ. JNK phosphorylation was significantly (p < 0.05) increased at 1 h after I/R injury by 2.87 (±1.87) fold compared to 0 h control. b Representative images of phosphorylated c-Jun (pc-Jun), total c-Jun, and loading control GAPDH as well as ratio of pc-Jun versus total c-Jun analyzed by ImageJ. Bars represent mean ± SEM (n = 3). Asterisk indicates statistical difference (p < 0.05) compared to the 0 h control group by One-way ANOVA then Dunnett’s test
In immunohistochemical analysis, basal level of JNK phosphorylation was observed in the same location with the RGC marker Tuj-1(magenta arrows) and OPL corresponding with our immunoblotting results. I/R injury induced drastic increase of JNK phosphorylation in Tuj-1 positive RGCs at early post-I/R injury times (1 h and 6 h) and detected in non-RGCs (white arrows) from 12 h to 72 h after I/R injury (Fig. 3). Notably, JNK phosphorylation was also observed in the INL from 6 h to 12 h after I/R injury, and in the IPL from 24 h to 72 h. In addition, strong JNK phosphorylation was continuously observed in OPL from 1 h to 72 h after I/R injury (Fig. 3). Phosphorylated c-Jun was also detected in a very similar manner to pJNK in I/R-injured retinas. Phosphorylated c-Jun was first observed in Tuj-1-positive RGCs at 1 h (yellow arrows) after I/R injury, and the presence of pc-Jun was continuously detected in both RGCs and non-RGCs (white arrow) from 6 h to 72 h in GCL after I/R injury (Fig. 4). As we also observed phosphorylation of pc-Jun in INL, we further stained phosphorylated c-Jun with bipolar cell (PKCα) and amacrine cell (syntaxin) markers at 24 h after I/R injury. Interestingly, phosphorylated JNK and c-Jun was not co-localized with either bipolar or amacrine cells (white arrow) (Fig. 5).
Fig. 3.
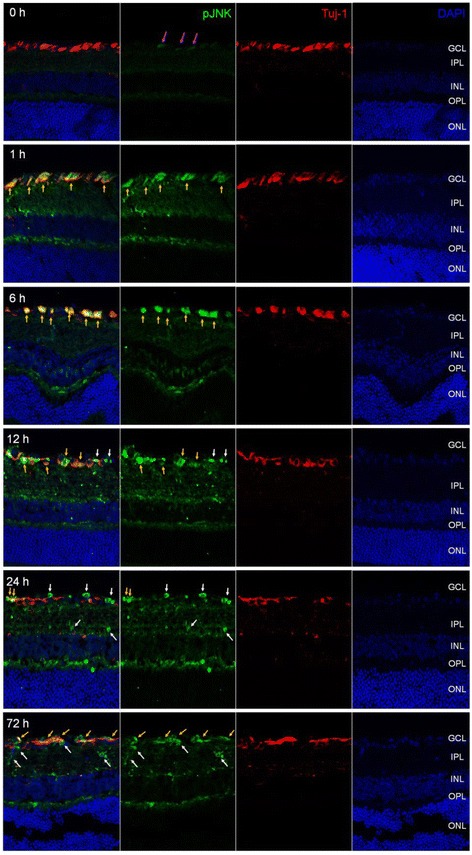
Phosphorylated JNK was detected in retina after I/R injury. Frozen-sectioned (10 μm) retina samples from 0, 1, 6, 12, 24 and 72 h after I/R injury were used for immunohistochemistry. Phosphorylated JNK was detected (green fluorescence). Tuj-1 immunofluorescence (red) was used as RGC marker. DAPI staining (blue) represents cell nuclei for counter staining. The magenta arrows represent basal JNK phosphorylation in 0 h retina. All yellow arrows represent phosphorylated JNK in RGC and white arrows represent non-RGC in GCL
Fig. 4.
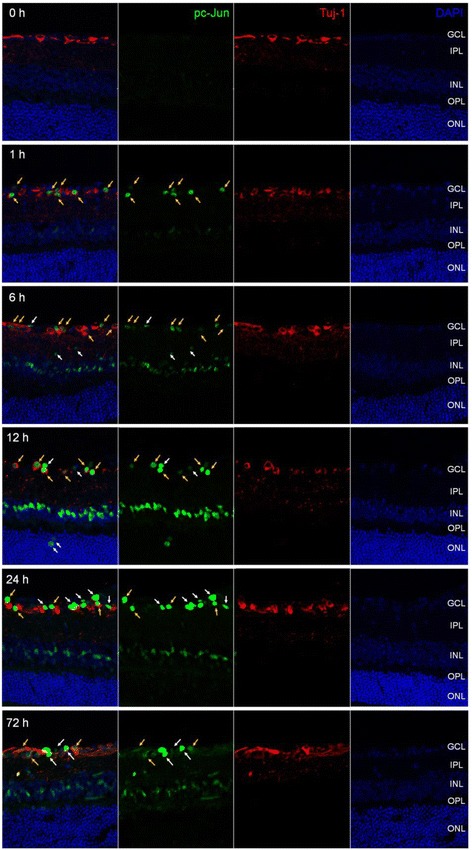
Phosphorylated c-Jun was detected in retina after I/R injury. Frozen-sectioned (10 μm) retina samples from 0, 1, 6, 12, 24 and 72 h after I/R injury were used for immunohistochemistry. As with JNK, phosphorylated c-Jun was detected (green fluorescence). Tuj-1 immunofluorescence (red) was used as RGC marker. DAPI staining (blue) represents cell nuclei for counter staining. All yellow arrows represent phosphorylated c-Jun in RGC and white arrows represent non-RGC in GCL
Fig. 5.
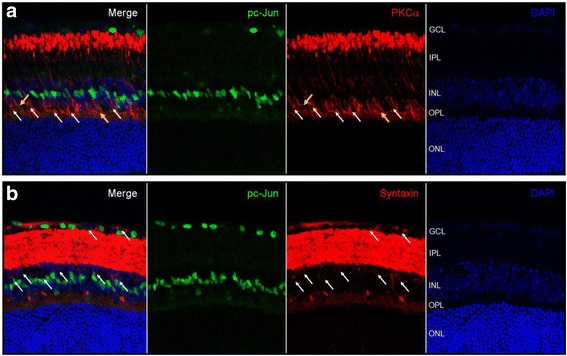
Phosphorylated c-Jun was not co-localized with both bipolar cell and amacrine cells in retina after I/R injury. 10 μm frozen retinal sections from 0, 1, 6, 12, 24 and 72 h after I/R injury were used to detect phosphorylated c-Jun (green fluorescence) in bipolar cells (a) and amacrine cells (b). PKCα (a) or syntaxin (b) immunofluorescence (red) were used to identify bipolar cells or amacrine cells in INL. DAPI staining (blue) represents cell nuclei for counter staining. All phosphorylated c-Jun in INL were indicated using white arrow (no co-localization) or yellow arrow (co-localization) to show its location in INL with amacrine cells (a) and bipolar cells (b)
Protection against I/R-induced retinal damage by SP600125
Previously, it was shown that systemic administration of SP600125 showed protection in neurodegenerative animal models such as focal brain ischemia, in which the JNK signaling pathway are involved [36]. Therefore, we administered SP600125 to mice in our model of retinal I/R to determine potential protective effects of JNK inhibition based on our in vitro data. We preliminarily verified in vivo inhibition of SP600125 against JNK and c-Jun phosphorylation after retinal I/R injury. SP600125 (30 mg/kg) was administered intraperitoneally 2 h prior to I/R injury followed by immunostaining analysis for phosphorylated JNK and c-Jun after 24 h. As expected, SP600125 administration decreased pc-Jun phosphorylation in both the GCL and INL (Additional file 1: Figure S9). Three different doses of SP600125 (5, 15, 30 mg/kg) or vehicle were administered intraperitoneally as described above followed by once daily administration for 28 days after I/R injury. Histological measurement of H&E stained retinas showed that I/R in the vehicle control group caused significant (p < 0.01) reduction of retinal layer thickness, particularly in the IPL and INL over 28 days (Fig. 6) as previously reported [27]. Importantly, administration of SP600125 at each of the three doses completely prevented I/R-induced thinning of the IPL and INL (Fig. 6). In addition to histology, we also examined morphological changes of retina using SD-OCT at various time points after I/R injury, with or without SP600125 (15 mg/kg) treatment. I/R injury caused progressive and significant (p < 0.05) thinning of the IPL and INL, whereas SP600125 treatment ameliorated these changes (Fig. 7). I/R-induced cell loss in the GCL was also significantly attenuated by all three doses of SP600125 (Fig. 8). In addition, I/R injury significantly (p < 0.05) reduced the scotopic ERG b-wave (Fig. 9b). The I/R-mediated reduction in b-wave amplitudes were also restored by SP600125 treatment (Fig. 9b).
Fig. 6.
