

Summary
Chronic inflammatory disorders are thought to arise due to an interplay between predisposing host genetics and environmental factors. For example, the onset of inflammatory bowel disease is associated with enteric proteobacterial infection, yet the mechanistic basis for this association is unclear. We have shown previously that genetic defiency in TLR1 promotes acute enteric infection by the proteobacteria Yersinia enterocolitica. Examining that model further, we uncovered an altered cellular immune response that promotes the recruitment of neutrophils which in turn increases metabolism of the respiratory electron acceptor tetrathionate by Yersinia. These events drive permanent alterations in anti-commensal immunity, microbiota composition and chronic inflammation, which persist long after Yersinia clearence. Deletion of the bacterial genes involved in tetrathionate respiration or treatment using a targeted probiotics could prevent microbiota alterations and inflammation. Thus, acute infection can drive long term immune and microbiota alterations leading to chronic inflammatory disease in genetic predisposed individuals.
Introduction
The human intestine is home to a microbial ecosystem containing 100 trillion bacteria, a number 10 times greater than our own eukaryotic cells (Eckburg et al., 2005; Savage et al., 1968) and play key roles in structural, immunological and metabolic functions (Cho and Blaser, 2012). The microbiota is not a static community, but rather dynamic in its ability to alter its composition, distribution and/or gene expression in response to factors such as antibiotics, diet and infection (Cho and Blaser, 2012). These changes have been associated with intestinal and systemic diseases, yet genetic and environmental signals that contribute to the outgrowth and colonization of opportunistic commensal species are not well understood.
The gastrointestinal (GI) tract represents one of the primary sites of exposure to pathogens. GI infections can cause damage to host tissues directly through expression of virulence genes, and indirectly by changing the interactions between the microbiota and mucosal tissues. Alterations in the microbiota can be both beneficial and deleterious. For example, the microbiota may promote adaptive immunity (Benson et al., 2012), colonization resistance (Ferreira et al., 2011) or the development of long-lasting anti-commensal memory cells (Hand et al., 2012) all of which are a benefit to the host. On the other hand, enteric pathogens can reduce commensal diversity (Behnsen et al., 2014; Fukuda et al., 2011; Lupp et al., 2007; Raffatellu et al., 2009; Stecher et al., 2007), induce new metabolic intermediates (Raffatellu et al., 2009; Winter et al., 2010), promote inflammation (Heimesaat et al., 2006; Lupp et al., 2007; Stecher et al., 2007) or cause long-lasting re-programming of immune cells and tissues that may set the stage for development of chronic inflammatory diseases, a phenomenon recently referred to as immunological scarring (Fonseca et al., 2015).
Toll-like receptors (TLRs) are pattern recognition receptors that sense and respond to broadly conserved microbial motifs (Beutler et al., 2006; Iwasaki and Medzhitov, 2004). TLRs are broadly expressed throughout the intestinal epithelium and the underlying cells of the lamina propria (LP) (Abreu, 2010). Functionally, TLRs have been shown to regulate spatial localization (Vaishnava et al., 2011) and composition (Larsson et al., 2012; Vijay-Kumar et al., 2007) of the microbiota. The contribution of individual TLRs to inflammation and commensal dysbiosis remains unclear as both protective (Cario et al., 2007; Katakura et al., 2005; Morgan et al., 2014) and deleterious (Heimesaat et al., 2010; Santaolalla et al., 2013) roles have been observed.
TLR1 recognizes triacylated lipoproteins when dimerized with TLR2 (Schumann and Tapping, 2007). We have previously demonstrated that TLR1 signaling is critical for mucosal protection against oral infection caused by the gram-negative pathogen Yersinia enterocolitica (DePaolo et al., 2012; Sugiura et al., 2013). Ingestion of Y. enterocolitica in contaminated food and water causes a self-limiting gastroenteritis characterized by colonization of the distal ileum and translocation to the Peyer’s patch and mesenteric lymph nodes (Trulzsch et al., 2007). While there have been reports of an association between patients with inflammatory bowel disease and prior Y. enterocolitica infection, molecular and cellular mechanisms supporting this association are not clear.
Here, we report that a genetic deficiency in TLR1 during acute GI infection caused by Y. enterocolitica results in an increase in an opportunistic commensal of the δ-Proteobacteria family, chronic inflammation and anti-commensal immunity despite pathogen clearance. Further, the outgrowth of δ-Proteobacteria was dependent upon the presence of neutrophils and tetrathionate respiration by Y. enterocolitica. These data indicate a complicated communication between host, pathogen and commensal bacteria, leading to changes in the microbiota and anti-commensal immunity.
Results
TLR1 signaling during acute GI infection regulates inflammation and the composition of the microbiota
TLR1 signaling is critical for protection against the enteric pathogen Yersinia enterocolitica via the recruitment of CCR6+ dendritic cells (DC) and induction of a TH17 response (Sugiura et al., 2013). Despite an increase in mortality approximately 40% of TLR1−/− mice can survive the infection (Sugiura et al., 2013). To further understand whether TLR1-deficiency during an acute gastroenteritis may have consequences post-infection, a cohort of survivors for was followed for 70 days (10 weeks) post-infection with Y. enterocolitica. Mice deficient in TLR1 lost more weight during the acute infection and were slower to gain back the same weight compared to wild type littermate control (WT) mice (Fig 1A). The inability to gain weight was not due to a persistent infection, as Y. enterocolitica was not detected in the lumen (data not shown), mesenteric lymph nodes (MLN) (data not shown) or LP (Fig 1B) using conventional plating, quantitative PCR (qPCR) (data not shown) or 16S sequencing (data not shown). Chronic inflammation has been shown to affect metabolic rate and weight gain in patients with IBD (Tigas and Tsatsoulis, 2012) and in animal models of intestinal inflammation (Melgar et al., 2007). Innate cytokines were evaluated over the 70 days in the MLN (data not shown) and the LP of post-infected mice. As previously reported, WT mice produced more IL-6 and IL-23 compared to TLR1−/− mice during the acute infection (Sugiura, Kamdar et al. 2013 and Fig 1C). While IL-6 and IL-23 returned to pre-infection levels in the WT mice, these cytokines were significantly elevated 70 days post-infection in the TLR1−/− mice, despite the absence of persistent Y. enterocolitica infection (Fig 1C).
Figure 1. TLR1 signaling during acute gastroenteritis prevents δ-Proteobacteria outgrowth.

(A) Weight change in mice after acute gastroenteritis. Data is the mean ± SEM of three independent experiments (n=7–9 mice/group). (B) Y. enterocolitica in the LP of WT and TLR1−/− mice over 70 days after initial infection. Data is pooled mean ± SEM from three independent experiments (n=6–8 mice/group). *, p < 0.05; **, p < 0.01 Student’s unpaired t-test. (C) IL-6 and IL-23 levels in the LP during acute infection and 70 days post-infection. Data is pooled mean ± SEM from three independent experiments (n=6–8 mice/group). *, p < 0.05; **, p < 0.01 Student’s unpaired t-test. (D) mRNA transcript levels of antimicrobial peptides in distal ileum. Data is the mean ± SEM CT compared to respective naïve samples (n=5–6 mice/group). **, p < 0.01 Student’s unpaired t-test. (E) Relative abundance of class and genera-based classifications of bacterial populations in cecum determined by full-length SSU sequence libraries from TLR1−/− or WT mice. Data is representative of two mice from each group. (F) 16S gene copy numbers of cecal bacteria harvested from WT and TLR1−/− mice 70 days post-GI infection. Copy number was determined using plasmids to create a standard curve. Data is mean ± SEM (n=7–10 mice/group). *, p < 0.05 Student’s unpaired t-test.