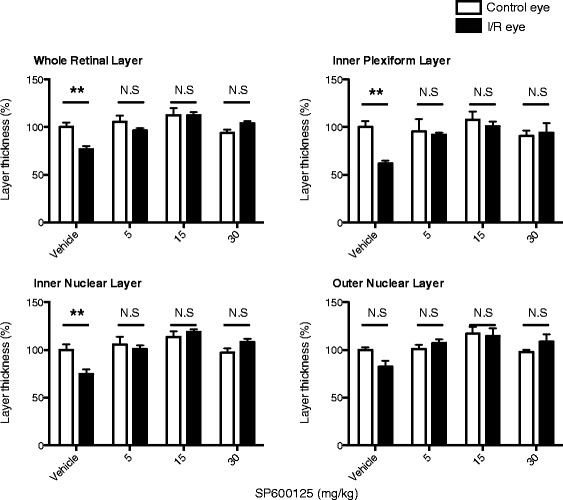
SP600125 protected against I/R-induced thinning of inner retinal layers. Three doses of SP600125 (5, 15, and 30 mg/kg) or vehicle were administered intraperitoneally 2 h prior to retinal I/R injury followed by once daily dosing. After 28 days, histological changes in retinal thickness, including whole retina (from NFL to ONL), IPL, INL and ONL were analyzed. Data represent mean ± SEM (n = 5) at each time point. The mean thickness of the uninjured, vehicle-treated retinas was set as 100 %. In vehicle-treated group, I/R injury significantly reduced whole retina thickness 23.2 ± 5.7 % compared to non-injured contralateral retinas. In particular, IPL was reduced 38.0 ± 6.7 % and INL reduced 25.1 ± 7.4 %. No significant layer change was observed in ONL. In contrast, no significant layer thickness reduction was observed from SP60025-administered groups at each dose (5, 15 and 30 mg/kg). **: p < 0.01; NS: non-significant between contralateral control and I/R eyes by two-tailed, paired t-test
Fig. 7.
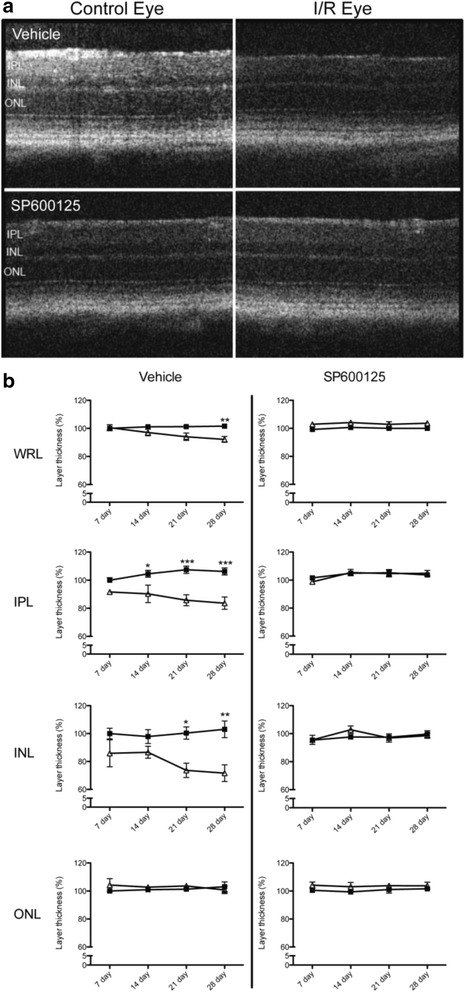
SP600125 ameliorated I/R-induced progressive retina thinning. Retinal I/R injured mice were treated with SP600125 (15 mg/kg) or vehicle for 28 days. SD-OCT was used to determine progressive changes in whole retinal layer (from NFL to ONL), and each of IPL, INL, and ONL. a Representative images showing layer changes after 28 days post I/R injury. b Quantitative analyses of progressive changes of thickness of each layer at different time points. Open symbols: I/R eyes; closed symbols: un-injured contralateral eyes. In vehicle-administered groups, whole retina thickness in I/R-injured eyes were progressively decreased at 28 days of injury compared to contralateral non-injured eyes. These changes were mainly observed in inner retinal layers, IPL and INL. In contrast, SP600125 administration inhibited I/R-induced layer reduction. Data are shown as mean and SEM (n = 4). Two-way ANOVA analyses indicate that there were no significant time-dependent changes in all SP600125 groups, but significant differences were detected between the control and I/R groups at certain time points (*: p < 0.05, **: p < 0.01, ***: p < 0.001 by Bonferroni’s post-hoc test). (WRL: whole retina layer, NFL: nerve fiber layer, IPL: inner plexiform layer, INL: inner nuclear layer, ONL: outer nuclear layer)
Fig. 8.
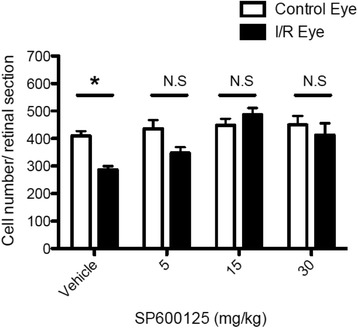
SP600125 protected against I/R-induced cell loss in the GCL. Total cell numbers in the GCL were counted from retinas of vehicle or SP600125-treated animals at 28 days after I/R injury. Non-injured contralateral eyes were also analyzed for comparison. I/R injury induced significant GCL cell loss (30.0 ± 5.6 %) compared to contralateral non-injured eyes whereas SP600125 administration showed total protection. Data represent mean ± SEM (n = 5). *: p < 0.05; NS: non-significant between contralateral control and I/R eyes by two-tailed, paired Student’s t-test
Fig. 9.
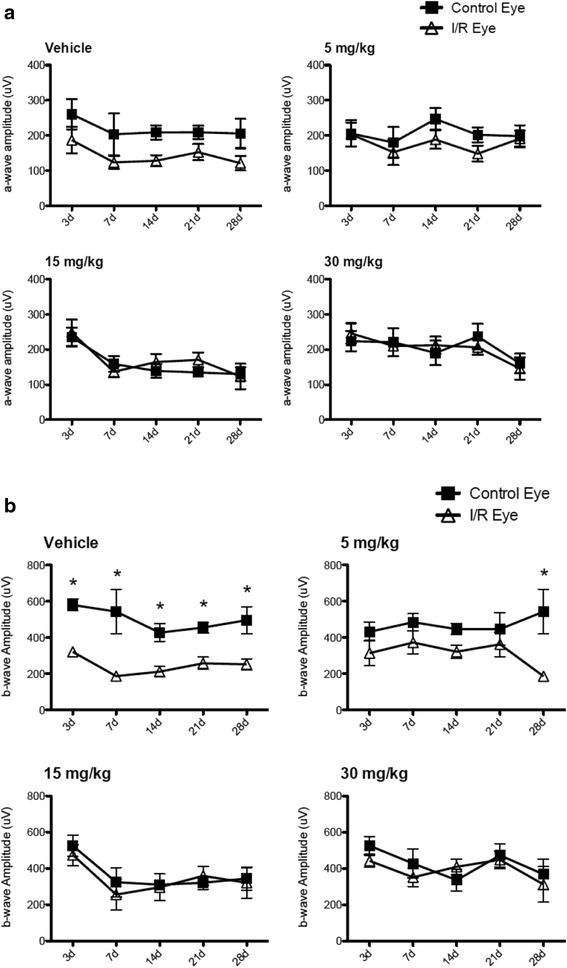
SP600125 ameliorated the I/R-induced reductions in ERG b-wave amplitudes. The a- and b- wave amplitudes of scotopic ERG at 10 cd light intensity were measured in the vehicle and SP600125-treated groups 3, 7, 14, 21, and 28 days after I/R. Data represent mean ± SEM (n = 5). I/R injury induced slight reductions of a-wave amplitudes (a) and significant (p < 0.05) impairment of b waves (b) over 28 days in the vehicle group. In contrast, daily administration of SP600125 prevented impairment of a-waves and b-waves (a and b). Two-way ANOVA analyses indicate that there were no significant time-dependent changes in all SP600125 groups, but significant differences were detected between the control and I/R groups at certain time points (*: p < 0.05 by Bonferroni’s post-hoc test)
Protection against I/R-induced degeneration of RGC axons in the SC by SP600125
In the mouse visual system, approximately 95 % of RGC axons project contralaterally [37], and at least 70 % of RGCs project to the superficial layer of SC [38]. As shown in Fig. 10, I/R injury caused a significant (p < 0.05) decrease (~20 %) in the superficial SC layer cross-sectional area in the contralateral side of the I/R injury compared to the ipsilateral side after 28 days. Importantly, administration of SP600125 (5, 15, 30 mg/kg) protected against this I/R-induced SC neuron decrease. We also assessed the synaptic terminal of RGC neurons in the SC superficial layer by examining expression of the presynaptic marker VGLUT2 (vesicular glutamate transporter 2). Furthermore, we also quantified intensity of the postsynaptic marker PSD95 to measure potential failure in post-synapses of RGC neurons in the SC. I/R injury significantly (p < 0.05) decreased expression of both VGLUT2 and PSD95 in the contralateral SC in comparison to the ipsilateral side after 28 days. Decreased expression of these two markers were attenuated by daily SP600125 administration for 28 days (Fig. 11).
Fig. 10.
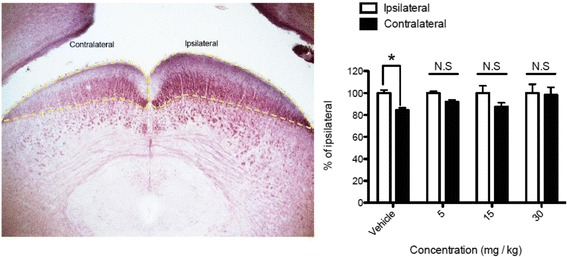
SP600125 protected retinal I/R-induced reduction in size of superior colliculus. Regions of mouse brains containing SC from vehicle or SP600125 (5, 15, 30 mg/kg)- treated groups after 28 days post retinal I/R injury were collected for serial frozen sectioning and black gold staining. Bright field images were taken and cross-sectional area of superficial layers from each contralateral or ipsilateral side were measured and averaged in series of all stained sections. In vehicle treated groups, I/R injury induced significant area volume loss (~16 %) in the contralateral SC region compared to the ipsilateral region. However, SP600125 administration protected SC from I/R-induced area volume loss in contralateral hemispheres. Data represent mean ± SEM (n = 4–5). The ipsilalateral area volume in each brain was set as a 100 %. *: p < 0.05 between contralateral and ipsilateral volumes of SC by two-tailed, paired t-test
Fig. 11.
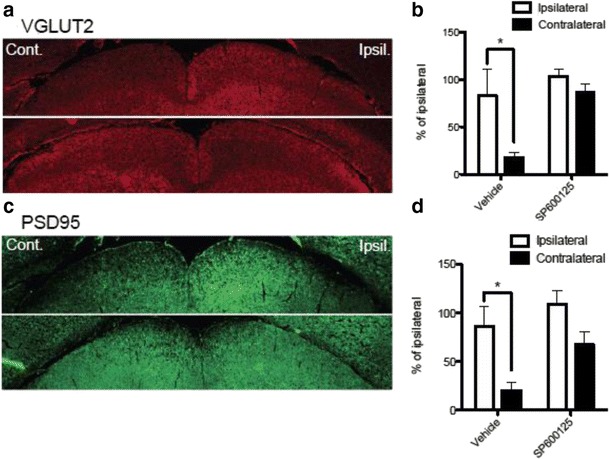
SP600125 prevented I/R-induced impaired expression of synaptic markers VGLUT2 and PSD95 in the superior colliculus. Mouse brains from vehicle or SP600125 (15 mg/kg) treatment groups were collected for immunohistochemistry at 28 days post retinal I/R injury. Each brain section was incubated with primary antibody for presynaptic marker VGLUT2 or post-synaptic marker PSD95. a and c Images including both ipsilateral (Ipsil) and contralateral (Cont) superficial layers of the superior colliculus from vehicle treated (above) and SP600125 treated (below) mice. b and d Data represent mean ± SEM (n = 4 - 5). I/R injury resulted in significant fluorescence intensity loss of VGLUT2 (84.4 ± 19.0 %) and PSD95 (76.8 ± 12.6 %) in the contralateral SC after 28 days in vehicle-administered group (b and d). In contrast, 28-day administration of SP600125 prevented the loss of both synaptic markers (b and d). Total area volume of the ipsilateral SC defines 100 %. *: p < 0.05 between contralateral and ipsilateral volumes by two-tailed, paired Student’s t-test
Protective effects in the retina and SC by only 7-day treatment with SP600125
The above results demonstrated that daily treatment with SP600125 for 28 days was protective against retinal I/R-induced damage to the retina and SC. To assess whether a shorter treatment period was also protective, SP600125 (15 mg/kg) was administered daily for only the first 7 days after I/R injury, and changes of the retina and SC were monitored for 28 days. I/R-induced degeneration of the INL and IPL was completely ameliorated by the 7-day administration of SP600125 as measured by SD-OCT (Fig. 12). Similarly, 7-day treatment with SP600125 also prevented the loss of synapse markers VGLUT2 and PSD95 in the SC, showing protection from RGC axon loss (Fig. 13).
Fig. 12.
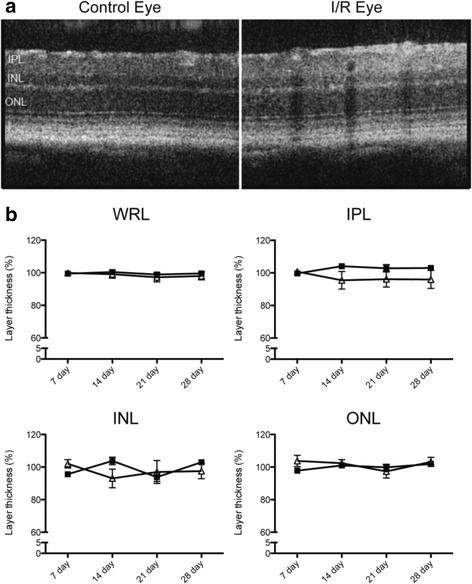
Seven-day administration of SP600125 also protected the retina from I/R-induced layer degeneration. SP600125 (15 mg/kg) was administered to retinal I/R injured mice for the first 7 days only. SD-OCT scanning was used to determine progressive changes in whole retinal layer (from NFL to ONL) or each sublayer including IPL, INL and ONL. Images were selected to show layer changes 28 days post I/R injury (a). Progressive changes of whole retinal layer or sub-layers were monitored (b). 7-day administration of SP600125 resulted in protection from retinal layer degeneration induced by I/R injury after 28 day. Open symbols: I/R eyes; closed symbols: un-injured contralateral eyes. Two-way ANOVA analyses indicate that there were no significant time-dependent or injury-dependent changes. (WRL: whole retina layer, IPL: inner plexiform layer, INL: inner nuclear layer, ONL: outer nuclear layer)
Fig. 13.
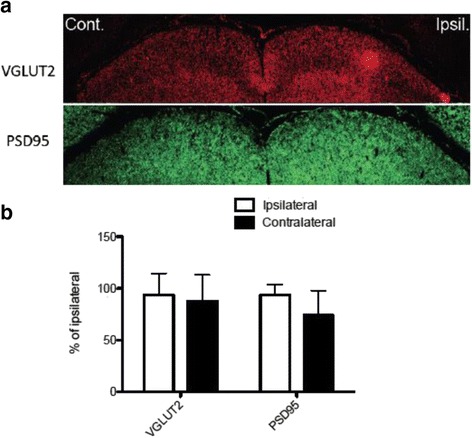
The 7-day administration of SP600125 protected I/R-induced reduction in VGLUT2 and PSD95 expression in the superior colliculus. (a) Mouse brains from 7-day SP600125 treatment (15 mg/kg) were collected for immunohistochemistry 28 days post retinal I/R injury. (b) Data represent mean ± SEM (n = 4) of percentages at each hemisphere from both contralateral (dark bars) and ipsilateral (open bars) sides compared to vehicle ipsilalateral volume, set as a 100 %. No significant difference was detected between contralateral and ipsilateral samples by two-tailed, paired Student’s t-test
Discussion
In this report, we demonstrated that JNK inhibitors were effective in protecting cultured adult rat RGCs from cytotoxicity, and also against neurodegeneration in the mouse of retinal I/R. In cultured adult rat retinal neurons, we confirmed previous findings that excitotoxicity, TFW, and TNFα result in loss of RGCs [30, 39]. We further showed that two selective JNK inhibitors, SP600125 and TAT-JNKi-III, protected RGCs in a dose-dependent manner against glutamate- and TFW-induced cell death, but not TNFα cytotoxicity. These findings agree with reports indicating the involvement of JNK following NMDA excitotoxicity in the rat retina [40, 41]. Furthermore, we established SP600125 as a neuroprotective agent to neurons in the mouse retina and SC following retinal I/R. To our knowledge, this is the first study to validate the involvement of the JNK pathway both in cytotoxic injury to cultured primary retinal neurons, as well as in vivo following retinal I/R, through multiple functional and morphological assessments of these neural tissues.
We previously reported retinal I/R injury caused progressive morphological and functional degeneration in the retina. We observed strong temporal correlation with various pathological markers, including those related to proinflammatory response and cell death [27]. Here, we extend those characterizations and further identify significant increases in phosphorylation of JNK and its downstream target c-Jun. Strong activation of JNK and c-Jun signaling has been reported in animal models of CNS ischemia. In transient forebrain ischemia induced by occlusion of the common carotid artery, there were significant increases in pJNK in hippocampal CA1 and CA3 neurons within 15 min of reperfusion [42–44]. Transient global ischemia induced by cardiac arrest in mice and rats similarly led to increased levels of active JNK in CA1 neurons [45, 46]. In a murine model of middle cerebral artery occlusion, pJNK was increased in the cerebral cortex and striatum [47–49]. Finally, in similar models of retinal I/R, active JNK signaling has been identified throughout the neural layers of the retina 24 h post ischemia [50–52].
Activation of JNK occurs in many neurodegenerative diseases in addition to ischemic damage. Cerebral cortex and hippocampal sections from Alzheimer’s disease patients have markedly increased pJNK staining co-localized with neurofibrillary tangles [53]. In tissue samples from Parkinson’s disease (PD) patients, pJNK staining was in close proximity to Lewy bodies. Likewise, in the MPTP mouse model of PD, there was significantly increased activation of the JNK pathway; and dual Jnk2/3 knockout mice were protected against the neurodegenerative effects of this toxin [2, 54]. Protective strategies aimed at the reduction or removal of JNK through the use of mouse genetics, viral vectors, and small molecule inhibitors support the involvement of JNK signaling in multiple neurodegenerative conditions.