Aberrant immune responses against an enteric pathogen in a host with genetic deficiency may lead to changes in the commensal population (Arthur et al., 2012; Couturier-Maillard et al., 2013). For example, Myd88-dependent pathways activated during pathogenic infection can induce an antimicrobial program that cause death of the pathogenic bacteria (Ayabe et al., 2000; Hooper et al., 2012; Vaishnava et al., 2011) but can also prevent the outgrowth of commensal species (Salzman et al., 2010). As TLR1 can activate intestinal epithelial cells (IEC) (Sugiura et al., 2013) the levels of antimicrobial peptide mRNA expression were evaluated two days after Y. enterocolitica infection. Expression of antimicrobial mRNA transcripts for angiogenin-4 (ANG4) and regenerating-islet derived protein 3-γ (REG-3γ) were significantly up-regulated in WT mice and unchanged in the TLR1−/− mice when compared to naïve controls (Fig 1D) and was specific to TLR1, as it was not observed in TLR6−/− mice (data not shown). By 70 days post-infection, the level of mRNA transcripts were similar between WT mice and uninfected controls, while there was a significant increase of the relative expression of these anti-microbial peptides in TLR1−/− mice (Fig 1D).
Dysregulation of antimicrobial peptide expression and inflammatory cytokine responses may consequently affect the composition of the commensal microbiota (Hooper et al., 2012). In order to determine whether there were changes in the microbial composition that may account for the induction of innate immune responses after the pathogen has been cleared we constructed and sequenced 16S rRNA clone libraries from the cecum of WT and TLR1−/− mice 70 days post-infection. Using a limited number of clones we observed an increase in the relative abundance of δ-Proteobacteria (family, Desulfovibrionaceae) while there was a decrease in Bacilli (family, Lactobacillaceae) in the cecum of TLR1−/− mice compared to WT controls (Fig 1E). These data were confirmed using qPCR, amplifying broad members of the following bacterial classes: Bacteroides, Clostridiales, Lactobacillus, γ- and δ-Proteobacteria (Fig 1F) (Barman et al., 2008; Winter et al., 2010). Altogether, these data suggest that the absence of TLR1 during acute GI infection leads to chronic immune activation and alterations in the composition of the commensal bacteria.
Altered microbiota in the absence of TLR1 confers inflammatory potential and increases susceptibility to tissue injury
Members of Desulfovibrionaceae are sulfate-reducing bacteria that have been shown to be associated with inflammation in mouse (Devkota et al., 2012) and human (Loubinoux et al., 2002); (Rowan et al., 2011) models of disease. In contrast, Lactobacillus bacteria are associated with important anti-bacterial (Heineman et al., 2012) and anti-inflammatory effects (Yan and Polk, 2011). To demonstrate that the altered microbiota, and not an overall defect in immunity conferred by the absence of TLR1 signaling, was responsible for the chronic inflammatory response observed following acute GI infection, we reconstituted WT germ-free (GF) mice with cecal contents from WT and TLR1−/− mice that were uninfected or 70 days post-infection. GF mice receiving cecal contents from TLR1−/− mice 70 days post-infection had significantly elevated IFN-γ and IL-17 and their respective transcription factors, RORC and TBET, in the distal ileum (not shown) and proximal colons (Fig 2A–B). In contrast, molecules more generally associated with immune-suppression and tolerance, such as IL-10 and mRNA transcripts of FOXP3 remained unchanged between GF mice reconstituted with cecal contents from infected TLR1−/− and WT mice (Fig 2A–B).
Figure 2. Altered microbiota drives an inflammatory phenotype and prevents intestinal healing.
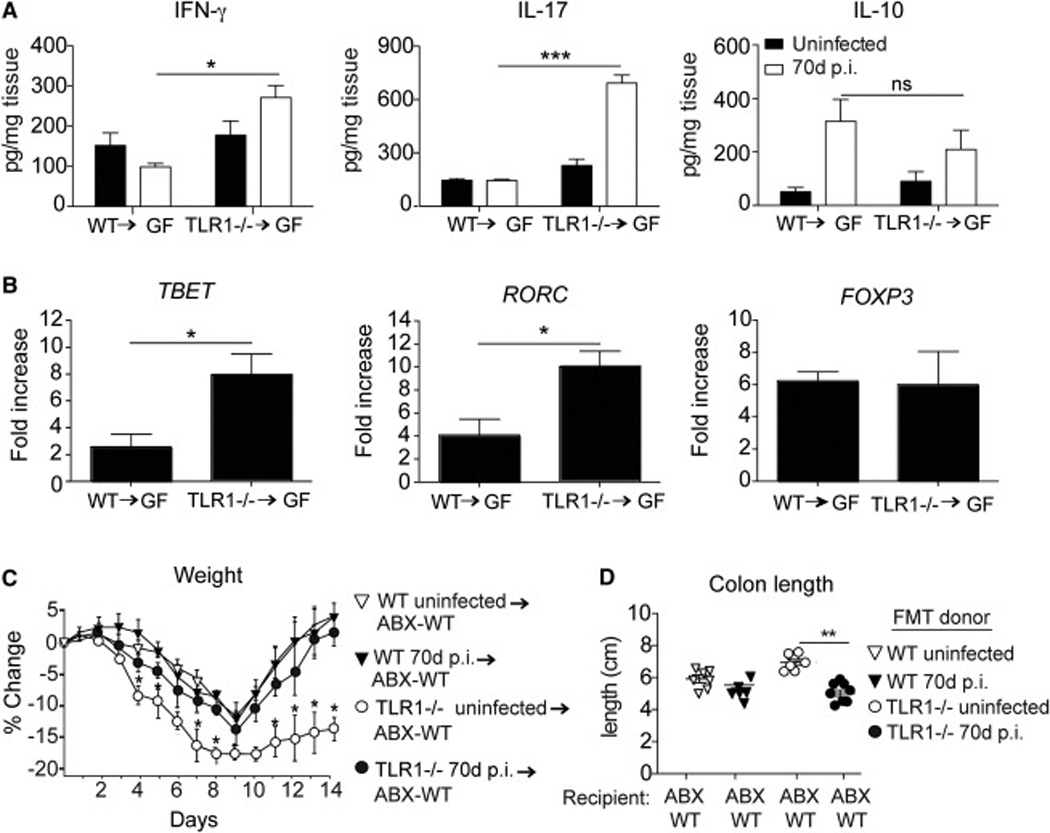
Germ-free (GF) mice were reconstituted with cecal contents from WT or TLR1−/− mice 70 days (70d) post-infection (p.i.). Two weeks after reconstitution mucosal scrapings were evaluated for indicated cytokines (A) and transcription factors (B). Data shown is the mean ± SEM protein concentration (A) or fold change (B) compared to untreated GF mice. Data is pooled from two independent experiments (n=6–7 mice/group). *, p < 0.05; **, p < 0.01; ***, p < 0.001 Student’s paired t-test. WT mice were treated with an antibiotic cocktail (ABX-WT) for two weeks and then reconstituted with cecal contents from WT or TLR1−/− mice 70 days post-infection. After two weeks the mice were placed on 2.5% dextran sodium sulfate DSS for 7 days followed by normal drinking water for 7 days. The percent change in weight of mice over the 14 days (C) and the individual length of the colon (D) are shown. Data is the average of n=7–9 mice collected from two separate experiments. *, p < 0.05 Student’s paired t-test.
An intestinal injury model was used to determine whether the increased inflammatory cytokines induced by the altered commensal bacteria would impact repair. Naïve WT mice were treated with broad-spectrum antibiotics (ABX-WT) for two weeks prior to reconstitution with WT or TLR1−/− cecal contents 70 days post-GI infection or uninfected WT and TLR1−/− controls. Two weeks following reconstitution, the mice were administered 2.5% dextran sodium sulfate (DSS) for seven days to cause acute epithelial injury, followed by seven days of normal drinking water to allow for repair. As predicted, ABX-WT mice receiving TLR1−/− microbiota post-infection had more severe weight loss and shorter colon lengths compared to the ABX-WT mice receiving cecal contents from WT mice post-infection (Fig 2C–D). Of note, there were no differences observed between ABX-WT mice receiving cecal contents from uninfected WT and TLR1−/− mice (Fig 2C). These data indicate that the altered microbiota from TLR1−/− mice is able to induce inflammation and damage independent of the expression of TLR1.