The biphasic increase in pJNK and pc-Jun in our study occurred over a 72 h time period. While our report nicely showed the sustained expression of pJNK several hours to days following reperfusion, it is interesting that the most significant increases of pJNK and pc-Jun occurred 1 h following reperfusion, which were observed mainly in the Tuj-1 positive RGC cell population. Among the earliest responses to reperfusion of occluded blood flow are influx of ions, excitatory neurotransmitter release, and free radical formation [23]. Reactive oxygen species (ROS) such as superoxide are potent activators of JNK signaling, acting far upstream on the MAP3K ASK1. This kinase-mediated activation by ROS facilitates apoptosis in the mitochondria, as JNKs are known to activate caspases and induce cytochrome c release [55, 56]. Thus, our observed early activation of JNK signaling may be attributed to the increase in free-radical production resulting from reperfusion, and RGC may be the most susceptible cell type to trigger JNK signaling by this microenvironmental stress. Previously, Roth and colleagues showed increased pJNK within 6 h after I/R [57]. This apparent discrepancy in temporal retinal JNK activation may be explained by slight differences between the models. Ross et al. used a milder elevation of IOP to induce retinal ischemia in rats, while our study was done in mice. Nonetheless, both studies indicate that JNK phosphorylation is widespread in many cell types in the inner retina, including the GCL, IPL and INL. In support, Schmid et al. showed that I/R injury induced loss of amacrine cells, but not rod bipolar cells in INL [58]. Together, these studies indicate that I/R injury may trigger the activation of JNK pathway in association with diverse microenvironmental changes in inner retina and promote the cellular degeneration based on the different susceptibility of each cells to specific initiating factors.
Notably, we also observed that phosphorylated c-Jun was not co-localized with amacrine cells and bipolar cells, although we observed strong c-Jun phosphorylation in INL from 6 h to 72 h after I/R injury (Fig. 5). In contrast, Gesslein et al. showed increased phosphorylated c-Jun signals in Müller cell bodies in the INL after I/R injury [59]. In retinal I/R injury, Müller cell gliosis is a major marker associated with their cellular changes causing pathological progression [27]. Importantly, several in vitro studies suggested that JNK pathway may be involved in Müller cell activation related to proinflammatory cytokine production and modified glutamate-uptake, which are also important I/R-induced molecular stressors to determine the fate of retinal pathology [60, 61]. Although further characterization is necessary, our observations suggest that the cells co-localized with phosphorylated c-Jun may be Müller cells based on their locational polarity in the INL toward vitreous side [62]. In addition, our data also suggest the diversity in the role of the JNK pathway in different retinal cell types that may result in different cellular fates such as programmed cell death or inflammatory activation.
A number of strategies have been developed for inactivation of the JNK pathway, either directly targeting one or all of the JNK isoforms [18], the adapter protein JIP1 [63], or an upstream kinase such as MLK1/2/3 [64]. Inhibition of JNK activation has been previously shown to be an effective therapeutic strategy in vitro and in vivo. Reduction of pJNK protected primary neuronal cultures from TFW [65]. In animal studies, JNK inhibition produced a marked reduction in lesion size resulting from brain ischemia and preserved neuronal health of affected tissues, decreased the number TUNEL-positive cells following excitotoxicity, and rescued against retinal thinning and cellular loss of RGCs [16–19, 41, 45, 65–67]. However, only a few studies have determined whether JNK signaling is the critical pro-apoptotic mediator of pathological structural and functional changes following retinal I/R injury.
SP600125 is a widely used inhibitor for all three JNK isoforms [68]. This compound showed significant protection in animal models of neurodegenerative disease [54], cerebral ischemia [69, 70], and retinal excitotoxicity [41]. We evaluated the neuroprotective effect of SP600125 in our model of retinal I/R injury. SP600125 protected against thinning of the inner neural retinal layers and cellular loss in the GCL. We used SD-OCT to temporally and non-invasively follow morphological changes in the retina of each mouse. Furthermore, we believe our study to be the first to also show complete preservation of retinal b-wave amplitude over a 28-day period after I/R injury. Our findings of neuroprotection are in disagreement with those previously published by Roth and colleagues, who reported a lack of protection by two different JNK inhibitors including SP600125 [57]. This discrepancy may be explained by the differences in dosing strategies. Our study effectively protected the retina through two different systemic dosing regimens that each required daily administration following reperfusion in addition to a dose given right before I/R injury. In contrast, Roth et al. used a single post-ischemic intravitreal dose of SP600125. A recent study showed JNK modulation and neuroprotection against retinal ischemia using crocetin, a carotenoid derivative [52]. However, their assessments were made only at 7 days post injury, and the long-term effectiveness was not reported.
Pathological changes related to inner retinal injury are not limited to just the retina because RGC axons form synapses with neurons in the visual centers of the brain. Degeneration of these brain neurons has been reported after injury to the retina [71, 72]. In the mouse, 90–95 % of axons in the optic nerve cross the optic chiasm and progress toward the contralateral side of the brain [37]. Among them, approximately 70 % of axon terminals are localized in the superficial layer of the SC [38]. Damage resulting from retinal ischemia leads to defects in anterograde transport and loss of innervation to the SC [73], causing a significantly decreased visual evoked potential response [74, 75]. Topical treatment with the α2-adrenergic agonist brimonidine has been shown to maintain innervation of the SC and protect against loss of neuronal volume up to 3 months following transient ligation of the ophthalmic vessels in rats [73]. In our study, we describe significant decreases in areas of myelination as well as loss of pre- and post-synaptic proteins only in the contralateral hemisphere of the SC. Several studies have shown the neuroprotective action of JNK inhibitors, including SP600125 in the hippocampus [69, 70]. However, we believe this to be the first report of protection in the SC, or any visual center in the brain, provided by JNK inhibition. Long-term administration of SP600125 efficaciously preserved myelination in the superficial SC as well as maintained expression of an RGC pre-synaptic marker. In addition, JNK inhibition may promote subsequent preservation of structural integrity of post-synapses, which is necessary for active neurotransmission and neurotrophic factor exchange. The same trend was observed with only 7 days of dosing.
Conclusions
In summary, we have shown JNK inhibitors protected RGCs against glutamate and TFW-induced cytotoxicity in primary cultured retinal neurons. Further, intraperitoneal injection of the JNK inhibitor SP600125 ameliorated retinal I/R-induced degeneration and functional loss of the inner neural retina and synaptic loss at RGC axon terminals in the SC. Our findings implicate that JNK activation, within hours immediately following reperfusion, plays a pivotal role in cell death and pathological progression in the retina and SC. This may be the critical therapeutic window for JNK inhibitors, at least in this acute model of retinal I/R injury. Together with previously published results, we suggest that the JNK signaling pathway is a prominent and critical path for neurodegeneration. Inhibition of this pathway represents a promising novel therapeutic target for preventing neurodegeneration in the visual axis related to retinal ischemic injury.
Methods
Animal
Female Sprague–Dawley rats (2–4 months of age) from Charles River Laboratories (Wilmington, MA) were used for retinal cell culture. Female C57BL/6J mice (10–12 weeks of age) from the Jackson Laboratory (Bar Harbor, ME) were used for transient retinal I/R studies. Animals were maintained in 12-h:12-h/light:dark cycle. All studies and animal care were performed as approved by the Institutional Animal Care and Use Committee at the University of North Texas Health Science Center and followed the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research.
Mixed retinal cell culture
Culture of retinal neurons was performed as previously reported [30]. Briefly, rats were euthanized by CO2 asphyxiation and their eyes enucleated. The retina from each eye was dissected and incubated in a papain solution, containing 2 mg/mL papain (Sigma, St. Louis, MO), 0.4 mg/mL DL-cysteine (Sigma), and 0.4 mg/mL bovine serum albumin (Sigma) in Neurobasal medium (Gibco/Invitrogen, Carlsbad, CA), for 25 min at 37 °C, then washed 3 times with RGC culture medium (Neurobasal/B27 medium containing 100 units/mL penicillin (Sigma), 100 μg/mL streptomycin (Sigma), 1 mM pyruvate (Gibco/Invitrogen), 2 mM glutamine (Gibco/Invitrogen), 5 μg/mL insulin (Sigma), 100 μg/mL transferrin (Sigma), 100 μg/mL bovine serum albumin (Sigma), 60 ng/mL progesterone (Sigma), 16 μg/mL putrescine (Sigma), 40 ng/mL sodium selenite (Sigma), 40 ng/mL thyroxine (Sigma), 40 ng/mL tri-iodothyronine (Sigma), 50 ng/mL brain-derived neurotrophic factor (BDNF; Biosource, Camarillo, CA), 10 ng/mL ciliary neurotrophic factor (CNTF; Biosource), 10 ng/mL basic fibroblast growth factor (bFGF; Biosource), 5 μM forskolin (Sigma), and 1 % fetal calf serum (Atlas Biologicals, Fort Collins, CO)). Retinal pieces were triturated by passing through a fire-polished disposable glass pipette until cells were dispersed. Cell density in the suspension was assessed with a Coulter counter (Beckman Coulter, Fullerton, CA). 1 × 106 to 3 × 106 cells/well were placed onto poly-D-lysine- and laminin-coated 8-well chambered culture slides (surface area = 0.69 cm2/well; Becton Dickinson, Franklin Lakes, NJ) and cultured in 95 % air/5 % CO2 at 37 °C. These cell cultures contained RGC-enriched retinal neurons [30]. More than 90 % of the cells were neurons, which were positively labeled with neuron-specific enolase antibody. Approximately 20–30 % of these cells expressed RGC markers, Thy-1 and neurofilament-L. Morphologically, the Thy-1-positive cells had the characteristic appearance of neurons. After 2–3 days in culture, neurite outgrowth typically had 2–4 main branches of approximately 20–50 μm in length. Cells in culture were negative for arrestin (photoreceptor), glial fibrillary acidic protein (astroglia and Müller cell), glutamine synthetase (Müller cell), or ED1 (microglia). Less than 10 % of the cells were labeled with the protein kinase Cα antibody (rod bipolar cells) [30]. Characterization of this cell culture system is shown in Additional file 1: Table S1 and Figures S1–S8. For glutamate-induced toxicity studies, cells were pre-treated with vehicle or the indicated JNK inhibitors, SP600125 (Sigma) or TAT-JNK-III, a cell-permeable peptide inhibitor specific for JNK [76] (EMD Chemicals, Billerica, MA), for 30 min, followed by 100 μM glutamate (Sigma) for three days. For TNFα-induced toxicity studies, cells were pre-treated with vehicle or the indicated compounds for 30 min, followed by 10 ng/mL TNFα (Sigma) for three days. For trophic factor withdrawal (TFW) studies, three trophic factors, bFGF, BDNF, and CNTF, were removed from the culture medium. Cells were cultured in this medium with the indicated compounds for three days. At the end of the incubation period, the cells were fixed and labeled for Thy-1, a RGC marker, by immunocytochemistry (primary antibody against Thy-1, diluted 1:500; Chemicon, Temecula, CA; secondary antibody: Alexa fluor 594-labeled goat anti-mouse IgG, 1:300; Invitrogen/Molecular Probes, Carlsbad, CA). Cell survival was quantified by counting the Thy-1-positive cells in each well.
Retinal I/R and SP600125 administration
Retinal I/R was induced as described previously [27]. Briefly, mice were anesthetized with a ketamine/xylazine/acepromazine cocktail (100/10/3 mg/kg) followed by cannulation of the anterior chamber with a 30 gauge needle connected to a reservoir containing sterile PBS. The reservoir was elevated to generate an intraocular pressure of 120 mmHg for 1 h to induce retinal ischemia. The JNK inhibitor SP600125 (5, 15, 30 mg/kg, 100 μL) or vehicle control (6 % DMSO in PBS, 100 μL) was intraperitoneally administered 2 h prior to retinal I/R, followed by once daily injection after I/R.
Western blotting of retinal proteins
Whole retina was gently removed at indicated time points after I/R injury and homogenized in mammalian protein extraction reagent (MPER, Thermo Fisher Scientific Inc, Rockford, IL). Total protein was quantified using DC™ Protein Assay (Bio-RAD, Life Science Research, Hercules, CA). Total retinal protein (50 μg) in Laemmli buffer (Bio-RAD) and 0.5 % β-mercaptoethanol was electrophoresed in 10 % SDS-PAGE gels. Gels were transferred to Immobilon-P PVDF transfer membrane (EMD Millipore, Billerica, MA) followed by blocking in 10 % non-fat milk. Membranes were incubated overnight in 4 °C with primary antibodies for pJNK (#9251), total JNK (#9252), phospho-c-Jun (pc-Jun) (#2631), total c-Jun (#9165), and GAPDH (#2118) from Cell Signaling Technology (Danvers, MA), followed by incubation with secondary antibody conjugated with horse radish peroxidase (Cell Signaling Technology) for 1 h at room temperature. Protein expression was detected using SuperSignal West Femto Chemiluminescent substrate (Thermo Fisher Scientific, Rockford, IL) and Alpha Innotech Gel Documentation System with Flourchem 8900 software (ProteinSimple, Alpha Innotech, Santa Clara, CA). Immunoblots were quantified using the ImageJ program (National Institutes of Health, Bethesda, MD) and expression was normalized to GAPDH.
Immunohistochemistry of retina
Ischemic or contralateral control eyes were gently removed from euthanized mice and placed in 4 % para-formaldehyde/PBS for 2 h followed by 20 % sucrose embedding. Eyes were placed in Tissue-Tek O.C.T compound (Sakura Finetek USA, Torrance, CA) and stored at −80 °C. Eyes were frozen-sectioned at 10 μm thickness and placed on Superfrost Plus Microscope Slides (Fisher Scientific, Hampton, NH). Slides were washed twice in PBS, blocked and permeabilized for 1 h using blocking buffer (PBS with 10 % horse serum and 0.1 % Triton X-100), then incubated with antibodies against pJNK (#4668), pc-Jun (#3270) (Cell Signaling Technology, Danvers, MA), and/or Tublin βIII (Tuj1 #801201, Biolegend, San Diego, CA), or PKCα (ab11723), Syntaxin (ab112198) (Abcam plc. Cambridge, MA) for 16 h at 4 °C, followed by treatment of secondary antibody conjugated with Alexaflour 488 and Alexaflour 568 (Invitrogen) for 1 h at room temperature. Slides were mounted with cover slips using ProlongGold anti-fade reagent with DAPI (Molecular Probes, Life Technologies, Grand Island, NY). All retinal images were taken using Zeiss LSM Meta Confocal microscope system (located in Johns Hopkins University, School of Medicine, Baltimore, MD) and Zen 2009 software (ZEISS, Thornwood, NY).
Histology of retina
Retinal layer thickness and cell numbers in ganglion cell layer (GCL) were assessed as previously described [27]. Briefly, thicknesses of individual layers or whole retina (from nerve fiber layer (NFL) to outer nuclear layer (ONL), excluding outer segments) were measured using the ImageJ program (NIH) at every quarter point of each entire retinal cross-section (from ora serrata to ora serrata) and averaged. Four retinal cross-sections were used per eye and their results averaged. Cell numbers in GCL were counted from each of the four cross-sections from each eye and averaged.
Spectral domain optical coherence tomography (SD-OCT)
The SD-OCT ophthalmic Imaging System (Bioptigen, Durham, NC) was used for retinal scanning. Mice were anesthetized using a ketamine/xylazine cocktail (100/10 mg/kg) and placed on a mouse holder. Using InVivoVue software (Bioptigen), each retina was scanned with rectangular scanning mode (1.2 × 1.2 mm) consisting of 1000 A scans per B scan × 100 B scans. Images from superior, central, and inferior regions were analyzed using ImageJ software with four measurements at quarter points for thicknesses of the whole retinal layer, inner plexiform layer (IPL), inner nuclear layer (INL), and ONL.
Scotopic electroretinography (ERG)
After 16 h of dark adaptation, mice were anesthetized with isoflurane and connected to the HMsERG system (Ocuscience, Rolla, MO) with body temperature controlled at 37 °C. The study eye was exposed to a series of light flashes of various intensities (0.1, 0.3, 1, 3, 10, and 25 cd/m2). Amplitudes and implicit times of ERG waveforms were measured and analyzed.
Brain histology and immunohistochemistry
For black gold staining, blocks of mice brains bracketing the SC (3 mm to 6 mm caudal to bregma) were frozen-sectioned at 40 μm thickness and placed on slides. Slides were incubated with 0.3 % Black Gold II solution (Histo-Chem, Jefferson, AR) at 60 °C as instructed by the manufacturer. Before mounting, slides were washed in PBS and dehydrated in a series of 95 % ethanol (2 min), 100 % ethanol (2 min) and xylene (2 min). All bright-field images were taken using Nikon eclipse Ti inverted microscope and CRi Nuance FX multispectral imaging system with Nuance 3.0 software. Cross-sectional areas of SC of all slides were measured from the myelinated SC layer III to superficial layer in each hemisphere as previously described [77] using the ImageJ polygonal selection method. The cross-sectional areas of SC of all slides from each hemisphere of the same brain were averaged and reported.
For immunohistochemistry, brains were frozen-sectioned at 10 μm thickness. Prior to applying primary antibody, slides were hydrated in PBS for 10 min and blocked with 10 % goat serum containing 0.1 % Triton X-100 in PBS. Anti-VGLUT2 (ab79157, Abcam) and anti-PSD95 (ab18258, Abcam) antibodies were applied at 4 °C overnight. Slides were washed and incubated with secondary antibodies conjugated with Alexaflour 488 or Alexaflour 568 for 1 h at room temperature. All fluorescence images were taken using the same system described above. Images were taken using Nikon eclipse Ti inverted microscope (Nikon) and CRi Nuance FX multispectral imaging system (Caliper Life Sciences, Hopkinton, MA). All background autofluorescence was subtracted using Nuance 3.0 software (Caliper Life Sciences).