Deficient TLR1 signaling is associated with the development of anti-commensal immunity
Histological examination of the small and large intestine revealed a large lymphocytic infiltration in the proximal colon of TLR1−/− mice 70 days post-infection suggesting the activation of immune cells within the LP (Fig 3A). Re-stimulation of isolated colonic LPLs with cecal contents revealed only a small but insignificant amount of commensal-specific IFN-γ and IL-17 compared to the robust production produced from the LPLs of TLR1−/− mice (Fig 3B). Re-stimulation of the TLR1−/− LPL’s with D. desulfuricans (DSV), induced significantly elevated levels of IFN-γ and IL-17 almost equal to stimulation with the cecal lysate, whereas DSV stimulation had no impact in cytokine production from WT LPL’s (Fig 3B). In contrast, re-stimulation of the LPLs of both WT and TLR1−/− mice with Yersinia lysate had a small effect on cytokine production, perhaps suggesting the re-activation of memory cells (Fig 3B). IL- 22 production was not induced or significantly different between groups (Fig S1).
Figure 3. TLR1 signaling during acute GI infection prevents development of anti-commensal immunity.
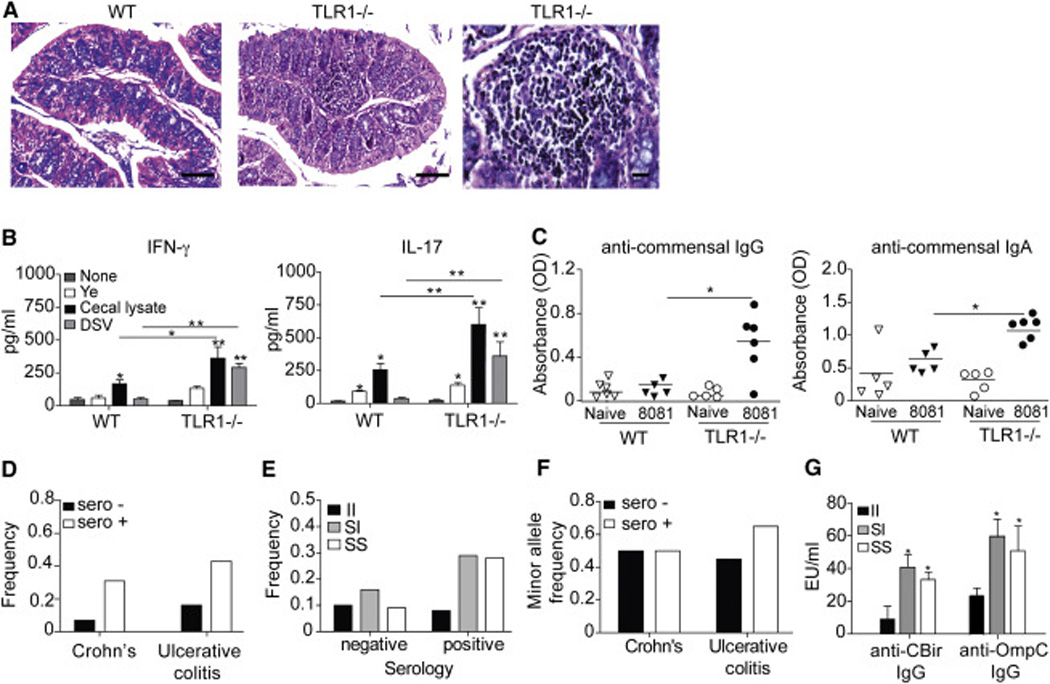
(A) Images of H&E stained proximal colon of WT (left) and TLR1−/− (center right) mice 70 days post-infection by Y. enterocolitica and TLR1−/− infiltrate (right) scale bars are 200 µM and 50 µM, respectively. Data shown are representative images from n=4 mice/group. (B) Mean concentration of IFN-γ and IL-17 production from colonic lamina propria lymphocytes (LPC) harvested 70 days after infection by Y. enterocolitica and re-stimulated with indicated bacterial lysate. Data is the mean ± SEM from three independent experiments (n=6–8 mice/group). *, p < 0.05; **, p < 0.01; ***, p < 0.001 Student’s unpaired t-test. (C) Optical density at 600 nm of individual mice (n=6 mice/group) assayed for anti-IgG and ant i-IgA serum reactivity against commensal lysate. *, p < 0.05 Student’s unpaired t-test. (D) The average frequency of anti-commensal serology positive and negative pediatric Crohn’s and ulcerative colitis patients. (E) The average frequency of TLR1 I602S genotypes in patients with positive and negative serology for anti-commensal antibodies. (F) The average minor allele (S-variant) frequency (MAF) in serology positive and negative Crohn’s disease and ulcerative colitis patients. (G) The average anti-CBir (flagellin) and anti-OmpC antibody concentrations in UC patients homozygous for the S-variant (TLR1 602S), homozygous for the non-variant (TLR1 602I) and heterozygous for the S-variant (TLR1 602S/I). Data is a composite of 220 patients in our pediatric IBD cohort that underwent serological testing. *, p < 0.05 Student’s paired t-test.
Chronic intestinal inflammation can disturb the epithelial barrier leading to bacterial translocation and the generation systemic antibody responses against the commensal bacteria (Zimmermann et al., 2012). Such responses have been observed in chronic intestinal diseases such as IBD (Prideaux et al., 2012) with up to 80% of IBD patients containing such antibodies (Mow et al., 2004). Analysis of the serum levels of anti-commensal antibodies showed that TLR1−/− mice had significantly more anti-commensal IgG and IgA than WT mice (Fig 3C). In humans, a common single nucleotide polymorphism (SNP) in the transmembrane region of TLR1 at position 602 replaces a hydrophobic isoleucine with a hydrophilic serine (Schumann and Tapping, 2007). Both the heterozygotic and homozygotic expression of the S-variant prevents TLR1 surface expression (Johnson et al., 2007) and causes defective NF-κB activation (Hawn et al., 2007), similar to the observed effects of TLR1-deletion in our animal model. Expression of the S-variant is common among Caucasian Americans and Europeans, with a minor allele frequency (MAF) of 0.75 in Europeans and 0.2 globally (Wong et al., 2010). As we observed that dysregulated TLR1 signaling in mice following acute GI infection leads to the development of anti-commensal antibodies, we hypothesized that expression of the S-variant may increase the incidence of anti-commensal antibodies, especially in persons with intestinal inflammation. To address this, we assessed expression of the TLR1 602S variant in DNA isolated from saliva samples of IBD patients from the Pediatric Gastroenterology Clinic at University of Chicago in a retroactive study (Fig 3D–3G). Patients that had gone for serological testing and had either positive or negative results for anti-commensal antibodies against bacterial surface proteins OmpC and CBir were further evaluated for expression of the variant. Patients who did not have serology performed were excluded from the analysis. We observed a higher frequency of serology positive patients with both Crohn’s disease (0.35) and Ulcerative colitis (UC) (0.42) than those that had negative serology for antibodies against OMPC and CBir (Fig 3D). Analysis of genotype frequencies demonstrated an increase in the frequency of serology positive patients with either homozygous or heterozygous expression of the S-variant (Fig 3E). While there was no difference in the expression of the S-variant (minor allele frequency (MAF)) between serology positive and negative patients with Crohn’s disease, UC patients with positive serology had a higher expression of the S-variant (minor allele frequency (MAF) of 0.64) (Fig 3F). Interestingly these same UC patients also had significantly higher concentrations of anti-commensal antibodies in their serum compared to patients expressing the non-variant allele (Fig 3G). Together, these data suggest that TLR1-deficiency in an inflamed gut is associated with a heightened anti-commensal antibody response in both mice and humans.
δ-Proteobacteria growth is dependent on tetrathionate respiration by Y. enterocolitica
CD11c+ DCs are important for the generation of protective IL-17-producing T cells during Y. enterocolitica infection and are recruited to the infected site via TLR1 signals from the IEC (Sugiura et al., 2013). However, DC are not required for immunity as ablation of the CD11c+ population does not cause complete mortality, instead protective immunity is shifted towards an increase in neutrophils that produce reactive oxygen species (ROS) (Autenrieth et al., 2012). Three days after Y. enterocolitica infection the Peyer’s patches were analyzed for Ly6G+ and CD11b+ cells to evaluate whether the protection observed in the TLR1−/− mice was being mediated by neutrophils. WT mice had an approximate three-fold increase in the frequency of Ly6G+CD11b+ cells compared to uninfected controls, whereas the TLR1−/− mice had a 10-fold increase in these cells (Fig 4A). In order to determine the contribution of neutrophils, as well as other Ly6G+ inflammatory monocytes, in protection of TLR1−/− mice against Y. enterocolitica infection these cells were depleted using a monoclonal antibody against Ly6G/Ly6C. While depletion had no effect in WT mice, it resulted in severe mortality in the TLR1−/− mice (Fig 4B).
Figure 4. Gastroenteritis in the absence of TLR1 causes an increase in Ly6G+CD11c+ cells.
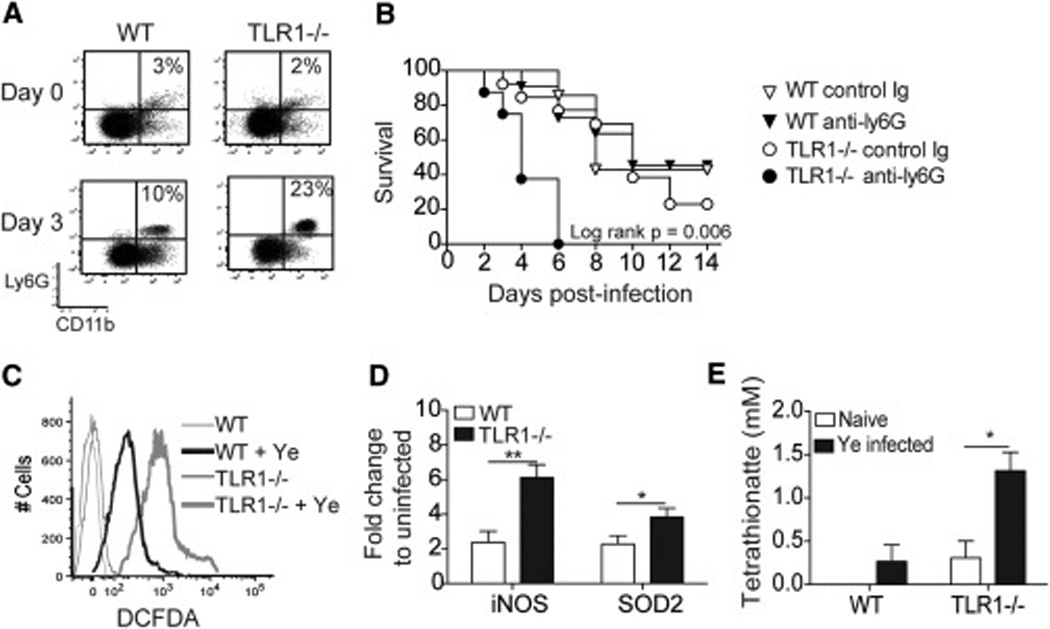
Analysis of Peyer’s patches from WT and TLR1−/− mice infected for 3 days with Y. enterocolitica. (A) Flow cytometry plots for Ly6G+CD11c+ cells. Data is representative from 5 individual mice collected from 2 separate experiments. (B) Percent survival of mice treated with depleting antibody against Ly6G during Y. enterocolitica infection. (C) Representative histogram of amine-reactive fluorescein (DCFDA) stained cells from the Peyer’s patches 3 days after infection. Data is a representative image from n=4 mice/group. (D) Fold increase of iNOS and superoxide dismutase (SOD2) from the Peyer’s patches 3 days after infection. Data is the mean ± SEM from three independent experiments (n=6–8 mice/group). *, p < 0.05; **, p < 0.01; Student’s unpaired t-test. (E) Tetrathionate levels determined by reverse-phase LC-MS from lamina propria of the terminal ileum. Data is the mean ± SEM from 3 individual mice/group.
Excessive neutrophil recruitment and elevated ROS production may have important consequences for the genetic and metabolic activity in Y. enterocolitica as well as the commensal microbiota. Similar to Salmonella typhimurium, Y. enterocolitica harbors a gene cluster that enables tetrathionate respiration, but this occurs only after commensal-produced thiosulfate is reduced to tetrathionate in the presence of ROS (Winter et al., 2010). Indeed, the increase in neutrophils observed in the TLR1−/− mice was also accompanied by a more robust expression of ROS (Fig 4C), ROS-associated gene expression (Fig 4D) and tetrathionate levels (Fig 4E) than WT mice.
To determine whether the increase in tetrathionate promotes its utilization by Y. enterocolitica and impacts the changes observed in the microbiota, we removed the ttrBCA genes from Y. enterocolitica strain 8081 (Fig S2A–C). Once the strain was created and verified using a tetrathionate agar plate test (Fig S2D), WT and TLR1−/− mice were infected with 1×105 cfu of 8081ΔttrBCA or the virulent 8081 strain. WT mice demonstrated a slight but insignificant increase in survival (Fig 5A) when infected with 8081ΔttrBCA, as well as a reduction in overall bacterial burden (Fig 5B). In contrast, infection with the 8081ΔttrBCA mutant significantly improved disease with 70% of mice surviving infection and a reduction in bacterial burden compared to TLR1−/− mice infected with the fully virulent 8081 strain (Fig 5A–B). Infection of the TLR1−/− mice with the 8081ΔttrBCA strain had no impact on ROS as indicated by gene expression of iNOS (Fig 5C) and tetrathionate levels (Fig 5D). In fact, tetrathionate was significantly increased in TLR1−/− mice infected with 8081ΔttrBCA, likely due to the inability of the mutant bacteria to utilize the anion and its accumulation in the tissue. Infection of the TLR1−/− mice with the 8081ΔttrBCA mutant significantly reduced the amount of δ-Proteobacteria (Fig 5E) suggesting that the tetrathionate respiration pathway, and not the increase in tetrathionate itself, is necessary for the outgrowth of δ-Proteobacteria (Fig 5E). In contrast, the abundance of Lactobacillus was not affected by the absence of tetrathionate respiration (Fig 5F).
Figure 5. Tetrathionate respiration is necessary for the outgrowth of D. desulfuricans in the absence of TLR1.
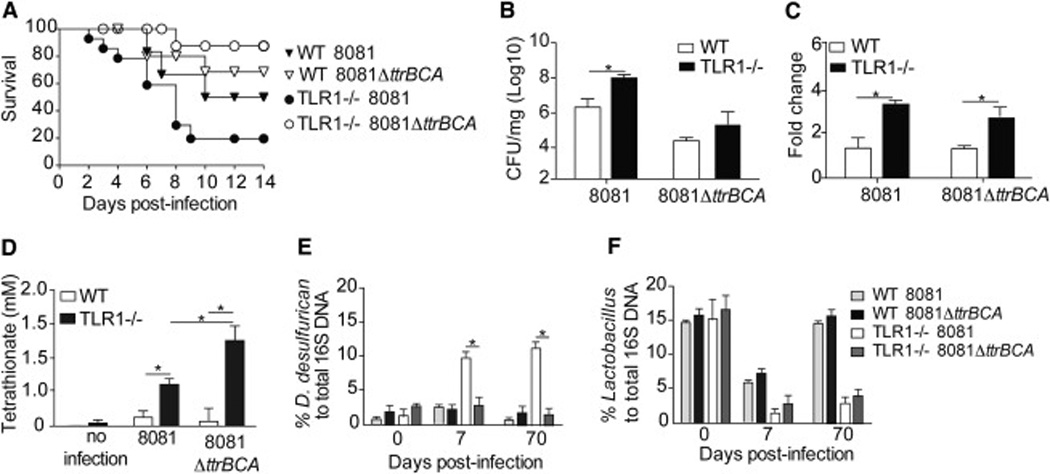
Analysis of WT and TLR1−/− mice after infection with Y. enterocolitica (8081) or the tetrathionate mutant (8081ΔttrBCA). (A) Survival (B) Bacteria load per mg of tissue in the LP 3 days post-infection. (C) Fold increase in mRNA transcripts of iNOS. Data is the mean ± SEM from two independent experiments (n=4–5 mice/group). *, p < 0.05; Student’s unpaired t-test. (D) Percent of total 16S DNA of D. desulfuricans and (E) Lactobacillus as determined by qPCR of the cecal contents of WT and TLR1−/− mice after infection with Y. enterocolitica or ΔttrBCA mutant. Data is the mean ± SEM pooled from two separate experiments (n=8–12 mice/group). *, p < 0.05; **, p < 0.01 Student’s paired t-test.