Statistics
Statistical analysis was performed using GraphPad Prism Version 5.0 (GraphPad Software, San Diego, CA). Student’s t-test was used to compare two experimental groups. One-way or two-way ANOVA was used to compare among three or more groups. The specific statistical method used is described in the respective figure legends. All data are expressed as means ± standard error of mean (SEM). A p value less than 0.05 is considered statistically significant.
Acknowledgement
This work was supported by a grant (W81XWH-10-20-0003) from the Department of Defense. We wish to thank Sandra Maansson for technical assistance. We also would like to thank Dr. Don Zack (Johns Hopkins) for his support of additional studies.
Abbreviations
- ANOVA
analysis of variance
- ASK1
apoptosis signal regulating kinase 1
- ATF-2
activating transcription factor 2
- BDNF
brain-derived neurotrophic factor
- bFGF
basic fibroblast growth factor
- CNTF
ciliary neurotrphic factor
- DAPI
4′ 6-diamido-2-phenylindole
- ELK-1
ETS domain containing protein
- ERG
electroretinography
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GCL
ganglion cell layer
- GFAP
glial fibrillar acidic protein
- Glu
glutamate
- I/R
ischemia/reperfusion
- INL
inner nuclear layer
- IOP
intraocular pressure
- IPL
inner plexiform layer
- JNK
cJun N-terminal kinase
- MAP3K
mitogen activated protein kinase kinase kinase
- MLK
mixed lineage kinase
- MPER
mammalian protein extraction reagent
- MPTP
1-methyl-4-phenol-1,2,3,6-tetrahydropyridine
- NF-L
neurofilament light
- NIH
National Institutes of Health
- NMDA
N-methyl-D-aspartate
- NSE
neuron specific enolase
- OCT
optimal cutting temperature compound
- OPL
outer plexiform layer
- pc-Jun
phopho-c-Jun
- PD
Parkinson’s Disease
- pJNK
phospho-c-Jun N-terminal kinase
- PKCα
protein kinase C alpha
- PSD95
postsynaptic density protein 95
- PVDF
polyvinylidine fluoride
- RGC
retinal ganglion cell
- ROS
reactive oxygen species
- SC
superior colliculus
- SD-OCT
spectral domain-optical coherence tomography
- SEM
standard error of the mean
- Stat3
Signal transducer and activator of transcription 3
- TFW
trophic factor withdrawal
- TNFα
tumor necrosis factor alpha
- Tuj-1
neuron specific class III β-tubulin
- VGTUT2
vesicular glutamate transporter 2
- WRL
whole retinal layer
Additional file
Table S1. Summary of distribution of cell markers in the mixed rat retinal cell culture. Figure S1. Representative images showing colocalization of Thy-1 and neurofilament-L (NF-L) by immunocytochemistry in cultured rat retinal cells. Figure S2. Representative images showing lack of colocalization of Thy-1 and arrestin by immunocytochemistry in cultured rat retinal cells. There were no arrestin positive cells in culture. Figure S3. Representative images showing lack of colocalization of Thy-1 and ED-1 (CD68) by immunocytochemistry in cultured rat retinal cells. There were no ED-1 positive cells in culture. Figure S4. Representative images showing lack of colocalization of Thy-1 and glutamine synthetase (GS) by immunocytochemistry in cultured rat retinal cells. There were no GS positive cells in culture. Figure S5. Representative images showing lack of colocalization of Thy-1 and GFAP by immunocytochemistry in cultured rat retinal cells. There were no GFAP positive cells in culture. The lower right-hand panel includes brain astrocytes as a positive control for the anti-GFAP antibody. Figure S6. Representative images showing lack of colocalization of Thy-1 and PKCα by immunocytochemistry in cultured rat retinal cells. There were very few PKCα positive cells in culture. Figure S7. Representative images showing partial colocalization of Thy-1 and neuron-specific enolase (NSE) by immunocytochemistry in cultured rat retinal cells. Figure S8. Effect of glutamate on RGC morphology. Cells were treated with glutamate (100 μM) or vehicle for 3 days. (Top panels) The cells were labeled with anti-Thy-1 antibody (red) and DAPI nuclear stain (blue). (Bottom panels) The corresponding phase-contrast image. Figure S9. Retinal c-Jun phosphorylated c-Jun was attenuated by SP600125 administration in 24 h after I/R injury. Phosphorylated c-Jun was detected (green fluorescence). Tuj-1 immunofluorescence (red) was used as RGC marker. DAPI staining (blue) represents cell nuclei for counter staining. (A) Vehicle, (B) SP600125. (PDF 362 kb)
Footnotes
Byung-Jin Kim and Sean M. Silverman are co-first authors.
Robert J. Wordinger is deceased
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BJK, AFC, RJW, IHP, YL, and SMS were involved in the conception and design of the study, as well as data analyses and interpretation of data. BJK and SMS generated the initial draft of the manuscript. AFC, IHP, YL, and RJW outlined and/or revised the manuscript for critical intellectual content. IHP conducted the in vitro studies. BJK and SMS generated the in vivo data. BJK, SMS, and YL conducted the immunofluorescent and immunohistochemical staining. All authors read and approved the final manuscript (with the exception of RJW, who recently passed away). All authors have agreed to be accountable for all aspects of this work.
Contributor Information
Byung-Jin Kim, Email: bkim68@jhmi.edu.
Sean M. Silverman, Email: ssilverm@live.unthsc.edu
Yang Liu, Email: yang.liu@unthsc.edu.
Iok-Hou Pang, Email: iok-hou.pang@unthsc.edu.
Abbot F. Clark, Phone: (817) 735-2094, Email: abe.clark@unthsc.edu
References
- 1.Savage MJ, Lin YG, Ciallella JR, Flood DG, Scott RW. Activation of c-Jun N-terminal kinase and p38 in an Alzheimer’s disease model is associated with amyloid deposition. J Neurosci. 2002;22(9):3376–85. doi: 10.1523/JNEUROSCI.22-09-03376.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hunot S, Vila M, Teismann P, Davis RJ, Hirsch EC, Przedborski S, et al. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2004;101(2):665–70. doi: 10.1073/pnas.0307453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwong JM, Caprioli J. Expression of phosphorylated c-Jun N-terminal protein kinase (JNK) in experimental glaucoma in rats. Exp Eye Res. 2006;82(4):576–82. doi: 10.1016/j.exer.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 4.Barr RK, Bogoyevitch MA. The c-Jun N-terminal protein kinase family of mitogen-activated protein kinases (JNK MAPKs) Int J Biochem Cell Biol. 2001;33(11):1047–63. doi: 10.1016/S1357-2725(01)00093-0. [DOI] [PubMed] [Google Scholar]
- 5.Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Genet Dev. 2002;12(1):14–21. doi: 10.1016/S0959-437X(01)00258-1. [DOI] [PubMed] [Google Scholar]
- 6.Jurewicz A, Matysiak M, Tybor K, Selmaj K. TNF-induced death of adult human oligodendrocytes is mediated by c-jun NH2-terminal kinase-3. Brain. 2003;126(Pt 6):1358–70. doi: 10.1093/brain/awg146. [DOI] [PubMed] [Google Scholar]
- 7.Leow-Dyke S, Allen C, Denes A, Nilsson O, Maysami S, Bowie AG, et al. Neuronal Toll-like receptor 4 signaling induces brain endothelial activation and neutrophil transmigration in vitro. J Neuroinflammation. 2012;9:230. doi: 10.1186/1742-2094-9-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X, Commane M, Jiang Z, Stark GR. IL-1-induced NFkappa B and c-Jun N-terminal kinase (JNK) activation diverge at IL-1 receptor-associated kinase (IRAK) Proc Natl Acad Sci U S A. 2001;98(8):4461–5. doi: 10.1073/pnas.071054198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, et al. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389(6653):865–70. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 10.Lim CP, Cao X. Serine phosphorylation and negative regulation of Stat3 by JNK. J Biol Chem. 1999;274(43):31055–61. doi: 10.1074/jbc.274.43.31055. [DOI] [PubMed] [Google Scholar]