Targeted microbiota therapy during acute GI infection prevents the bloom of δ-Proteobacteria and anti-commensal immunity
Lactobacillus are important probiotic bacteria that have also been used to treat intestinal inflammatory disease in animal models (Yan and Polk, 2011), yet the therapeutic potential of these bacteria have not been as clearly delineated in human clinical trials (Kuisma et al., 2003). Initial observations showed a decrease in the abundance of Lactobacillus in TLR1−/− mice 70 days post-GI infection (Fig 1B). In the studies using the ΔttrBCA mutant, both the TLR1−/− and the WT mice had a decrease in the abundance of Lactobacillus during the acute infection, yet the Lactobacillus were able to re-colonize in WT but not in TLR1−/− mice (Fig 5F), independent of tetrathionate respiration. Using sets of primers for different Lactobacillus species, we examined their abundance over the course of the 70 days following Y. enterocolitica infection. We were able to detect L. johnsonii, L. reuteri, L. casei, L. murinus and L. intestinalis in uninfected WT and TLR1−/− mice, while L. crispatus was not present in the naïve animals (Fig 6A and Fig S3A). Interestingly, L. johnsonii, L. casei, L. murinus and L. intestinalis decreased during the acute infection but began to recolonize at approximately 14 days in both WT and TLR1−/− mice (Fig 6A and Fig S3A). L. reuteri and L. johnsonii decreased in both WT and TLR1−/− mice during the acute infection, and both were able to re-colonize in WT mice. In the TLR1−/− mice L. johnsonii but not L. reuteri was present at its pre-infection levels (Fig 6A). From these initial observations, the absence of L. reuteri in TLR1−/− mice may also contribute to the persistence of δ-Proteobacteria. To test this, TLR1−/− mice were infected with Y. enterocolitica and four days following infection, when the mice began to exhibit symptoms of gastroenteritis (blood in stool, weight loss and lethargy), they received 109 CFU of either L. johnsonii or L. reuteri. The mice received either probiotic or PBS treatment every other day for a total of 3 feedings and cecal levels of D. desulfuricans were assessed over the 70 days by qPCR. Probiotic treatment with L. johnsonii had no effect on the levels of D. desulfuricans (Fig 6B), while treatment with L. reuteri significantly reduced the amount of δ-Proteobacteria even 48 days after the last administration (Fig 6B). Reconstitution of antibiotic treated WT mice with the cecal lysates of L. reuteri treated TLR1−/− mice prevented the dramatic weight loss seen after DSS administration of mice receiving cecal contents from control infected TLR1−/− mice (Fig S3B). The specific restoration of L. reuteri during the acute GI infection prevented the weight loss and reduced bacterial burden (Fig 6C). Analysis of the anti-microbial peptide response (Fig 6D) and IL-22 cytokine levels (Fig S3C) showed that L. reuteri treatment had no effect during the acute infection but was able to reduce IL-22 and Reg3γ levels 70 days post-infection, likely due to the overall reduction on δ-Proteobacteria at this time point. L. reuteri treatment also reduced anti-commensal antibodies (Fig 6E) innate cytokines (Fig 6F) and TH17 responses (Fig 6G). These data suggest that a targeted approach in which restoring specific species of commensals lost during GI infection or inflammation can prevent and/or mitigate commensal outgrowth and anti-commensal immunity.
Figure 6. Targeted microbiota therapy during acute GI infection prevents growth of δ-Proteobacteria and generation of anti-commensal immunity in the absence of TLR1.
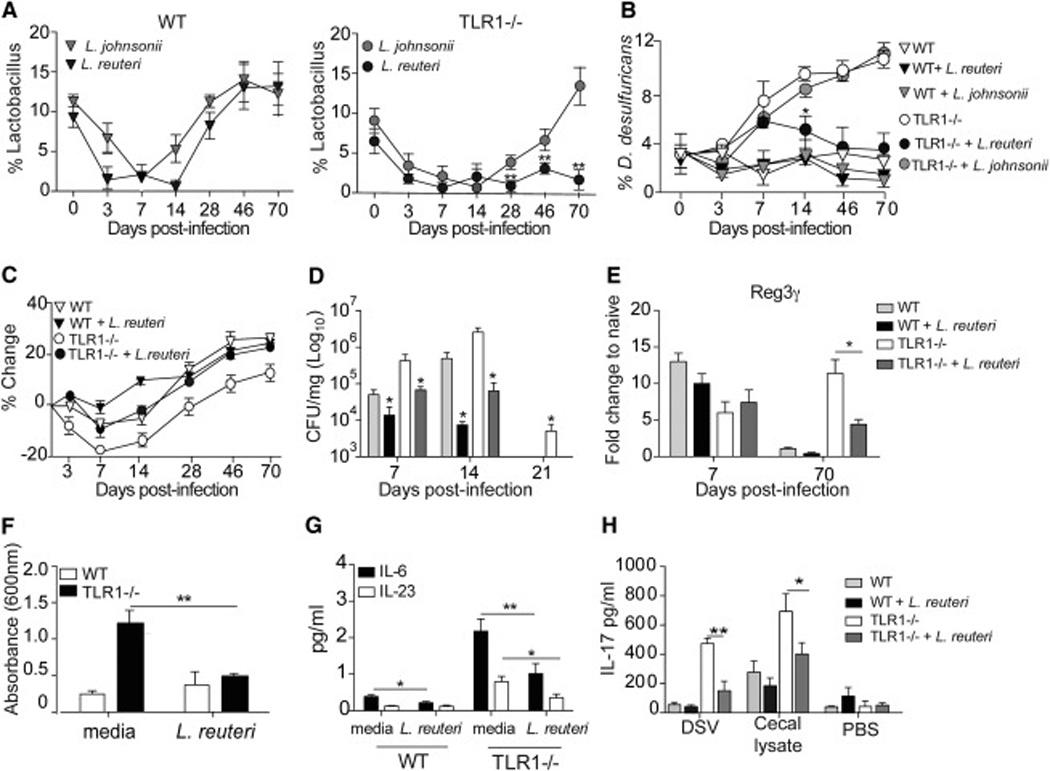
WT and TLR1−/− mice were infected with Y. enterocolitica. (A) Percent of Lactobacillus species to total 16S DNA over the 70 days following infection in WT (left) and TLR1−/− (right) mice. Data is the mean ± SEM of n=4–6 mice/group. (B) WT and TLR1−/− mice were infected with Y. enterocolitica and treated orally with 109 L. reuteri, L. johnsonii or media control on days 4, 6 and 8. D. desulfuricans DNA was measured by qPCR and compared to total 16S DNA. Data is the mean ± SEM pooled from two separate experiments (n=7–9 mice/group). (C) Average weight change, (D) Bacterial burden and (E) Reg3γ mRNA transcript levels of WT and TLR1−/− mice infected with Y. enterocolitica and treated with L. reuteri or media control on days 4, 6,and 8. Data is the mean ± SEM pooled from two separate experiments (n=5–7 mice/group). (F) OD at 600 nm of anti-commensal IgA antibodies in the serum and (G) average IL-6, IL-23 and (H) IL-17 cytokine production in the LP of L. reuteri treated WT and TLR1−/− mice. Data is the mean ± SEM pooled from two independent experiments (n=6 mice/group). *, p < 0.05; **, p < 0.01 Student’s paired t-test
Discussion
It is well established that the complex etiology of chronic intestinal inflammatory diseases is due to the involvement of environmental, microbial and immunological cues occurring within the genetic context of an individual. This scenario is further complicated by the close proximity of intestinal tissue to the trillions of resident microbiota. Despite research establishing an association of chronic illnesses and dysbiosis within the gut microbial community, it is still unclear whether these shifts are a cause of disease or a consequence of chronic inflammation. Much of our current knowledge regarding the changes in the composition of the microbiota is from studies inducing disease in a specific genetically deficient animal. Here, our data reveals that it is not any individual factor but the amalgamation of genetic and metabolic signals coming from both host and microbes that influence the extent of damage. In this report we demonstrate that genetic variations within the innate immune system reshape local tissue responses against an enteric pathogen creating an environment permissive to colonization by opportunistic commensal species, development of chronic inflammation and anti-commensal immunity.
An emerging concept is that the immune response can be scarred from an encounter with a pathogen. Specifically, work by Belkaid and colleagues (Fonseca et al., 2015) shows that in a small percentage of animals infected by a close relative to Y. enterocolitica, Y. psuedotuberculosis, there is a re-programming of mucosal DC responses and infiltration into local fat depots despite the absence of significant changes in the microbiota. The authors refer to this altered immunological state after infection as an immunological scar and predict that a subsequent trigger in these individuals may cause progression to chronic inflammatory disease. Our work provides evidence that the genetic context in which an environmental trigger occurs plays an essential role in the progression from an “immunological scar” to chronic inflammatory disease.
Here, we show that TLR1-deficient mice surviving Y. enterocolitica infection develop a neutrophil-mediated immune response setting the stage for the development of chronic inflammation. Previous studies have shown that reactive oxygen species (ROS) produced by neutrophils provide a growth advantage to Salmonella via the conversion of commensal derived thiosulfate to tetrathionate (Winter et al., 2010). We found that Yersinia-infected TLR1-deficient mice have elevated levels of ROS and tetrathionate that are abrogated upon depletion of Ly6G+ cells (data not shown) or when the genes involved in the reduction of tetrathionate by Y. enterocolitica are deleted. In both cases the bloom of δ-Proteobacteria is also abrogated linking alterations of the microbiota with a pathogenic metabolic pathway. These data suggest the intriguing possibility the underlying genetics of an individual will influence the virulence and metabolic pathways utilized by a pathogen, which will select the outgrowth of the commensal species that can best take advantage of these new metabolic products.
Additionally, our data indicate that Lactobacillus play an important role in the process of regulating the commensal microbiota. L. reuteri was the only Lactobacillus species that was unable to recover from the Yersinia-dependent decrease in the TLR1−/− mice. Interestingly, while infection with the tetrathionate mutant strain was able to prevent the δ-Proteobacteria bloom, it had no effect on levels of L. reuteri. These data suggest that the initial and sustained decrease of L. reuteri observed during Y. enterocolitica infection, while dependent upon TLR1 expression, is independent of tetrathionate respiration. The specific restoration of L. reuteri to mice during acute infection was able to impede the expansion of δ-Proteobacteria in contrast to mice receiving L. johnsonii. Whether the inability of L. reuteri to recolonize the TLR1−/− intestine is due to a competition amongst the Lactobacillus species, the δ-Proteobacteria species or another unidentified factor regulated by TLR1 is not yet known. Further, the exact mechanism by which L. reuteri inhibits the δ-Proteobacteria expansion and whether this is a specific attribute of L. reuteri is also not known. In a recent study L. reuteri could protect against mucosal fungal infections via IL-22 (Zelante et al., 2013), however in the present study analysis of IL-22 levels showed no difference between WT and TLR1−/− mice (SA Fig 3B) and was reduced in TLR1−/− mice following L. reuteri treatment (SA Fig 3B). Two other studies have shown promise using L. reuteri to reduce inflammation in vitro (Lin et al., 2008) and in pediatric patients with ulcerative colitis (Oliva et al., 2012). However, the anti-bacterial effect of L. reuteri on commensal species was not assessed in either study. Our data suggests therapeutic success is determined by whether an individual’s microbiota has contained the probiotic species or their intestinal microenvironment is at least conducive to its colonization.
An important physiopathological implication identified in this study is that disruption of a single gene is enough to cause progression to chronic inflammatory disease following a GI infection. This disease was characterized by the generation of anti-commensal immunity, infiltration of activated leukocytes in the mucosa and the production of IL-17, all of which are hallmarks of IBD (Kaser et al., 2010). These data also provide a framework that can help explain the association of IBD and enteric infections caused by members of the Proteobacteria family (Mukhopadhya et al., 2012). While the current view is that IBD is caused by dysregulated immunity against commensal bacteria, historically IBD was linked to enteric infections caused by different classes of Proteobacteria, such as Salmonella, Campylobacter and Yersinia (Mukhopadhya et al., 2012). Studies identifying Proteobacteria in IBD patients used either direct identification of the pathogen in the tissue or identification indirectly through pathogen-specific recall responses. Our data suggests that in a genetically susceptible individual the immune response against the enteric pathogen may be altered thereby preventing or reducing the pathogen-specific memory response. Thus patients that had been infected previously by a GI pathogen may not have been identified through evaluation of memory responses and the results within these reports re-evaluated.
The development of anti-commensal antibodies is thought to be a consequence of chronic intestinal inflammation that leads to the break down of the mucosal barrier and immune activation (Zimmermann et al., 2012). Our data suggests that the absence of TLR1 signaling during GI infection also leads to the development of systemic anti-commensal IgG and IgA. To understand whether TLR1 signaling may have an impact on the development of anti-commensal antibodies in human disease, we correlated the presence of anti-commensal antibodies and the expression of a common SNP in TLR1 that reduces surface expression (Johnson et al., 2007) and activation (Hawn et al., 2007; Schumann and Tapping, 2007). We found a significantly higher allele frequency of the TLR1 602 variant in UC patients with positive serology for anti-commensal antibodies compared to CD patients. Interestingly, overall serum concentrations of anti-commensal antibodies were significantly higher in UC patients with either hetero- or homo-zygous expression of the variant allele.
Taken all together, our study illustrates a complex relationship in which genetic polymorphisms within the innate immune system impact virulence and metabolic pathways used by enteric pathogens, thus resulting in shifts within the commensal microbiome. These data are interesting as genome-wide association studies of IBD identified many polymorphisms involved in innate sensing and handling of microbes (Duerr et al., 2006; Hampe et al., 2007; Rioux et al., 2007). Furthermore, our data suggest that using targeted microbiota therapy during acute GI infection may be a therapeutic strategy to prevent dysbiosis in individuals with these polymorphisms, while also preventing or treating chronic intestinal inflammatory disease.
Experimental Procedures
Mice
TLR1−/− mice and littermate wild type mice were used as controls and caged together throughout the duration of all experiments. All mice were maintained at University of Southern California. Experiments were performed following experimental protocol review and approval by the Institutional Biosafety Committee and the Institutional Animal Care and Use Committee.
Bacterial administration
Yersinia enterocolitica 8081 and ΔttrBCA were grown overnight at 26°C in Tryptic soy broth (BD Biosciences, San Jose CA), diluted to 2×106 CFU/mL in sterile PBS and 100 µl was administered by gastric gavagae. In some experiments when mice began exhibiting loose stools they were administered 100 µl of L. johnsonii and L. reuteri that had been grown overnight at 37°C in Lactobacilli MRS broth (EMD Chemicals, Gibbstown, NJ) and diluted to 1010 CFU/ by intragastric gavage.
Lamina propria isolation
Terminal small intestine or proximal colon was removed, flushed with ice cold PBS and 1 mM DTT and were shaken at 37°C in HBSS with 2 mM EDTA (Sigma- Aldrich, St. Louis, MO) and 2 mM DTT (Sigma-Aldrich). Tissue was then digested with collagenase type IV (Sigma) and supernatants were passed through 70 uM mesh filters (BD Biosciences, San Jose, CA) and washed in PBS.