- 11.Del Villar K, Miller CA. Down-regulation of DENN/MADD, a TNF receptor binding protein, correlates with neuronal cell death in Alzheimer’s disease brain and hippocampal neurons. Proc Natl Acad Sci U S A. 2004;101(12):4210–5. doi: 10.1073/pnas.0307349101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le-Niculescu H, Bonfoco E, Kasuya Y, Claret FX, Green DR, Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol Cell Biol. 1999;19(1):751–63. doi: 10.1128/MCB.19.1.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizuno N, Kokubu H, Sato M, Nishimura A, Yamauchi J, Kurose H, et al. G protein-coupled receptor signaling through Gq and JNK negatively regulates neural progenitor cell migration. Proc Natl Acad Sci U S A. 2005;102(35):12365–70. doi: 10.1073/pnas.0506101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, Flavell R, et al. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci. 2001;21(19):7551–60. doi: 10.1523/JNEUROSCI.21-19-07551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Podkowa M, Zhao X, Chow CW, Coffey ET, Davis RJ, Attisano L. Microtubule stabilization by bone morphogenetic protein receptor-mediated scaffolding of c-Jun N-terminal kinase promotes dendrite formation. Mol Cell Biol. 2010;30(9):2241–50. doi: 10.1128/MCB.01166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tezel G, Yang X, Yang J, Wax MB. Role of tumor necrosis factor receptor-1 in the death of retinal ganglion cells following optic nerve crush injury in mice. Brain Res. 2004;996(2):202–12. doi: 10.1016/j.brainres.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 17.Bessero AC, Chiodini F, Rungger-Brandle E, Bonny C, Clarke PG. Role of the c-Jun N-terminal kinase pathway in retinal excitotoxicity, and neuroprotection by its inhibition. J Neurochem. 2010;113(5):1307–18. doi: 10.1111/j.1471-4159.2010.06705.x. [DOI] [PubMed] [Google Scholar]
- 18.Fernandes KA, Harder JM, Fornarola LB, Freeman RS, Clark AF, Pang IH, et al. JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death. Neurobiol Dis. 2012;46(2):393–401. doi: 10.1016/j.nbd.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balaiya S, Ferguson LR, Chalam KV. Evaluation of sirtuin role in neuroprotection of retinal ganglion cells in hypoxia. Invest Ophthalmol Vis Sci. 2012;53(7):4315–22. doi: 10.1167/iovs.11-9259. [DOI] [PubMed] [Google Scholar]
- 20.Welsbie DS, Yang Z, Ge Y, Mitchell KL, Zhou X, Martin SE, et al. Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc Natl Acad Sci U S A. 2013;110(10):4045–50. doi: 10.1073/pnas.1211284110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bresnick GH, De Venecia G, Myers FL, Harris JA, Davis MD. Retinal ischemia in diabetic retinopathy. Arch Ophthalmol. 1975;93(12):1300–10. doi: 10.1001/archopht.1975.01010020934002. [DOI] [PubMed] [Google Scholar]
- 22.Goldblum D, Mittag T. Prospects for relevant glaucoma models with retinal ganglion cell damage in the rodent eye. Vision Res. 2002;42(4):471–8. doi: 10.1016/S0042-6989(01)00194-8. [DOI] [PubMed] [Google Scholar]
- 23.Osborne NN, Casson RJ, Wood JP, Chidlow G, Graham M, Melena J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res. 2004;23(1):91–147. doi: 10.1016/j.preteyeres.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 24.Lam TT, Abler AS, Tso MO. Apoptosis and caspases after ischemia-reperfusion injury in rat retina. Invest Ophthalmol Vis Sci. 1999;40(5):967–75. [PubMed] [Google Scholar]
- 25.Piras A, Gianetto D, Conte D, Bosone A, Vercelli A. Activation of autophagy in a rat model of retinal ischemia following high intraocular pressure. PLoS One. 2011;6(7):e22514. doi: 10.1371/journal.pone.0022514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, et al. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res. 2010;88(7):1569–76. doi: 10.1002/jnr.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim BJ, Braun TA, Wordinger RJ, Clark AF. Progressive morphological changes and impaired retinal function associated with temporal regulation of gene expression after retinal ischemia/reperfusion injury in mice. Mol Neurodegener. 2013;8:21. doi: 10.1186/1750-1326-8-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeuchi O, Akira S. Toll-like receptors; their physiological role and signal transduction system. Int Immunopharmacol. 2001;1(4):625–35. doi: 10.1016/S1567-5769(01)00010-8. [DOI] [PubMed] [Google Scholar]
- 29.Otori Y, Wei JY, Barnstable CJ. Neurotoxic effects of low doses of glutamate on purified rat retinal ganglion cells. Invest Ophthalmol Vis Sci. 1998;39(6):972–81. [PubMed] [Google Scholar]
- 30.Pang IH, Zeng H, Fleenor DL, Clark AF. Pigment epithelium-derived factor protects retinal ganglion cells. BMC Neurosci. 2007;8:11. doi: 10.1186/1471-2202-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tezel G, Wax MB. Increased production of tumor necrosis factor-alpha by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J Neurosci. 2000;20(23):8693–700. doi: 10.1523/JNEUROSCI.20-23-08693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bogoyevitch MA, Ngoei KR, Zhao TT, Yeap YY, Ng DC. c-Jun N-terminal kinase (JNK) signaling: recent advances and challenges. Biochim Biophys Acta. 2010;1804(3):463–75. doi: 10.1016/j.bbapap.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 33.Johnson GL, Nakamura K. The c-jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim Biophys Acta. 2007;1773(8):1341–8. doi: 10.1016/j.bbamcr.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watkins TA, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, Modrusan Z, Kaminker JS, Tessier-Lavigne M, Lewcock JW. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc Natl Acad Sci U S A. 2013;110(10):4039–44. doi: 10.1073/pnas.1211074110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huntwork-Rodriguez S, Wang B, Watkins T, Ghosh AS, Pozniak CD, Bustos D, Newton K, Kirkpatrick DS, Lewcock JW. JNK-mediated phosphorylation of DLK suppresses its ubiquitination to promote neuronal apoptosis. J Cell Biol. 2013;202(5):747–63. doi: 10.1083/jcb.201303066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao Y, Signore AP, Yin W, Cao G, Yin XM, Sun F, et al. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J Cereb Blood Flow Metab. 2005;25(6):694–712. doi: 10.1038/sj.jcbfm.9600062. [DOI] [PubMed] [Google Scholar]
- 37.Drager UC, Olsen JF. Origins of crossed and uncrossed retinal projections in pigmented and albino mice. J Comp Neurol. 1980;191(3):383–412. doi: 10.1002/cne.901910306. [DOI] [PubMed] [Google Scholar]
- 38.Hofbauer A, Drager UC. Depth segregation of retinal ganglion cells projecting to mouse superior colliculus. J Comp Neurol. 1985;234(4):465–74. doi: 10.1002/cne.902340405. [DOI] [PubMed] [Google Scholar]
- 39.Ma J, Yu W, Wang Y, Cao G, Cai S, Chen X, et al. Neuroprotective effects of C-type natriuretic peptide on rat retinal ganglion cells. Invest Ophthalmol Vis Sci. 2010;51(7):3544–53. doi: 10.1167/iovs.09-5049. [DOI] [PubMed] [Google Scholar]
- 40.Munemasa Y, Ohtani-Kaneko R, Kitaoka Y, Kumai T, Kitaoka Y, Hayashi Y, et al. Pro-apoptotic role of c-Jun in NMDA-induced neurotoxicity in the rat retina. J Neurosci Res. 2006;83(5):907–18. doi: 10.1002/jnr.20786. [DOI] [PubMed] [Google Scholar]
- 41.Munemasa Y, Ohtani-Kaneko R, Kitaoka Y, Kuribayashi K, Isenoumi K, Kogo J, et al. Contribution of mitogen-activated protein kinases to NMDA-induced neurotoxicity in the rat retina. Brain Res. 2005;1044(2):227–40. doi: 10.1016/j.brainres.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 42.Shackelford DA, Yeh RY. Modulation of ERK and JNK activity by transient forebrain ischemia in rats. J Neurosci Res. 2006;83(3):476–88. doi: 10.1002/jnr.20747. [DOI] [PubMed] [Google Scholar]
- 43.Sugino T, Nozaki K, Takagi Y, Hattori I, Hashimoto N, Moriguchi T, et al. Activation of mitogen-activated protein kinases after transient forebrain ischemia in gerbil hippocampus. J Neurosci. 2000;20(12):4506–14. doi: 10.1523/JNEUROSCI.20-12-04506.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tian H, Zhang G, Li H, Zhang Q. Antioxidant NAC and AMPA/KA receptor antagonist DNQX inhibited JNK3 activation following global ischemia in rat hippocampus. Neurosci Res. 2003;46(2):191–7. doi: 10.1016/S0168-0102(03)00057-9. [DOI] [PubMed] [Google Scholar]
- 45.Shioda S, Ozawa H, Dohi K, Mizushima H, Matsumoto K, Nakajo S, et al. PACAP protects hippocampal neurons against apoptosis: involvement of JNK/SAPK signaling pathway. Ann N Y Acad Sci. 1998;865:111–7. doi: 10.1111/j.1749-6632.1998.tb11169.x. [DOI] [PubMed] [Google Scholar]
- 46.Ozawa H, Shioda S, Dohi K, Matsumoto H, Mizushima H, Zhou CJ, et al. Delayed neuronal cell death in the rat hippocampus is mediated by the mitogen-activated protein kinase signal transduction pathway. Neurosci Lett. 1999;262(1):57–60. doi: 10.1016/S0304-3940(99)00034-8. [DOI] [PubMed] [Google Scholar]