Detection of bacteria using RT-PCR
Composition of the microbiota was analyzed as described (Barman et al., 2008; Winter et al., 2010). The DNA from cecal contents was extracted using the QIAamp DNA stool kit (Qiagen) and two microliters of bacterial DNA was used as a template. The 16S gene copy numbers per microliter of DNA was determined using plasmids as previously described (Table S1) (Winter et al., 2010) or specific bacterial primers designed against each bacteria (Table S2) and normalized against total 16S.
Microbiota reconstitution of germ-free mice and antibiotic-treated mice
Wild type mice were treated with a cocktail of five antibiotics (ABX-WT), in their drinking water for two weeks and received 150 µL of cecal contents from indicated donor by oral gavage every other day for a total of 3 total gavages. Germ-free mice received a single 150 µL gavage of cecal material and were maintained within a plastic film isolator 4 weeks post-conventionalization. Four weeks after reconstitution intestinal tissue was harvested from germ-free mice while four weeks post reconstitution ABX-WT received 2.5% dextran sodium sulfate (DSS) (Affymetrix, Santa Clara, CA) in their drinking water for 7 days followed by 7 days of regular water.
Anti-bacterial and commensal T cell and antibody responses
Lamina propria lymphocytes were restimulated with syngeneic antigen presenting cells (CD3 and CD19-depeleted splenocytes) pulsed with 10 µg/mL bacterial lysates and cytokines measured by ELISA 48 hours later. For antibody levels, serum (diluted 1:2) was added to cecal lysate (10 µg/mL) coated plates for 2 hours at room temperature followed by HRP-conjugated secondaries: anti-mouse IgA (Santa Cruz Biotechnology Inc, Santa Cruz, CA), rabbit anti-mouse IgG1, rabbit anti-mouse IgG2a, and rabbit anti-mouse IgM followed by anti-Rabbit HRP (BD Biosciences). TMB substrate (Dako, Carpinteria, CA) was used for detection and absorbance was read at 495 nm OD.
Anti-Ly6G treatment
Mice were treated i.p. with 500 µg anti-Ly6G (Gr-1) (RB6-8C5, Bio X Cell, West Lebanon, NH) or rat IgG2b isotype control (Bio X Cell) every other day from the day of infection for 8 days.
Tetrathionate measurement
The ileal mucosa was harvested at day 3 after infection and washed 5 times with ultra-pure water. All samples were centrifuged at 10,000 g for 10 min =, filter sterilized, and analyzed using ion pairing RP-LC-MS (HPLC-LTQ coupling (Surveyor HPLC module – Linear Ion Trap [ThermoFischer]; single reaction monitoring in negative mode).
Statistics
Paired and unpaired Student’s t-tests were used when noted in figure legends. Tests that had an interaction of p < 0.05 were considered significant. Log rank test was used for analysis of survival.
Supplementary Material
Acknowledgments
We thank Denise Chac and Robert Rankin for critical reading and editing of the manuscript. This research was supported by NIH grants K01082725 and R03DK097442 to RWD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Investigation, KK, SK, VL; investigation, visualization, JC, TM, SK and JB; Supervision, and methodology, BK, SK, EC, DA and GY; Conceptualization, methodology, writing, visualization and funding acquisition RWD.
References
- Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol. 2010;10:131–144. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ. New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Appl Environ Microbiol. 2006;72:5734–5741. doi: 10.1128/AEM.00556-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autenrieth SE, Warnke P, Wabnitz GH, Lucero Estrada C, Pasquevich KA, Drechsler D, Gunter M, Hochweller K, Novakovic A, Beer-Hammer S, et al. Depletion of dendritic cells enhances innate anti-bacterial host defense through modulation of phagocyte homeostasis. PLoS Pathog. 2012;8:e1002552. doi: 10.1371/journal.ppat.1002552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol. 2000;1:113–118. doi: 10.1038/77783. [DOI] [PubMed] [Google Scholar]
- Barman M, Unold D, Shifley K, Amir E, Hung K, Bos N, Salzman N. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun. 2008;76:907–915. doi: 10.1128/IAI.01432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnsen J, Jellbauer S, Wong CP, Edwards RA, George MD, Ouyang W, Raffatellu M. The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity. 2014;40:262–273. doi: 10.1016/j.immuni.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson A, Murray S, Divakar P, Burnaevskiy N, Pifer R, Forman J, Yarovinsky F. Microbial infection-induced expansion of effector T cells overcomes the suppressive effects of regulatory T cells via an IL-2 deprivation mechanism. J Immunol. 2012;188:800–810. doi: 10.4049/jimmunol.1100769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler B, Jiang Z, Georgel P, Crozat K, Croker B, Rutschmann S, Du X, Hoebe K. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu Rev Immunol. 2006;24:353–389. doi: 10.1146/annurev.immunol.24.021605.090552. [DOI] [PubMed] [Google Scholar]
- Cario E, Gerken G, Podolsky DK. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology. 2007;132:1359–1374. doi: 10.1053/j.gastro.2007.02.056. [DOI] [PubMed] [Google Scholar]
- Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;13:260–270. doi: 10.1038/nrg3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, Huot L, Grandjean T, Bressenot A, Delanoye-Crespin A, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest. 2013;123:700–711. doi: 10.1172/JCI62236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePaolo RW, Kamdar K, Khakpour S, Sugiura Y, Wang W, Jabri B. A specific role for TLR1 in protective T(H)17 immunity during mucosal infection. J Exp Med. 2012;209:1437–1444. doi: 10.1084/jem.20112339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature. 2012;487:104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira RB, Gill N, Willing BP, Antunes LC, Russell SL, Croxen MA, Finlay BB. The intestinal microbiota plays a role in Salmonella-induced colitis independent of pathogen colonization. PLoS One. 2011;6:e20338. doi: 10.1371/journal.pone.0020338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca DM, Hand TW, Han SJ, Gerner MY, Glatman Zaretsky A, Byrd AL, Harrison OJ, Ortiz AM, Quinones M, Trinchieri G, et al. Microbiota-Dependent Sequelae of Acute Infection Compromise Tissue-Specific Immunity. Cell. 2015;163:354–366. doi: 10.1016/j.cell.2015.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, Tobe T, Clarke JM, Topping DL, Suzuki T, et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature. 2011;469:543–547. doi: 10.1038/nature09646. [DOI] [PubMed] [Google Scholar]
- Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- Hand TW, Dos Santos LM, Bouladoux N, Molloy MJ, Pagan AJ, Pepper M, Maynard CL, Elson CO, 3rd, Belkaid Y. Acute gastrointestinal infection induces long-lived microbiota-specific T cell responses. Science. 2012;337:1553–1556. doi: 10.1126/science.1220961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawn TR, Misch EA, Dunstan SJ, Thwaites GE, Lan NT, Quy HT, Chau TT, Rodrigues S, Nachman A, Janer M, et al. A common human TLR1 polymorphism regulates the innate immune response to lipopeptides. Eur J Immunol. 2007;37:2280–2289. doi: 10.1002/eji.200737034. [DOI] [PubMed] [Google Scholar]
- Heimesaat MM, Bereswill S, Fischer A, Fuchs D, Struck D, Niebergall J, Jahn HK, Dunay IR, Moter A, Gescher DM, et al. Gram-negative bacteria aggravate murine small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii. J Immunol. 2006;177:8785–8795. doi: 10.4049/jimmunol.177.12.8785. [DOI] [PubMed] [Google Scholar]
- Heimesaat MM, Nogai A, Bereswill S, Plickert R, Fischer A, Loddenkemper C, Steinhoff U, Tchaptchet S, Thiel E, Freudenberg MA, et al. MyD88/TLR9 mediated immunopathology and gut microbiota dynamics in a novel murine model of intestinal graft-versus-host disease. Gut. 2010;59:1079–1087. doi: 10.1136/gut.2009.197434. [DOI] [PubMed] [Google Scholar]