- 47.Hayashi T, Sakai K, Sasaki C, Zhang WR, Warita H, Abe K. c-Jun N-terminal kinase (JNK) and JNK interacting protein response in rat brain after transient middle cerebral artery occlusion. Neurosci Lett. 2000;284(3):195–9. doi: 10.1016/S0304-3940(00)01024-7. [DOI] [PubMed] [Google Scholar]
- 48.Wu X, Li L, Zhang L, Wu J, Zhou Y, Zhou Y, et al. Inhibition of thioredoxin-1 with siRNA exacerbates apoptosis by activating the ASK1-JNK/p38 pathway in brain of a stroke model rats. Brain Res. 2015;1599:20–31. doi: 10.1016/j.brainres.2014.12.033. [DOI] [PubMed] [Google Scholar]
- 49.Guo Z, Cao G, Yang H, Zhou H, Li L, Cao Z, et al. A combination of four active compounds alleviates cerebral ischemia-reperfusion injury in correlation with inhibition of autophagy and modulation of AMPK/mTOR and JNK pathways. J Neurosci Res. 2014;92(10):1295–306. doi: 10.1002/jnr.23400. [DOI] [PubMed] [Google Scholar]
- 50.Dreixler JC, Bratton A, Du E, Shaikh AR, Savoie B, Alexander M, et al. Mitogen-activated protein kinase phosphatase-1 (MKP-1) in retinal ischemic preconditioning. Exp Eye Res. 2011;93(4):340–9. doi: 10.1016/j.exer.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fang IM, Yang CM, Yang CH. Chitosan oligosaccharides prevented retinal ischemia and reperfusion injury via reduced oxidative stress and inflammation in rats. Exp Eye Res. 2015;130:38–50. doi: 10.1016/j.exer.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 52.Ishizuka F, Shimazawa M, Umigai N, Ogishima H, Nakamura S, Tsuruma K, et al. Crocetin, a carotenoid derivative, inhibits retinal ischemic damage in mice. Eur J Pharmacol. 2013;703(1–3):1–10. doi: 10.1016/j.ejphar.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 53.Zhu X, Raina AK, Rottkamp CA, Aliev G, Perry G, Boux H, et al. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J Neurochem. 2001;76(2):435–41. doi: 10.1046/j.1471-4159.2001.00046.x. [DOI] [PubMed] [Google Scholar]
- 54.Philpott KL, Facci L. MAP kinase pathways in neuronal cell death. CNS Neurol Disord Drug Targets. 2008;7(1):83–97. doi: 10.2174/187152708783885129. [DOI] [PubMed] [Google Scholar]
- 55.Shen HM, Liu ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med. 2006;40(6):928–39. doi: 10.1016/j.freeradbiomed.2005.10.056. [DOI] [PubMed] [Google Scholar]
- 56.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288(5467):870–4. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 57.Roth S, Shaikh AR, Hennelly MM, Li Q, Bindokas V, Graham CE. Mitogen-activated protein kinases and retinal ischemia. Invest Ophthalmol Vis Sci. 2003;44(12):5383–95. doi: 10.1167/iovs.03-0451. [DOI] [PubMed] [Google Scholar]
- 58.Schmid H, Renner M, Dick HB, Joachim SC. Loss of inner retinal neurons after retinal ischemia in rats. Invest Ophthalmol Vis Sci. 2014;55(4):2777–87. doi: 10.1167/iovs.13-13372. [DOI] [PubMed] [Google Scholar]
- 59.Gesslein B, Håkansson G, Carpio R, Gustafsson L, Perez MT, Malmsjö M. Mitogen-activated protein kinases in the porcine retinal arteries and neuroretina following retinal ischemia-reperfusion. Mol Vis. 2010;16:392–407. [PMC free article] [PubMed] [Google Scholar]
- 60.Zhong Y, Li J, Chen Y, Wang JJ, Ratan R, Zhang SX. Activation of endoplasmic reticulum stress by hyperglycemia is essential for Müller cell-derived inflammatory cytokine production in diabetes. Diabetes. 2012;61(2):492–504. doi: 10.2337/db11-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Namekata K, Harada C, Kohyama K, Matsumoto Y, Harada T. Interleukin-1 stimulates glutamate uptake in glial cells by accelerating membrane trafficking of Na+/K + −ATPase via actin depolymerization. Mol Cell Biol. 2008;28(10):3273–80. doi: 10.1128/MCB.02159-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gu D, Beltran WA, Li Z, Acland GM, Aguirre GD. Clinical light exposure, photoreceptor degeneration, and AP-1 activation: a cell death or cell survival signal in the rhodopsin mutant retina? Invest Ophthalmol Vis Sci. 2007;48(11):4907–18. doi: 10.1167/iovs.07-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Whitmarsh AJ, Kuan CY, Kennedy NJ, Kelkar N, Haydar TF, Mordes JP, et al. Requirement of the JIP1 scaffold protein for stress-induced JNK activation. Genes Dev. 2001;15(18):2421–32. doi: 10.1101/gad.922801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harris CA, Deshmukh M, Tsui-Pierchala B, Maroney AC, Johnson EM., Jr Inhibition of the c-Jun N-terminal kinase signaling pathway by the mixed lineage kinase inhibitor CEP-1347 (KT7515) preserves metabolism and growth of trophic factor-deprived neurons. J Neurosci. 2002;22(1):103–13. doi: 10.1523/JNEUROSCI.22-01-00103.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coffey ET, Smiciene G, Hongisto V, Cao J, Brecht S, Herdegen T, et al. c-Jun N-terminal protein kinase (JNK) 2/3 is specifically activated by stress, mediating c-Jun activation, in the presence of constitutive JNK1 activity in cerebellar neurons. J Neurosci. 2002;22(11):4335–45. doi: 10.1523/JNEUROSCI.22-11-04335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sun H, Wang Y, Pang IH, Shen J, Tang X, Li Y, et al. Protective effect of a JNK inhibitor against retinal ganglion cell loss induced by acute moderate ocular hypertension. Mol Vis. 2011;17:864–75. [PMC free article] [PubMed] [Google Scholar]
- 67.Irving EA, Bamford M. Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab. 2002;22(6):631–47. doi: 10.1097/00004647-200206000-00001. [DOI] [PubMed] [Google Scholar]
- 68.Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98(24):13681–6. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guan QH, Pei DS, Liu XM, Wang XT, Xu TL, Zhang GY. Neuroprotection against ischemic brain injury by SP600125 via suppressing the extrinsic and intrinsic pathways of apoptosis. Brain Res. 2006;1092(1):36–46. doi: 10.1016/j.brainres.2006.03.086. [DOI] [PubMed] [Google Scholar]
- 70.Guan QH, Pei DS, Zhang QG, Hao ZB, Xu TL, Zhang GY. The neuroprotective action of SP600125, a new inhibitor of JNK, on transient brain ischemia/reperfusion-induced neuronal death in rat hippocampal CA1 via nuclear and non-nuclear pathways. Brain Res. 2005;1035(1):51–9. doi: 10.1016/j.brainres.2004.11.050. [DOI] [PubMed] [Google Scholar]
- 71.Aviles-Trigueros M, Mayor-Torroglosa S, Garcia-Aviles A, Lafuente MP, Rodriguez ME. Miralles de Imperial J et al. Transient ischemia of the retina results in massive degeneration of the retinotectal projection: long-term neuroprotection with brimonidine. Exp Neurol. 2003;184(2):767–77. doi: 10.1016/S0014-4886(03)00298-X. [DOI] [PubMed] [Google Scholar]
- 72.Ito Y, Shimazawa M, Chen YN, Tsuruma K, Yamashima T, Araie M, et al. Morphological changes in the visual pathway induced by experimental glaucoma in Japanese monkeys. Exp Eye Res. 2009;89(2):246–55. doi: 10.1016/j.exer.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 73.Mayor-Torroglosa S, De la Villa P, Rodriguez ME, Lopez-Herrera MP, Aviles-Trigueros M, Garcia-Aviles A, et al. Ischemia results 3 months later in altered ERG, degeneration of inner layers, and deafferented tectum: neuroprotection with brimonidine. Invest Ophthalmol Vis Sci. 2005;46(10):3825–35. doi: 10.1167/iovs.05-0392. [DOI] [PubMed] [Google Scholar]
- 74.Li X, Kang Q, Gao S, Wei T, Liu Y, Chen X, et al. Transplantation with retinal progenitor cells repairs visual function in rats with retinal ischemia-reperfusion injury. Neurosci Lett. 2014;558:8–13. doi: 10.1016/j.neulet.2013.09.024. [DOI] [PubMed] [Google Scholar]
- 75.Jehle T, Wingert K, Dimitriu C, Meschede W, Lasseck J, Bach M, et al. Quantification of ischemic damage in the rat retina: a comparative study using evoked potentials, electroretinography, and histology. Invest Ophthalmol Vis Sci. 2008;49(3):1056–64. doi: 10.1167/iovs.07-1050. [DOI] [PubMed] [Google Scholar]
- 76.Holzberg D, Knight CG, Dittrich-Breiholz O, Schneider H, Dörrie A, Hoffmann E, Resch K, Kracht M. Disruption of the c-JUN-JNK complex by a cell-permeable peptide containing the c-JUN delta domain induces apoptosis and affects a distinct set of interleukin-1-induced inflammatory genes. J Biol Chem. 2003;278(41):40213–23. doi: 10.1074/jbc.M304058200. [DOI] [PubMed] [Google Scholar]
- 77.Crish SD, Sappington RM, Inman DM, Horner PJ, Calkins DJ. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc Natl Acad Sci U S A. 2010;107(11):5196–201. doi: 10.1073/pnas.0913141107. [DOI] [PMC free article] [PubMed] [Google Scholar]