- Heineman J, Bubenik S, McClave S, Martindale R. Fighting fire with fire: is it time to use probiotics to manage pathogenic bacterial diseases? Curr Gastroenterol Rep. 2012;14:343–348. doi: 10.1007/s11894-012-0274-4. [DOI] [PubMed] [Google Scholar]
- Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- Johnson CM, Lyle EA, Omueti KO, Stepensky VA, Yegin O, Alpsoy E, Hamann L, Schumann RR, Tapping RI. Cutting edge: A common polymorphism impairs cell surface trafficking and functional responses of TLR1 but protects against leprosy. J Immunol. 2007;178:7520–7524. doi: 10.4049/jimmunol.178.12.7520. [DOI] [PubMed] [Google Scholar]
- Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katakura K, Lee J, Rachmilewitz D, Li G, Eckmann L, Raz E. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Invest. 2005;115:695–702. doi: 10.1172/JCI22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuisma J, Mentula S, Jarvinen H, Kahri A, Saxelin M, Farkkila M. Effect of Lactobacillus rhamnosus GG on ileal pouch inflammation and microbial flora. Aliment Pharmacol Ther. 2003;17:509–515. doi: 10.1046/j.1365-2036.2003.01465.x. [DOI] [PubMed] [Google Scholar]
- Larsson E, Tremaroli V, Lee YS, Koren O, Nookaew I, Fricker A, Nielsen J, Ley RE, Backhed F. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut. 2012;61:1124–1131. doi: 10.1136/gutjnl-2011-301104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YP, Thibodeaux CH, Pena JA, Ferry GD, Versalovic J. Probiotic Lactobacillus reuteri suppress proinflammatory cytokines via c-Jun. Inflamm Bowel Dis. 2008;14:1068–1083. doi: 10.1002/ibd.20448. [DOI] [PubMed] [Google Scholar]
- Loubinoux J, Bronowicki JP, Pereira IA, Mougenel JL, Faou AE. Sulfate-reducing bacteria in human feces and their association with inflammatory bowel diseases. FEMS Microbiol Ecol. 2002;40:107–112. doi: 10.1111/j.1574-6941.2002.tb00942.x. [DOI] [PubMed] [Google Scholar]
- Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2:119–129. doi: 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Melgar S, Bjursell M, Gerdin AK, Svensson L, Michaelsson E, Bohlooly YM. Mice with experimental colitis show an altered metabolism with decreased metabolic rate. Am J Physiol Gastrointest Liver Physiol. 2007;292:G165–G172. doi: 10.1152/ajpgi.00152.2006. [DOI] [PubMed] [Google Scholar]
- Morgan ME, Koelink PJ, Zheng B, den Brok MH, van de Kant HJ, Verspaget HW, Folkerts G, Adema GJ, Kraneveld AD. Toll-like receptor 6 stimulation promotes T-helper 1 and 17 responses in gastrointestinal-associated lymphoid tissue and modulates murine experimental colitis. Mucosal Immunol. 2014;7:1266–1277. doi: 10.1038/mi.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mow WS, Vasiliauskas EA, Lin YC, Fleshner PR, Papadakis KA, Taylor KD, Landers CJ, Abreu-Martin MT, Rotter JI, Yang H, et al. Association of antibody responses to microbial antigens and complications of small bowel Crohn's disease. Gastroenterology. 2004;126:414–424. doi: 10.1053/j.gastro.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Mukhopadhya I, Hansen R, El-Omar EM, Hold GL. IBD-what role do Proteobacteria play? Nat Rev Gastroenterol Hepatol. 2012;9:219–230. doi: 10.1038/nrgastro.2012.14. [DOI] [PubMed] [Google Scholar]
- Oliva S, Di Nardo G, Ferrari F, Mallardo S, Rossi P, Patrizi G, Cucchiara S, Stronati L. Randomised clinical trial: the effectiveness of Lactobacillus reuteri ATCC 55730 rectal enema in children with active distal ulcerative colitis. Aliment Pharmacol Ther. 2012;35:327–334. doi: 10.1111/j.1365-2036.2011.04939.x. [DOI] [PubMed] [Google Scholar]
- Prideaux L, De Cruz P, Ng SC, Kamm MA. Serological antibodies in inflammatory bowel disease: a systematic review. Inflamm Bowel Dis. 2012;18:1340–1355. doi: 10.1002/ibd.21903. [DOI] [PubMed] [Google Scholar]
- Raffatellu M, George MD, Akiyama Y, Hornsby MJ, Nuccio SP, Paixao TA, Butler BP, Chu H, Santos RL, Berger T, et al. Lipocalin-2 resistance confers an advantage to Salmonella enterica serotype Typhimurium for growth and survival in the inflamed intestine. Cell Host Microbe. 2009;5:476–486. doi: 10.1016/j.chom.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan F, Docherty NG, Murphy M, Murphy TB, Coffey JC, O'Connell PR. Ileal pouch microbial diversity. Ann Surg. 2011;254:669. doi: 10.1097/SLA.0b013e3182306578. author reply 669–670. [DOI] [PubMed] [Google Scholar]
- Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjoberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol. 2010;11:76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaolalla R, Sussman DA, Ruiz JR, Davies JM, Pastorini C, Espana CL, Sotolongo J, Burlingame O, Bejarano PA, Philip S, et al. TLR4 activates the beta-catenin pathway to cause intestinal neoplasia. PLoS One. 2013;8:e63298. doi: 10.1371/journal.pone.0063298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage DC, Dubos R, Schaedler RW. The gastrointestinal epithelium and its autochthonous bacterial flora. J Exp Med. 1968;127:67–76. doi: 10.1084/jem.127.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt TM, Relman DA. Phylogenetic identification of uncultured pathogens using ribosomal RNA sequences. Methods Enzymol. 1994;235:205–222. doi: 10.1016/0076-6879(94)35142-2. [DOI] [PubMed] [Google Scholar]
- Schumann RR, Tapping RI. Genomic variants of TLR1--it takes (TLR-)two to tango. Eur J Immunol. 2007;37:2059–2062. doi: 10.1002/eji.200737604. [DOI] [PubMed] [Google Scholar]
- Stecher B, Robbiani R, Walker AW, Westendorf AM, Barthel M, Kremer M, Chaffron S, Macpherson AJ, Buer J, Parkhill J, et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007;5:2177–2189. doi: 10.1371/journal.pbio.0050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura Y, Kamdar K, Khakpour S, Young G, Karpus WJ, DePaolo RW. TLR1-induced chemokine production is critical for mucosal immunity against Yersinia enterocolitica. Mucosal Immunol. 2013;6:1101–1109. doi: 10.1038/mi.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigas S, Tsatsoulis A. Endocrine and metabolic manifestations in inflammatory bowel disease. Ann Gastroenterol. 2012;25:37–44. [PMC free article] [PubMed] [Google Scholar]
- Trulzsch K, Oellerich MF, Heesemann J. Invasion and dissemination of Yersinia enterocolitica in the mouse infection model. Adv Exp Med Biol. 2007;603:279–285. doi: 10.1007/978-0-387-72124-8_25. [DOI] [PubMed] [Google Scholar]
- Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, Ley R, Wakeland EK, Hooper LV. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay-Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV, Neish AS, Uematsu S, Akira S, Williams IR, et al. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest. 2007;117:3909–3921. doi: 10.1172/JCI33084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature. 2010;467:426–429. doi: 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SH, Gochhait S, Malhotra D, Pettersson FH, Teo YY, Khor CC, Rautanen A, Chapman SJ, Mills TC, Srivastava A, et al. Leprosy and the adaptation of human toll-like receptor 1. PLoS Pathog. 2010;6:e1000979. doi: 10.1371/journal.ppat.1000979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F, Polk DB. Probiotics and immune health. Curr Opin Gastroenterol. 2011;27:496–501. doi: 10.1097/MOG.0b013e32834baa4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young VB, Schmidt TM. Antibiotic-associated diarrhea accompanied by large-scale alterations in the composition of the fecal microbiota. J Clin Microbiol. 2004;42:1203–1206. doi: 10.1128/JCM.42.3.1203-1206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, Zecchi R, D'Angelo C, Massi-Benedetti C, Fallarino F, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. 2013;39:372–385. doi: 10.1016/j.immuni.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Zimmermann K, Haas A, Oxenius A. Systemic antibody responses to gut microbes in health and disease. Gut Microbes. 2012;3:42–47. doi: 10.4161/gmic.19344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.